

Summary
Background and Aims
The nuclear localization of β‐catenin, a mediator of canonical Wnt signaling, has been indicated in a variety of cancers and is frequently related to tumor progression and metastasis. Therefore, targeting β‐catenin is an attractive therapeutic strategy for cancers.
Methods
Herein, we identified a natural, small molecule inhibitor of β‐catenin signaling, BASI, and evaluated its therapeutic efficacy both in vitro and in orthotopic mouse models of glioma.
Results
BASI significantly suppressed proliferation and invasion and induced apoptosis in glioblastoma cells and resulted in the remarkable attenuation of orthotopic tumor growth in vivo. Furthermore, we found that BASI altered the expression of several microRNAs, which mediated the posttranscriptional silencing of β‐catenin expression either directly or indirectly through a von Hippel‐Lindau (VHL)‐mediated β‐catenin degradation pattern.
Conclusions
Taken together, our findings offer preclinical validation of BASI as a promising new type of β‐catenin inhibitor with a mechanism of inhibition that has broad potential for the improved treatment of glioblastoma.
Keywords: BASI, Glioblastoma, Inhibitor, microRNA, β‐Catenin
Introduction
Glioblastoma multiforme (GBM) is the most aggressive and common primary malignant brain tumor. It is characterized by high levels of proliferation, invasion, and migration. The standard treatment consists of maximal safe resection, followed by concurrent radiation therapy and chemotherapy; however, the prognosis remains poor, with an median survival of 14 months on average 1, 2. Genetic heterogeneity in GBM has been proposed to explain the limitations of current therapies and indicates a need for prognostic gene signatures 3. Therefore, identifying and comprehending the functional significance of genetic alterations that promote the GBM initiation and progression and subsequent development of novel therapeutics that target these alterations are crucial for developing effective therapies 4.
Currently, several pathways amenable to targeted therapy have been uncovered in GBM, and one of them is the Wnt/β‐catenin signaling pathway. Wnt/β‐catenin pathway is usually activated aberrantly and has been reported to be the driver of many human cancers including glioma 5. β‐Catenin is an oncogenic protein and acts as an intracellular signal transducer in the Wnt signaling pathway. Nuclear β‐catenin forms a transcriptional complex with TCF4, Lef‐1, and CBP to activate the downstream genes transcription, which is related to cell proliferation, differentiation, and apoptosis 6, 7. Our previous study indicated that the nuclear accumulation of β‐catenin was an emerging key regulator of the gliomas malignant progression and was associated with a poor prognosis, highlighting it as a potential therapeutic target 8. Furthermore, we found that the inhibition of β‐catenin transactivation by FH535 and aspirin increased the efficacy of TMZ chemotherapy for glioblastoma 9.
microRNAs, a class of endogenous noncoding small RNAs, modulate various biological processes by binding to partially complimentary sequences in the 3′‐untranslated regions (3′‐UTRs) of mRNA targets, which results in posttranscriptional inhibition and sometimes mRNA cleavage 10. The altered expression of some microRNAs is related to various neoplasms due to their interaction with many signaling pathways, especially the Wnt/β‐catenin pathway 11. microRNAs can regulate β‐catenin‐dependent transcription in some ways, including the direct inhibition of β‐catenin transcriptional activation and expression or the indirect suppression of positive or negative modulators of β‐catenin activity 12. On the basis of previous observations, we demonstrated that miR‐200a directly interacted with the 3′‐UTR of CTNNB1 (the gene that encodes β‐catenin) to suppress the Wnt/β‐catenin pathway and subsequently inhibit the growth of U251 glioblastoma cells 13. Additionally, the downregulation of miR‐21 and miR‐23b inhibited glioblastoma cell survival by inducing von Hippel‐Lindau (VHL) expression, leading to the suppression of the β‐catenin‐mediated Wnt pathway 14, 15.
In this study, we indicated that miR‐181d suppressed glioblastoma progression by directly targeting CTNNB1 and CREBBP (the gene that encodes CBP). Furthermore, we demonstrated that BASI, a small molecular inhibitor identified by previous high‐throughput screening 16, exhibited remarkable antiglioma effects through the alteration of miR‐21, miR‐23b, miR‐200a, and miR‐181d levels, which ultimately led to the blockage of β‐catenin transactivation. Consequently, BASI is capable of targeting β‐catenin signaling and is a potentially attractive antineoplastic agent that may be used in the future.
Materials and methods
Cell Culture and Treatment
Human glioblastoma cell lines (LN229, SNB19, U87, A172, and LN308) and a low‐grade glioma cell line H4 were purchased from ATCC (the American Type Culture Collection, Manassas, VA, USA). The cells were cultured in Dulbecco's modified Eagle's medium (Gibco, Uxbridge, UK) supplemented with 10% FBS (Gibco) and were incubated in a humidified 5% CO2 atmosphere at 37°C. BASI was obtained from the NCI diversity dataset, and a 10 mM stock solution was prepared in DMSO. The miR‐181d mimic and As‐miR‐181d were generated from GenePharma (Shanghai, China) and were transiently transfected with Lipofectamine 2000 at the dose of 200 pM at 70% confluence (Invitrogen, Carlsbad, CA, USA) as instructed by the manufacturer to overexpression or downregulation of miR‐181d expressions, respectively. Two hundred pico molar nonsense oligonucleotides was used as a nonsense control (NC). The glioma cells were treated with 40 μM BASI for 48 h for in vitro experiments.
