

Summary
Aims
Glial cell‐derived neurotrophic factor (GDNF) is emerging as a potent neurotrophic factor with therapeutic potential against a range of neurodegenerative conditions including Alzheimer's disease (AD). We assayed the effects of GDNF treatment in AD experimental models through gene‐therapy procedures.
Methods
Recombinant lentiviral vectors were used to overexpress GDNF gene in hippocampal astrocytes of 3xTg‐AD mice in vivo, and also in the MC65 human neuroblastoma that conditionally overexpresses the 99‐residue carboxyl‐terminal (C99) fragment of the amyloid precursor protein.
Results
After 6 months of overexpressing GDNF, 10‐month‐old 3xTg‐AD mice showed preserved learning and memory, while their counterparts transduced with a green fluorescent protein vector showed cognitive loss. GDNF therapy did not significantly reduce amyloid and tau pathology, but rather, induced a potent upregulation of brain‐derived neurotrophic factor that may act in concert with GDNF to protect neurons from atrophy and degeneration. MC65 cells overexpressing GDNF showed an abolishment of oxidative stress and cell death that was at least partially mediated by a reduced presence of intracellular C99 and derived amyloid β oligomers.
Conclusions
GDNF induced neuroprotection in the AD experimental models used. Lentiviral vectors engineered to overexpress GDNF showed to be safe and effective, both as a potential gene therapy and as a tool to uncover the mechanisms of GDNF neuroprotection, including cross talk between astrocytes and neurons in the injured brain.
Keywords: Alzheimer disease, Gene therapy, Glial cell line‐derived neurotrophic factor, Neuroblastoma, Transgenic mice
Introduction
Alterations in the neurotrophic levels, due to either age, genetic background, or other factors have been reported to lead to neurodegeneration. Glial cell‐derived neurotrophic factor (GDNF) gene expression is reduced in the cerebral cortex of aged mice 1 and in the hippocampus of the murine model of accelerated aging SAMP8 2. Analysis of GDNF in human brain has demonstrated a decrease in substantia nigra in Parkinson's disease (PD) 3, but its regulation in Alzheimer's disease (AD) brain is poorly documented. However, GDNF decreases in plasma from patients with mild cognitive impairment and AD were found to be a precursor to progression into AD 4. Furthermore, the expression patterns of novel human GDNF isoforms were found to be deregulated in AD brains 5.
Neurotrophic factors have been assayed in aged rodents and primates in order to search for effective and harmless treatments so as to reverse mild memory loss of normal aging and also mitigate memory loss in pathological aging. As regards GDNF, we previously showed that lentiviral‐induced GDNF overexpression in hippocampal astrocytes improved the cognitive deficits of aged rats 6. The role of GDNF against AD pathogenesis has not been previously analyzed in any AD transgenic mouse model. However, two previous studies show encouraging protective effects of GDNF against Aβ‐induced neuronal cell death in rabbit hippocampus 7 and in cultured septal neurons 8. There is also suggestive evidence that several pharmacological agents of proposed use against AD 9 may partially work by way of GDNF enhancement in the brain 10. In the case of the clinical drugs amantadine and memantine, brain astrocytes are proposed as operating as the mediators of amantadine and memantine‐elicited neurotrophic effects through an increased production of GDNF by these glial cells 11, 12.
GDNF and its receptor complex GFRα‐1 are expressed in both neurons and astrocytes 13, although GDNF is mainly produced in neurons in the normal mature brain 14. The ability of activated astrocytes of up‐regulating GDNF production after brain injury is believed to play an active role in neuron survival and plasticity 14. In fact, activated astrocytes have been observed to up‐regulate neurotrophic factors, antioxidants, and other key molecules, all of which support neurons and oligodendrocytes survival as well as tissue repair 15. This cross talk between neurons and astrocytes after brain injury provides an avenue for new therapeutic approaches targeting these glial cells in age‐related neurodegeneration and AD.
In this study, we investigated the neuroprotective effects of GDNF against AD‐related neuropathology using a gene‐therapy approach in the 3xTg‐AD mouse model 16. This transgenic mouse model presents progressive AD‐like pathology, including Aβ deposits and hyperphosphorylated tau (p‐tau), oxidative stress, disorders of brain physiology and behavior, and cognitive loss 16, 17. As viral vectors can be engineered to selectively transduce astrocyte cells, we addressed the feasibility of potentiating the neuroprotective role of these glial cells in the mouse brain. Furthermore, we analyzed the neuroprotection afforded by lenti‐GDNF transduction against cell damage induced by conditioned overexpression of the 99‐residue carboxyl‐terminal (C99) fragment of the amyloid precursor protein (APP) in the human neuroblastoma MC65 18. MC65 is an established AD neuronal model, where cell death is induced by intracellular accumulation of C99 and its derived cleavage fragments such as Αβ‐peptide with a prominent cytotoxic role of oxidative stress 18, 19.
Methods
Animals and Treatment Groups
Male 3xTg‐AD mice and nontransgenic (NTg) mice with the same genetic background were used for this study. The 3xTg‐AD mouse strain harboring human transgenes for presenilin‐1 (PS1) with the M146V mutation, APP with the Swedish mutation, and tau with the P301L mutation was genetically engineered at the University of California Irvine 16. Animals were maintained in Macrolon cages under standard laboratory conditions of food and water ad libitum, 22 ± 2°C and 12‐h light/dark cycle. Genotypes were confirmed by PCR analysis of DNA obtained from tail biopsies.
At 4 months of age, 3xTg‐AD and NTg mice were assigned to either GDNF treatment by gene overexpression in the hippocampus or control group which expressed green fluorescent protein (GFP) gene. The treatment groups (n = 8–9) were thus: NTg‐GFP, NTg‐GDNF, Tg‐GFP, and Tg‐GDNF. Animals were individually housed in cages with filters, fulfilling level 2 safety requirements. The study was terminated at 10 months of age, following a 6 month period of gene overexpression.
Animal handling and procedures were approved by the local animal ethics committee (Ref: DAAM 4664, CEEA, UB), in accordance with Spanish legislation and the EU Directive 2010/63/EU for animal experiments.
Lentiviral Vectors
Recombinant lentiviral vectors encoding human GDNF were constructed for the specific transduction of mouse astrocytes in vivo or MC65 cells in vitro. The combination of the lyssavirus Mokola glycoprotein (Mokola‐G) pseudotype with the human cytomegalovirus promoter (CMV) allowed efficient transgene expression in astrocytes after intrahippocampal injection, as previously reported 6. Furthermore, the viral particles used for transduction of neuroblastoma MC65 cells were pseudotyped with the vesicular stomatitis virus G glycoprotein (VSV‐G). Vectors encoding GFP instead of GDNF were used as control vectors. We used the plasmids Flap‐CMV‐GDNF‐WPRE and Flap‐CMV‐GFP‐WPRE. For details of obtaining and handling the viral stocks see 6. The viral titers were obtained using a real‐time quantitative PCR (qPCR)‐based method previously described 20.
Levels of GDNF in the hippocampus of an additional group of mice injected with lenti‐GDNF and in the culture medium of transduced MC65 cells were determined by ELISA with the GDNF Emax ImmunoAssay System kit (Promega, Madison, WI, USA), following the manufacturer's instructions.
Surgical Procedures and Sacrifice
Mice were anesthetized with 10 mg/kg xylazine (Rompun 2%, Bayer, Leverkusen, Germany) i.p. and 80 mg/kg ketamine (Ketolar 50 mg/mL, Pfizer, Alcobendas, Madrid, Spain) i.p., and placed in a stereotactic apparatus. Bilateral infusions of lenti‐GDNF and lenti‐GFP were performed into the CA1 area of the mouse hippocampus by stereotactic procedures. Injections of lentiviral vectors were performed at a rate of 1 μL/min and at coordinates relative to bregma of −2.0 mm A/P, ±1.2 mm M/L, +2 mm V/D. Coordinates were selected in preliminary studies with fast green colorant injection (not shown). One microliter per side of the GDNF vector solution, containing 8.81 × 109 vector genomes/mL, was delivered to the application point with a 25‐gauge stainless steel cannula (Small Parts Inc., Miami, FL, USA) connected to a Hamilton syringe through a Teflon tube. The syringe was attached to a micro‐infusion pump (Bioanalytical systems Inc., West Lafayette, IN, USA). The cannula was left in position for 5 min after delivery to prevent the solution from surging back. The incision was sutured, and the mice were allowed to recover from anesthesia on a thermal pad and placed back into their cages. Control animals transduced with lenti‐GFP received 1 μL injections of 6.28 × 109 vector genomes/mL solution.