3‐(4,5‐Dimethylthiazole)‐2,5‐diphenyltetrazolium bromide (MTT) Assay
U87 and LN229 cells were plated in 96‐well plates at 5 × 103 cells per well in a final volume of 100 mL and treated with BASI at a series of dose for 48 h. Cell proliferation was quantified by the MTT assay. The optical densities of the samples were measured at the wavelength of 570 nm. The cell survival rate was calculated as follows: BASI treatment group values/DMSO treatment group values ×100%.
Cell Cycle and Colony Formation Analysis
Cell cycle distribution was analyzed by propidium iodide (PI) staining and flow cytometry after 40 μM BASI or DMSO treatment for 48 h 17. For the colony formation analysis, 40 μM BASI‐ or miR‐181d mimic‐treated cells was plated in 6‐well plates at 200 cells per well and grown for 2 weeks. Afterward, the cells were washed twice with PBS, fixed in methanol, and stained with 0.5% crystal violet (Sigma, St. Louis, MO, USA). The colony formation assay was repeated at least three times.
Apoptosis and Transwell Invasion Assay
Cell apoptosis was quantified 48 h after treatment with 40 μM BASI or miR‐181d mimic using an annexin V‐FITC Apoptosis Detection Kit (BioVision, Palo Alto, CA, USA). Matrigel‐coated transwell inserts (BD Biosciences, Bedford, MA, USA) were used for the transwell invasion assay. Cells that passed through the Matrigel‐coated membrane were fixed in methanol and stained with 0.1% crystal violet. The average numbers of cells per field were determined by counting the cells in five random fields per well. All tests were performed in triplicate.
RNA Isolation and Quantitative Real‐time PCR Assay
Total RNA was extracted using Trizol reagent (Invitrogen). Real‐time quantification for gene and miRNA expressions was carried out using the TaqMan assay kit (Applied Biosystems, Foster City, CA, USA) and Hairpin‐it™ miRNAs qPCR Quantitation kit (GenePharma), respectively. All samples were normalized to internal controls, and fold changes were performed using the method. Specific RT‐PCR primers for CTNNB1 and CREBBP were obtained from Fulen Gene BiolEngineering Inc. (Guangdong, China). The primers for miR‐21, miR‐23b, miR‐200a, and miR‐181d were synthesized by GenePharma.
Western Blot Analysis
Total protein and nuclear proteins were isolated from cell lines by lysis in 1% Nonidet P‐40 lysis buffer. Western blotting assay was performed as previously described (8). The antibodies used were anti‐β‐catenin and anti‐CBP from Cell Signaling Technology, Danvers, MA, USA (1:1000 dilutions). The antibodies against histone H2A.X (Signalway Antibody, Pearland, TX, USA; 1:1000 dilution), β‐actin, or GAPDH (Santa Cruz Biotechnology, CA, USA; 1:1000 dilution) were used as a control. The band density of specific proteins was quantified after normalization with the density of GAPDH or histone H2A.X.
Luciferase Reporter Assay
To detect β‐catenin/TCF4 transcriptional activity, a pair of luciferase reporter constructs, TOP‐FLASH and FOP‐FLASH (Upstate Biotechnology, Lake Placid, NY, USA), were used. TOP‐FLASH consisted of three copies of the TCF binding sites upstream of a thymidine kinase promoter and the Firefly luciferase gene. FOP‐FLASH consisted of three mutated copies of TCF binding sites and was used as a control for measuring nonspecific reporter activation. Briefly, 105 cells were seeded in a 48‐well plate before transfection with TOP‐FLASH or FOP‐FLASH constructs. All transfections were performed using 0.8 mg plasmid and 2 μL Lipofectamine 2000. Forty‐eight hours after transfection, luciferase activity was detected with the dual‐luciferase reporter assay system (Promega, Madison, WI, USA). The Renilla luciferase activity was used as an internal control. Relative luciferase activity was reported as the fold induction after normalization for transfection efficiency. In addition, the wild type of pGL3‐CTNNB1‐3′UTR‐Subcloning and pGL3‐CREBBP‐3′UTR‐Subcloning were generated from GenScript (Nanjing, China) to detect whether CTNNB1 and CREBBP are functional targets of miR‐181d. The mutant reporter was generated from pGL3‐CTNNB1‐3′UTR‐Subcloning and pGL3‐CREBBP‐3′UTR‐Subcloning by replacing the predicted binding site of miR‐181d with restriction enzyme.
Immunofluorescence Staining and miRNA Locked Nucleic Acid In Situ Hybridization (FISH)
Immunofluorescence staining was conducted with an antibody against β‐catenin, and confocal images were acquired using an FV‐500 laser‐scanning confocal microscope and FluoView software (Olympus, Tokyo, Japan). To detect the expression of microRNAs, an antisense locked nucleic acid (LNA/DNA)‐modified oligonucleotide probe was used and in situ hybridization was performed with fluorescence in situ hybridization kit (CYBRDI, Xi'an, Shanxi, China) according to the manufacturer's protocol 18.
Nude Mouse Models and Treatment
Female BALB/c nude mice aged 4–5 weeks were purchased from the Animal Center of the Cancer Institute, Chinese Academy of Medical Science. Animal handling and experimental procedures were performed according to the regulations and internal biosafety and bioethics guidelines of Tianjin Medical University. Xenografts were initiated by subcutaneous injection of 5 × 106 U87 cells into the right hind flank region of each mouse, when tumors reached a volume of about 100 mm3; animals were randomized and divided into two groups of six mice per group. Each group was treated with 200 pM miR‐181d mimic or nonsense oligonucleotides in 10 μL Lipofectamine 2000 by local injection at multiple sites of tumors every 3 days for 24 days. Tumor lengths and widths were measured every 3 days using a digital caliper, and tumor volumes were calculated using the equation volume (mm3) = length × width2/2. At the end of the experiment, mice were anesthetized and sacrificed, and tumors were removed, photographed, and weighed. To establish intracranial gliomas, 0.5 × 105 U87 cells transduced with a luciferase‐encoding lentivirus were implanted into mice (n = 6 per group) stereotactically. Seven days postimplantation, BASI (40 mg/kg) or DMSO was administered by intraperitoneal injection every 2 days during the survival period. Fluc activity was detected by bioluminescence imaging (BLI) on days 0 (the first day of drug administration), 7, 14, 21, 28, 35, and 47 using the IVIS Spectrum Live Imaging System (PerkinElmer, Branford, CT, USA). Additionally, the body weight and overall survival time of mice were also monitored.
Hematoxylin and Eosin Staining and TUNEL and Immunohistochemistry Analysis
Tumors were removed and processed for paraffin embedding. Serial sections, obtained by cutting 4–5 μm of tissue, were analyzed by hematoxylin and eosin (H&E) staining and TUNEL for the presence of pathological and morphological characteristics. For immunohistochemistry (IHC) analysis, the sections were subject to deparaffinization and antigen retrieval in citrate buffer, blocked with 5% normal goat serum in PBS Triton‐X, and immunohistochemically analyzed using anti‐β‐catenin, anti‐CBP, and anti‐VHL antibodies (Cell Signaling Technology), respectively. The resultant immunostaining images were captured using a light microscopy (Olympus, Tokyo, Japan) computerized Image‐pro Plus6.0 system (Media Cybernetics Inc., Bethesda, MD, USA).
Statistical Analysis
Data were analyzed by Student's t‐test when comparing the two groups. All data were represented as the means ± SD, and differences were considered statistically significant at the level of P < 0.05.
Results
BASI Inhibits the Tumorigenic Properties of Glioma Cells
Utilizing a computational high‐throughput screening method for small molecules targeting miR‐21 based on the 3D structure of the Dicer binding site on pre‐miR‐21, we recently reported a selective and potent miR‐21 small molecular inhibitor, AC1MMYR2. Meanwhile, we found that BASI (known as NSC141562, and its chemical structure is shown in Figure 1A) also exhibited miR‐21 inhibition effect 16. To further evaluate whether BASI had tumor‐suppressive activity, we treated U87 and LN229 glioblastoma cells with BASI and tested the cells in functional assays.
Figure 1.
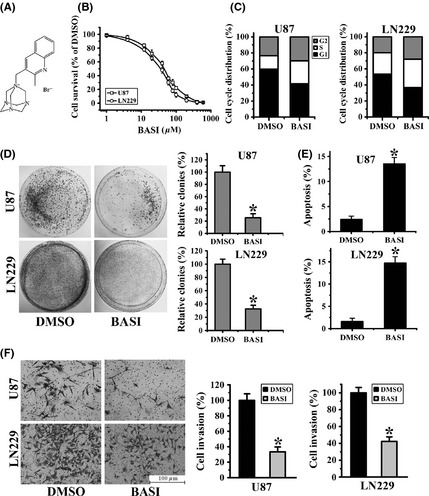
BASI inhibits glioma cells growth and invasion. (A) The chemical structure of BASI. (B) U87 and LN229 cells survival at 48 h following treatment with different doses of BASI, as determined by the MTT assay. (C) The cell cycle distribution was detected by flow cytometry after 40 μM BASI treatment. (D) Colony formation assays for U87 and LN229 cells (left). A relative quantification indicating the number of colonies formed by BASI‐treated cells, which was standardized to DMSO‐treated cells (right). (E) Effects of BASI and DMSO on cell apoptosis after 48‐h treatment. (F) Representative micrographs (left) and histograms (right) of transwell assays after treatment with BASI and DMSO. Data represent the mean ± SD of three independent experiments (*P < 0.05).
As analyzed by an MTT assay, BASI inhibited cell proliferation in a dose‐dependent manner, with approximately 50% inhibition at a concentration of 40 μM in both U87 and LN229 cells (Figure 1B). Moreover, cell cycle analysis showed that BASI treatment induced cell arrest in the G0/G1 phase (Figure 1C), decreased colony formation rates (Figure 1D), and promoted more cell apoptosis (Figure 1E) compared with the DMSO treatment group. In addition, as shown in Figure 1F, fewer glioma cells invaded across the Matrigel‐coated membranes following BASI treatment, which indicated that tumor cell invasion and migration were significantly inhibited by BASI. Collectively, these observations suggested that the tumorigenic properties of glioma cells, specifically proliferation, migration, and apoptosis, are affected by BASI.
BASI Blocks β‐Catenin Transcriptional Activity by Modulating the Expression Levels of Multiple microRNAs
Given that the Wnt/β‐catenin signaling pathway has recently emerged as a significant determinant of glioma initiation and progression, especially cell survival, proliferation, and migration 19, we next examined whether β‐catenin transcriptional activity was affected by BASI treatment. As shown in Figure 2A, TOP‐FLASH activity, which correlates with β‐catenin transcription activity, decreased by approximately 62 and 58% in BASI‐treated U87 and LN229 cells, respectively, compared with DMSO‐treated cells. There was no alteration in FOP‐FLASH activity. Furthermore, as nuclear expression of β‐catenin is related to its transcriptional activity, we investigated the nuclear levels and the subcellular localization of β‐catenin after BASI treatment. As shown in Figure 2B, nuclear expression levels of β‐catenin were reduced in BASI‐treated U87 and LN229 cells, as detected by Western blot. Additionally, confocal microscopy indicated that BASI treatment inhibited β‐catenin nuclear translocation from the cytoplasm, as evidenced by lower amounts of fluorescence intensity in the nucleus of BASI‐treated cells versus DMSO‐treated cells (Figure 2C).