At the end of the study, animals were sacrificed by decapitation and the brains were removed and processed as described below.
Behavioral Testing
Testing of cognitive behavior was performed with the Morris water maze test (MWM), as previously described 17. MWM was used to determine acquisition and retention of a hippocampus‐related spatial task.
Additional tests to check for changes in general behavior were performed with the corner test and the open field test, in which the former is used to measure neophobia to a new home cage, and the latter to measure exploration, emotionality and locomotor behavior 17.
MC65 Cell Line
The stably transformed MC65 cell line is derived from the human neuroblastoma SK‐N‐MC and conditionally expresses a partial APP fusion protein including the C99 fragment 18. Once the promoter is activated by the absence of tetracycline (TC), the C99, and derived fragments accumulate intracellularly leading to Aβ‐related cytotoxicity 18, 19, 21, 22.
MC65 cells were routinely cultured in DMEM supplemented with 10% FBS (Gibco‐BRL, Carlsbad, CA, USA) (DMEM10) and 2 μL g/mL TC (Sigma, St Louis, MO, USA) (TC+). For activation, cells were seeded at a density of 1.2–1.5 × 105 cells/cm2 in 24‐well plates, without TC (TC−). After 2–3 days, the media was changed to OptiMEM (Gibco‐BRL) TC−. Control cells were maintained in TC+ media. For GDNF transduction, 0.75 μL of the vector solution containing 8.9 × 109 vector genomes/mL was added to each culture well 24 h before inducing C99 expression. Well cultures transduced with GFP received 5.55 × 109 vector genomes/mL of lenti‐GFP. Upon infection, culture wells contained 300 μL DMEM10. All experiments were terminated after 24 h of MC65 cell activation (i.e., after 48 h of viral infection). Results were obtained from 4 to 6 independent experiments performed in duplicate wells. Transduction efficiency was assessed by analyzing the number of cells that showed positive for GFP fluorescence after fixation (see below) and by staining all cell nuclei with Hoechst bisbenzimide (Sigma). For each experiment, more than 200 cells were counted in each of two microphotographs, using ImageJ software (http://rsb.info.nih.gov/ij/).
Cytotoxicity and Oxidative Stress Tests
Cell death in MC65 cell cultures was measured by the spectrophotometric assay of lactate dehydrogenase (LDH) leakage following standard procedures.
Intracellular reactive oxygen species (ROS) production of MC65 cells was determined using 2′,7′‐dichlorofluorescin diacetate (DCFH‐DA) (Molecular Probes, Leiden, the Netherlands). Cultures were loaded with 10 μM of DCFH‐DA and maintained in the cell incubator for 24 h. DCFH oxidation yielded the fluorescent molecule 2′,7′‐dichlorofluorescein (DCF). For a direct measure of intracellular DCF, cells were washed with PBS and lysed in 10 mM Tris–HCl with 0.5% Tween‐20. The homogenates were centrifuged at 10,000 × g for 10 min and the supernatants measured for DCF fluorescence in a plate reader (Spectramax Gemini XS, Molecular Devices, Wokingham, UK) at 485 nm excitation/530 nm emission. Results were expressed as the percentage of DCF compared to control cultures wells.