Figure 2.
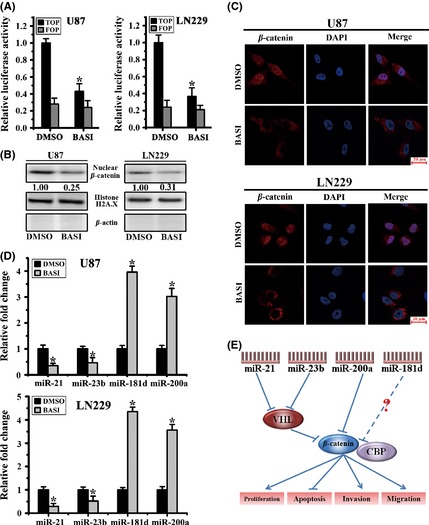
BASI blocks β‐catenin transcriptional activity via altering the expression of several miRNAs. (A) Luciferase reporter assays for U87 and LN229 cells cotransfected with Top and Fop plasmids (*P < 0.05). (B) Nuclear expression levels of β‐catenin were examined by Western blot after BASI treatment. Histone H2A.X and β‐actin were used as protein controls for the nuclear and cytoplasmic fractions, respectively. (C) β‐Catenin expression and subcellular location were confirmed by confocal microscopy. (D) miR‐21, miR‐23b, miR‐200a, and miR‐181d levels in cells treated with 40 μM BASI for 48 h (*P < 0.05). (E) The underlying mechanism of the antitumor effects of miR‐21, miR‐23b, miR‐200a, and miR‐181d occurs through the modulation of β‐catenin signaling.
As documented previously, miR‐21, miR‐23b, and miR‐200a are microRNAs that are commonly abnormally expressed in glioblastomas 13, 14, 15. Additionally, miR‐181d was reported to function as a tumor suppressor in glioma 20, 21. Therefore, we examined whether the expression levels of these microRNAs were altered after BASI treatment. We found that miR‐21 and miR‐23b were significantly decreased, whereas miR‐200a and miR‐181d levels were upregulated (Figure 2D). Because miR‐21, miR‐23b, and miR‐200a are all capable of modulating β‐catenin‐dependent transcription, we concluded that BASI inhibited β‐catenin transcriptional activity partly by altering the expression of these microRNAs. However, whether β‐catenin transcription was also modulated by miR‐181d requires further study (Figure 2E).
miR‐181d Binds the 3′‐UTRs of CTNNB1 and CREBBP and Blocks β‐catenin/CBP Transcriptional Activity
To examine miR‐181d expression in glioma cells, qRT‐PCR was used to measure miR‐181d levels in five glioblastoma cell lines and one low‐grade glioma cell line. Low expression levels of miR‐181d were observed in all glioblastoma cell lines. Specifically, miR‐181d expression was 1.54‐ to 3.65‐fold lower than that observed in a control H4 cell line (Figure 3A). To further assess whether β‐catenin transcription was modulated by miR‐181d, we used microRNA.org, a bioinformatic tool for microRNA target screening. We found that both CTNNB1 and CREBBP were putative targets of miR‐181d (Figure 3B). Therefore, we constructed the pGL3‐WT‐CTNNB1‐3′UTR, pGL3‐MUT‐CTNNB1‐3′UTR, pGL3‐WT‐CREBBP‐3′UTR, and pGL3‐MUT‐CREBBP‐3′UTR reporter plasmids to confirm whether CTNNB1 and CREBBP are direct targets of miR‐181d. Luciferase reporter assay indicated that upregulation of miR‐181d markedly reduced the pGL3‐WT‐CTNNB1‐3′UTR and pGL3‐WT‐CREBBP‐3′UTR luciferase activity in U87 and LN229 cells, both of which possess low endogenous miR‐181d expression. In contrast, reduced miR‐181d expression increased the luciferase activity of the reporter constructs in LN308 and A172 cells, which express relatively high levels of endogenous miR‐181d. No alterations in luciferase activity were observed when mutated reporter plasmids were used (Figure 3C). Furthermore, when cells were transfected with a miR‐181d mimic, we observed significantly decreased mRNA and protein levels of both CTNNB1 and CREBBP. In contrast, the inhibition of miR‐181d by transfection with As‐miR‐181d facilitated the elevated expression of these targets at both the mRNA and protein levels (Figure 3D and E).
Figure 3.