Immunofluorescence
At termination, the brain of all mice was bisected sagittally at the midline and a hemibrain of each mouse was processed for immunofluorescence. Tissue was fixed by immersion in 4% paraformaldehyde solution for 24 h, followed by an additional 24‐h period in fresh fixative solution. It was then cryopreserved in successive 10, 20, and 30% sucrose solutions and frozen on dye ice. Sagittal slices of 25 μm were stained with the following primary antibodies: anti‐GFP 1:100 from Abcam, (Cambridge, UK); anti‐GFAP, clone GA5, 1:400, from Sigma; anti‐GDNF 1:100 from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti‐Aβ, clone 6E10, 1:100, from Covance (Emerville, CA, USA); anti‐p‐tau, clone AT8 specific for double phosphorylated tau at Ser202/Thr205 (p‐tau), 1:50, from Pierce (Rockford, IL, USA); anti‐NeuN (1:100) from Millipore (Bedford, MA, USA); and anti‐phospho‐Ca2+ calmodulin‐dependent protein kinase II, α‐subunit, phosphorylated at Thr286 (p‐CaMKII), 1:100, from Abcam. Secondary antibodies used were as follows: Alexa Fluor 488 1:1000 and Alexa Fluor 546 1:1000 (Molecular Probes). The fluorescence intensity of the CA1 pyramidal neurons after staining with anti‐p‐CaMKII was measured by image analysis using ImageJ software. In brief, fluorescence intensities of histological slide images obtained in a Leica TCS SP2 confocal microscope were transformed to grayscale intensities, the background was subtracted, and the average values of the integrated optic densities of body neurons in each slide were registered.
MC65 cells grown onto glass coverslips were processed for immunofluorescence using anti‐APP C‐terminal fragment raised against residues 676–695 of APP695 (APP‐CTF) 1:100, from Covance. Cell nuclei were counterstained with Hoechst bisbenzimide.
Western Immunoblotting
Hippocampi were quickly dissected from the nonfixed hemibrain of each mouse and snap frozen in liquid nitrogen. Frozen tissues were crushed into a fine power under liquid nitrogen and aliquoted for subsequent Western blotting and qPCR analyses.
Freshly collected MC65 cells and frozen tissue powder were homogenized in ice‐cold RIPA buffer supplemented with protease and phosphatase inhibitors and processed for Western blot analysis by standard procedures. The following primary antibodies were used: anti‐Aβ 1:1000, clone 6E10, and anti‐APP‐CTF 1:1000 from Covance; anti‐p‐tau 1:1000, clone AT8, from Pierce; and anti‐actin (20–33, pan‐actin) 1:10,000 from Sigma. Secondary antibody was a horseradish peroxidase‐conjugated antibody (BD Amersham, Arlington Heights, IL, USA). Immunoreactive bands were detected with a chemiluminescence reaction and digitized. Densitometric results were normalized to actin.
Quantitative Real‐Time PCR
Total RNA was isolated from cells with TRIzol reagent (Life Technologies, Paisley, UK) and from mouse tissue using mirVana™ RNA Isolation Kit (Applied Biosystems Foster City, CA, USA) following the manufacturer's instructions. RNA samples were analyzed using a NanoDrop spectrophotometer (ND1000, Fisher Scientific, Waltham, MA, USA) and stored at −80°C until assay. Random‐primed cDNA synthesis was performed using the High‐Capacity cDNA Archive kit (Applied Biosystems). Gene expression was measured in an ABI Prism 7900HT qPCR system using TaqMan FAM labeled specific probes (Applied Biosystems). Gene expression of GDNF, disintegrin and metalloproteinase domain‐containing protein 10 (ADAM10), beclin 1 and neuritin was analyzed in MC65 cells, and that of GDNF, brain‐derived neurotrophic factor (BDNF), tyrosine receptor kinase B (TrkB) and NAD‐dependent deacetylase sirtuin‐1 (SIRT1) was analyzed in mouse hippocampus. Data were normalized to TATA‐binding protein (Tbp) gene expression.
Statistics
Results are expressed as mean ± SEM. Statistical analyses were performed using GraphPad Prism v4.02 (La Jolla, San Diego, CA, USA). Repeated‐measures ANOVA was used for the analysis of the acquisition curves in the MWM. Column statistics was used in the probe trial of the MWM. Results of the p‐CaMKII immunostaining were analyzed with one‐way ANOVA followed by Newman‐Keuls multiple comparison test. C99 Western blots immunolabelled with anti‐APP‐CTF antibody were analyzed with unpaired Student's t‐test. All other data were analyzed by two‐way ANOVA, while Bonferroni's post hoc test was used for comparison of means.