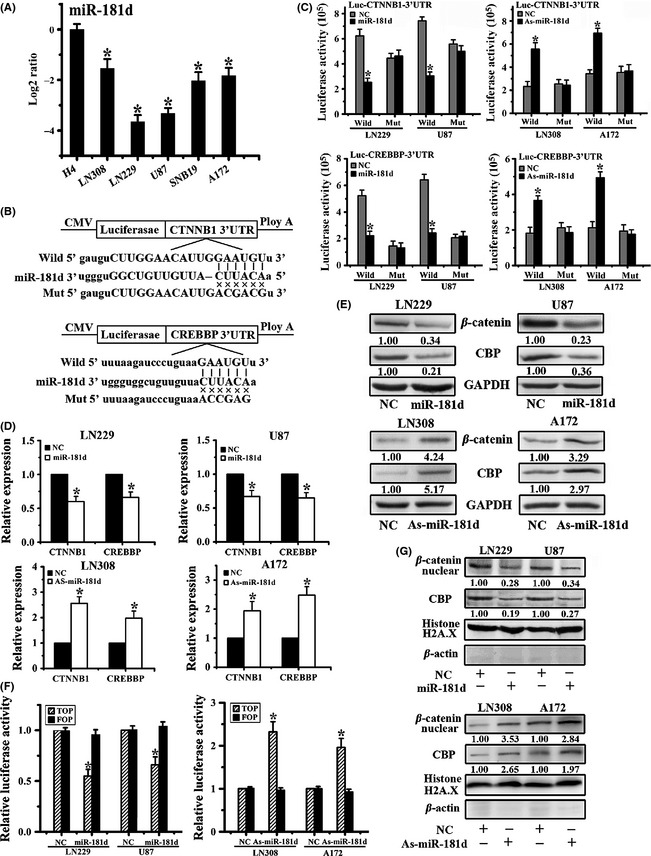
CTNNB1 and CREBBP are direct targets of miR‐181d, and ectopic miR‐181d expression disturbs β‐catenin/CBP transcriptional activity. (A) miR‐181d expression levels in U87, LN229, A172, LN308, and SNB19 glioblastoma cells and H4 cell. (B) Schematic representation of the putative binding sites in 3′‐UTRs of the CTNNB1 and CREBBP for miR‐181d and the designs of the wild‐type and mutant CTNNB1 or CREBBP 3′‐UTR‐containing reporter constructs. (C) Luciferase reporter assays after transfection with the wild‐type or mutant 3′‐UTRs of CTNNB1 or CREBBP. The mRNA (D) and protein levels (E) of CTNNB1 and CREBBP were detected after transfection with miR‐181d mimic or As‐miR‐181d. (F) Luciferase reporter assays were performed after Top or Fop reporter plasmid transfection following miR‐181d or As‐miR‐181d treatment. (G) Nuclear expression of β‐catenin and CBP was assayed by Western blot after transfection with an miR‐181d mimic or As‐miR‐181d. Histone H2A.X and β‐actin were used as protein loading controls for the nuclear and cytoplasmic fractions, respectively. Data represent the mean ± SD of three independent experiments (*P < 0.05).
We next investigated β‐catenin/CBP transcriptional activity after miR‐181d transfection. As shown in Figure 3F and G, ectopic miR‐181d expression resulted in a reduction in TOP‐FLASH activity and nuclear expression of β‐catenin and CBP. However, miR‐181d depletion increased TOP activity and the nuclear levels of β‐catenin and CBP, as detected by the TOP/FOP luciferase assay and Western blot, respectively. Taken together, these data strongly suggested that miR‐181d posttranscriptionally inhibits the transcriptional activity and expression of β‐catenin and CBP by directly binding to the 3′‐UTRs of their mRNAs.
Restoration of miR‐181d Suppresses Cellular Proliferation and Invasion and Induces Apoptosis
To investigate whether miR‐181d targeting β‐catenin signaling caused biological suppression of the malignant phenotype of glioma cells, we overexpressed miR‐181d in LN229 and U87 glioma cell lines and performed gain‐of‐function experiments in vitro. As shown in Figure 4A, cell cycle assay revealed that miR‐181d overexpression resulted in cell cycle block in the G0/G1 phase. Furthermore, the ability of both cell lines to form colonies was robustly compromised by miR‐181d transduction compared with corresponding NC cells (Figure 4B). Moreover, the number of apoptotic cells increased prominently after treatment with the miR‐181d mimic compared with NC (Figure 4C). In addition, as evidenced by the less number of tumor cells that invaded across the membranes precoated with matrigels, miR‐181d inhibited glioma cell invasion and migration activity (Figure 4D). Collectively, these data demonstrated that miR‐181d suppressed glioblastoma cell proliferation and invasiveness and induced apoptosis partly through the inhibition of β‐catenin/CBP transactivation.
Figure 4.
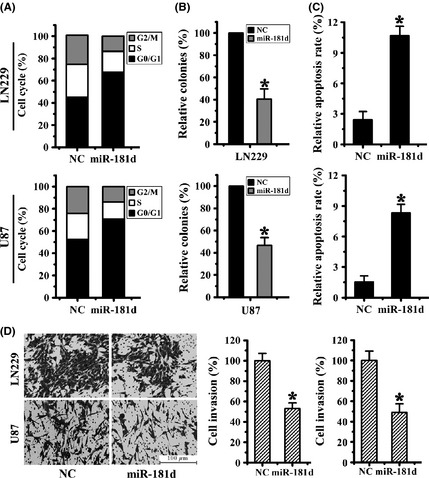
miR‐181d induced glioma cell proliferation and invasion suppression and cell apoptosis. (A) Representative histograms for cell cycle distribution after transfection with a miR‐181d mimic. (B) Representative pictures of colony formation assay of miR‐181d mimic or NC‐treated cells. (C) Staining with Annexin V and propidium iodide (PI) revealed greater amounts of apoptosis induced by miR‐181d compared with controls. (D) Representative histograms of transwell assays with U87 and LN229 cells after transfection with miR‐181d and NC. All data are representative of those obtained in three independent experiments (*P < 0.05).
miR‐181d Inhibits Glioma Tumor Growth in a U87 Subcutaneous Glioma Model
Next, we detected whether miR‐181d‐mediated β‐catenin/CBP inhibition was involved in tumorigenesis and invasiveness of glioma cells in vivo. An orthotopic glioma xenograft model was employed by xenotransplanting U87 cells. As early as 6 days postimplantation, the growth of transplanted tumors between the two groups became statistically significant. At the termination of the study, the average tumor volume and weight of the miR‐181d‐treated group were markedly reduced compared with NC (Figure 5A–C). At this point, the tumors were collected and prepared for H&E, FISH, and IHC assays. The tumor mass was greater, and chromatin staining was significantly stronger in the NC tumors compared with miR‐181d‐treated tumors. FISH detection indicated the re‐expression of miR‐181d after miR‐181d treatment. Moreover, miR‐181d‐treated gliomas displayed decreased nuclear expression of β‐catenin and CBP compared with NC tumors (Figure 5D). These data confirmed that re‐introduction of miR‐181d in vivo functions in a manner similar to that observed in vitro.
Figure 5.
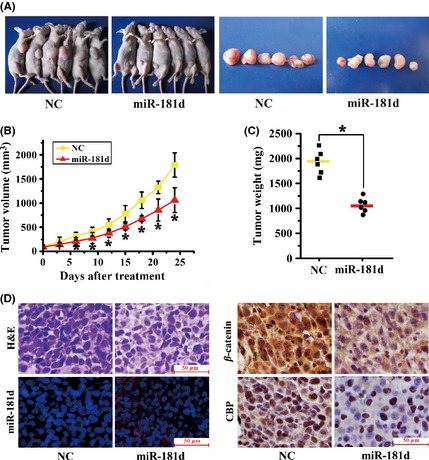
miR‐181d expression impairs U87 subcutaneous tumor growth in vivo. (A) Images of mice and tumors from miR‐181d and NC treatment groups. (B) Tumor volumes and (C) weights of mice in the miR‐181d and NC treatment groups in an in vivo proliferation assay. Data represent the mean ± SD (*P < 0.05). (D) Representative photomicrographs of H&E staining, FISH for miR‐181d, and immunohistochemistry (IHC) for β‐catenin and CBP on xenograft tumor sections.
BASI Inhibits Tumorigenesis and Invasiveness in a U87 Orthotopic Intracranial Model
Our in vitro experiments suggested that BASI was a potent inhibitor of β‐catenin signaling that suppressed tumor cell growth and invasion via the alteration of several microRNAs, including miR‐21, miR‐23b, miR‐200a, and miR‐181d. To further confirm this, an orthotopic glioma model was employed by xenotransplantation of luciferase‐expressing U87 glioma cells into the cortices of nude mice, followed by BASI or DMSO treatment. Tumor growth was monitored with a live animal BLI system during the experiment. As indicated in Figure 6A and B, tumor growth was markedly delayed in BASI treatment group relative to the animals receiving DMSO. The body weights of the BASI‐treated group increased constantly during their overall survival time, whereas the DMSO‐treated mice lost weight over this time (Figure 6C). Furthermore, comparing the overall survival of mice in the DMSO and BASI treatment groups demonstrated a substantial survival benefit for the BASI‐treated mice (Figure 6D). H&E staining and TUNEL analysis indicated that BASI induced more tumor cell apoptosis with decreased proliferation. Meanwhile, the miR‐21, miR‐23b, and β‐catenin expression levels were significantly reduced, whereas the miR‐200a, miR‐181d, and VHL expression levels were increased. Taken together, these data confirmed that BASI exhibited antitumor effects via the induction of microRNAs that subsequently mediated β‐catenin inhibition in vivo.
Figure 6.
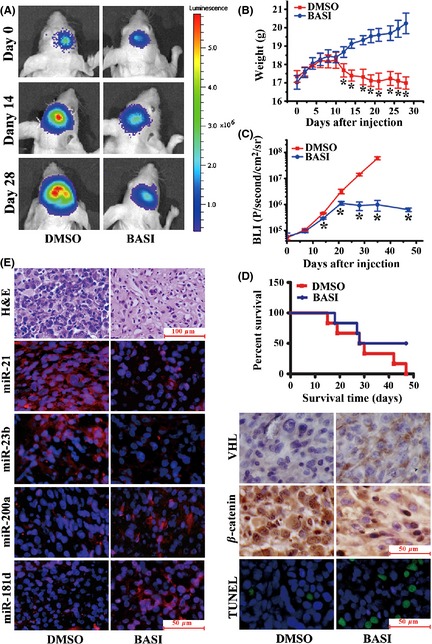
Antitumor effects of BASI in a U87 orthotopic intracranial model. (A) Representative pseudocolor bioluminescence images of mice that were implanted with intracranial tumors and treated intraperitoneally with 40 mg/kg BASI or DMSO on days 0, 14, and 28. (B) A plot depicting the Fluc activity measured bioluminescence imaging for the BASI and DMSO treatment groups. Data represent the mean ± SD (*P < 0.05). (C) A plot showing the body weight changes of nude mice bearing U87 orthotopic tumors. Weights were measured every 2 days following the intraperitoneal injection. Data represent the mean ± SD (*P < 0.05). (D) The overall survival of mice in the DMSO and BASI treatment groups for this same experiment. There was a substantial survival benefit for the BASI‐treated mice. (E) Representative photomicrographs of H&E staining, FISH for miR‐21, miR‐23b, miR‐200a, and miR‐181d, immunohistochemistry (IHC) for VHL and β‐catenin, and TUNEL staining on orthotopic tumor sections.
Discussion
Given the crucial role of the Wnt/β‐catenin pathway in cancer progression, including tumor initiation, tumor growth, cell death, and metastasis, the Wnt signaling pathway has emerged as a promising therapeutic target in cancers in recent years 19. Improved drug discovery platforms and updated technologies have contributed to the development of novel agents that alter Wnt signaling in preclinical models, numerous studies have reported antibody and siRNA approaches, and conventional agents that inhibit Wnt/β‐catenin signaling have shown considerable efficacy. Nonetheless, clinical application of these inhibitors remains elusive, demanding unremitting effort and new approaches 19, 22, 23. In the present work, we identified a small molecular compound, BASI, through the high‐throughput structure‐based silicon screening method. BASI is cytotoxic to glioblastoma cells at submicromolar concentrations. Incubation of glioblastoma cells with BASI led to cellular proliferation and invasion inhibition and increased apoptosis. BASI also exhibited antitumor efficacy and improved prognosis in glioma orthotopic models. Furthermore, β‐catenin transcriptional activity was repressed by BASI. Thus, we provided the preclinical evidence that BASI is a potentially valuable therapeutic β‐catenin inhibitor.
Numerous studies have revealed that microRNAs regulate the Wnt/β‐catenin signaling pathway to form a Wnt/β‐catenin–microRNA regulatory network 11, 12. Therefore, we investigated whether BASI could impact the expression of several microRNAs. In accordance with previous studies, we concluded that BASI reduced miR‐21 and miR‐23b levels and thus increased the levels of their functional target VHL 14, 15. BASI also directly upregulated miR‐200a levels 13 to inhibit β‐catenin activity. Furthermore, in the present study, we newly identified CTNNB1 and CREBBP as targets of miR‐181d in glioblastomas. CBP, as a transcriptional coactivator of β‐catenin via binding to its COOH‐terminal region, cooperates with β‐catenin to activate downstream genes expression 24. Targeted suppression of β‐catenin/CBP signaling has been demonstrated to ameliorate renal interstitial fibrosis 25, pulmonary fibrosis 26, and, importantly, to overcome therapy resistance and disease progression in acute lymphoblastic leukemia 27 and breast and bladder cancers 28. In this study, miR‐181d exerted a similar antitumor role in glioblastoma by inhibiting β‐catenin/CBP expression and activity through directly targeting the 3′‐UTRs of CTNNB1 and CREBBP. Therefore, we confirmed that BASI inhibited the Wnt/β‐catenin signaling pathway by upregulating miR‐181d, which subsequently blocked β‐catenin/CBP activity.
In contrast to the β‐catenin inhibitors, we previously used recombinant nucleic acids (siRNAs) and small molecule compounds (FH535 and aspirin) that directly suppressed β‐catenin expression or transcriptional activity of the β‐catenin/TCF4 complex 8, 9, 29; the underlying mode of action of BASI is to target β‐catenin via the alteration of several microRNAs. Because a single microRNA often has multiple targets and that many of these targets are related to diverse signaling pathways, their influence to gene expression can be amplified prominently. Therefore, we speculated that miR‐21, miR‐23b, miR‐200a, and miR‐181d might regulate β‐catenin activity indirectly by modulating other targets or signaling pathways. A microRNA‐mediated posttranscriptional silencing mechanism may play a critical role in regulating the expression of β‐catenin, which allows BASI to be a potent antitumor compound. Intriguingly, a recent study employed a molecular dynamics approach to demonstrate that NSC141562 may be a drug candidate for the treatment of the 2009 swine A/H1N1 virus 30.
In summary, the crosstalk between the Wnt/β‐catenin signaling pathway and microRNAs may help in the search for better strategies to treat tumors. Our preliminary evidence that BASI is capable of significant antitumor effects and is well tolerated in mice indicates that BASI may be an attractive antineoplastic agent that could be further developed in future studies.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported financially by grants from the China National Natural Scientific Fund (no. 81302185), the National High Technology Research and Development Program 863 (no. 2012AA02A508), the Natural Science Foundation of Tianjin Municipal Science and Technology Commission (no. 12ZCDZSY17300). The authors thank Ren Yu, Sun Ting, and Song Xin (Tianjin Research Center of Basic Medical Science, Tianjin Medical University) for their excellent technical assistance in the bioluminescent imaging assay and confocal microscopy imaging.
The first two authors contributed equally to this work.
References
- 1. Van Meir EG, Hadjipanayis CG, Norden AD, et al. Exciting new advances in neuro‐oncology: The avenue to a cure for malignant glioma. CA Cancer J Clin 2010;60:166–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tu Y, Gao X, Li G, et al. MicroRNA‐218 inhibits glioma invasion, migration, proliferation, and cancer stem‐like cell self‐renewal by targeting the polycomb group gene Bmi1. Cancer Res 2013;73:6046–6055. [DOI] [PubMed] [Google Scholar]
- 3. Shukla S, Pia Patric IR, Thinagararjan S, et al. A DNA methylation prognostic signature of glioblastoma: Identification of NPTX2‐PTEN‐NF‐κB nexus. Cancer Res 2013;73:6563–6573. [DOI] [PubMed] [Google Scholar]
- 4. Frattini V, Trifonov V, Chan JM, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet 2013;45:1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pu P, Zhang Z, Kang C, et al. Downregulation of Wnt2 and beta‐catenin by siRNA suppresses malignant glioma cell growth. Cancer Gene Ther 2009;16:351–361. [DOI] [PubMed] [Google Scholar]
- 6. CarcinomaPolakis P. Drugging Wnt signalling in cancer. EMBO J 2012;31:2737–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clevers H, Nusse R. Wnt/beta‐catenin signaling and disease. Cell 2012;149:1192–1205. [DOI] [PubMed] [Google Scholar]
- 8. Shi Z, Qian X, Li L, et al. Nuclear translocation of beta‐catenin is essential for glioma cell survival. J Neuroimmune Pharmacol 2012;7:892–903. [DOI] [PubMed] [Google Scholar]
- 9. Shi ZD, Qian XM, Liu CY, et al. Aspirin‐/TMZ‐coloaded microspheres exert synergistic antiglioma efficacy via inhibition of beta‐catenin transactivation. CNS Neurosci Ther 2013;19:98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Esquela‐Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer 2006;6:259–269. [DOI] [PubMed] [Google Scholar]
- 11. Sun X, He Y, Huang C, Ma TT, Li J. Distinctive microRNA signature associated of neoplasms with the Wnt/beta‐catenin signaling pathway. Cell Signal 2013;25:2805–2811. [DOI] [PubMed] [Google Scholar]
- 12. Huang K, Zhang JX, Han L, et al. MicroRNA roles in beta‐catenin pathway. Mol Cancer 2010;9:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Su J, Zhang A, Shi Z, et al. MicroRNA‐200a suppresses the Wnt/beta‐catenin signaling pathway by interacting with beta‐catenin. Int J Oncol 2012;40:1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang KL, Han L, Chen LY, et al. Blockage of a miR‐21/EGFR regulatory feedback loop augments anti‐EGFR therapy in glioblastomas. Cancer Lett 2014;342:139–149. [DOI] [PubMed] [Google Scholar]
- 15. Chen L, Han L, Zhang K, et al. VHL regulates the effects of miR‐23b on glioma survival and invasion via suppression of HIF‐1alpha/VEGF and beta‐catenin/Tcf‐4 signaling. Neuro Oncol 2012;14:1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shi Z, Zhang J, Qian X, et al. AC1MMYR2, an inhibitor of dicer‐mediated biogenesis of Oncomir miR‐21, reverses epithelial‐mesenchymal transition and suppresses tumor growth and progression. Cancer Res 2013;73:5519–5531. [DOI] [PubMed] [Google Scholar]
- 17. Zhang J, Huang K, Shi Z, et al. High β‐catenin/Tcf‐4 activity confers glioma progression via direct regulation of AKT2 gene expression. Neuro Oncol 2011;13:600–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Obernosterer G, Martinez J, Alenius M. Locked nucleic acid‐based in situ detection of microRNAs in mouse tissue sections. Nat Protoc 2007;2:1508–1514. [DOI] [PubMed] [Google Scholar]
- 19. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 2013;13:11–26. [DOI] [PubMed] [Google Scholar]
- 20. Wang XF, Shi ZM, Wang XR, et al. MiR‐181d acts as a tumor suppressor in glioma by targeting K‐ras and Bcl‐2. J Cancer Res Clin Oncol 2012;138:573–584. [DOI] [PubMed] [Google Scholar]
- 21. Zhang W, Zhang J, Hoadley K, et al. miR‐181d: A predictive glioblastoma biomarker that downregulates MGMT expression. Neuro Oncol 2012;14:712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yao H, Ashihara E, Maekawa T. Targeting the Wnt/beta‐catenin signaling pathway in human cancers. Expert Opin Ther Targets 2011;15:873–887. [DOI] [PubMed] [Google Scholar]
- 23. Watanabe K, Dai X. Winning WNT: Race to Wnt signaling inhibitors. Proc Natl Acad Sci USA 2011;108:5929–5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takemaru KI, Moon RT. The transcriptional coactivator CBP interacts with beta‐catenin to activate gene expression. J Cell Biol 2000;149:249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hao S, He W, Li Y, et al. Targeted inhibition of beta‐catenin/CBP signaling ameliorates renal interstitial fibrosis. J Am Soc Nephrol 2011;22:1642–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Henderson WR Jr, Chi EY, Ye X, et al. Inhibition of Wnt/beta‐catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci USA 2010;107:14309–14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gang EJ, Hsieh YT, Pham J, et al. Small‐molecule inhibition of CBP/catenin interactions eliminates drug‐resistant clones in acute lymphoblastic leukemia. Oncogene 2013. doi: 10.1038/onc.2013.169. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. He K, Xu T, Xu Y, et al. Cancer cells acquire a drug resistant, highly tumorigenic, cancer stem‐like phenotype through modulation of the PI3K/Akt/beta‐catenin/CBP pathway. Int J Cancer 2014;134:43–54. [DOI] [PubMed] [Google Scholar]
- 29. Yue X, Lan F, Yang W, et al. Interruption of beta‐catenin suppresses the EGFR pathway by blocking multiple oncogenic targets in human glioma cells. Brain Res 2010;1366:27–37. [DOI] [PubMed] [Google Scholar]
- 30. Mai BK, Viet MH, Li MS. Top leads for swine influenza A/H1N1 virus revealed by steered molecular dynamics approach. J Chem Inf Model 2010;50:2236–2247. [DOI] [PubMed] [Google Scholar]