Results
Expression of GFP and GDNF in 3xTg‐AD Mice
The injection of lenti‐GFP and lenti‐GDNF particles into the dorsal CA1 hippocampus yielded a selective expression of transduced proteins in astrocytes, as demonstrated by the overlapping of GFAP immunostaining with either GFP or GDNF immunostaining (Figure 1A,B respectively). Protein expression was restricted to the injected area (Figure S1). Mean levels of GDNF determined by ELISA of the whole hippocampus of an additional group of 3xTg‐AD sacrificed 1 week after the injection were 0.7 ± 0.04 pg/mg tissue in GFP mice and 18.5 ± 2.02 pg/mg tissue in GDNF mice (n = 4). Similar levels of GDNF attained in the striatum through engineered macrophages, exerted neuroprotective effects in a mouse model of PD 23.
Figure 1.
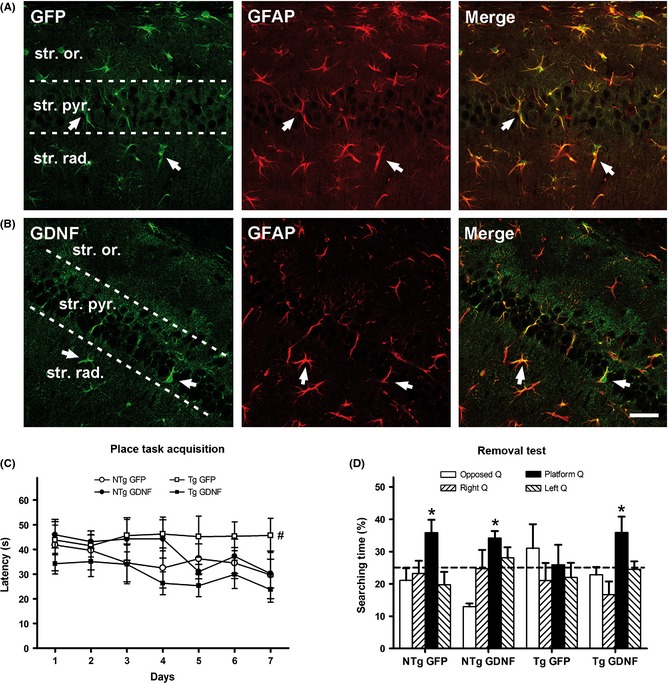
GDNF gene therapy and its effects on spatial learning and memory in the 3xTgAD mice. (A–B) Transgene expression of GFP and GDNF driven by lentiviral vectors stereotactically injected in the hippocampus. Both, GFP (A) and GDNF expression (B) in transduced astrocytes of the CA1 hippocampal area, were confirmed by co‐localization with the astrocyte marker GFAP in the merge images for GFP plus GFAP and GDNF plus GFAP, respectively. Representative confocal images of double immunohistochemistry. Abbreviations: str. or., stratum oriens; str. pyr., stratum pyramidale; str. rad., stratum radiatum. Arrows indicate the same astrocyte cells along each row. Scale bar = 50 μm. (C–D) Behavioral testing in the Morris water maze of 3xTgAD mice (Tg) and nontransgenic mice (NTg) treated with GFP or GDNF. (C) Latency to reach the escape platform during the 6 days of training indicating lower spatial learning in the Tg mice treated with GFP than in the other groups. (D) Time spent swimming in the platform quadrant of the pool (platform Q) when the platform was removed to test the retention of learning was different from chance in NTg mice and Tg mice treated with GDNF, but not in Tg‐GFP mice. Dotted line indicates chance performance. Values are the mean ± SEM, n = 8–9. Statistics: (C) #P < 0.01 compared to the acquisition curve of NTg‐GFP and Tg‐GDNF; (D) *P < 0.05 compared to chance value; see text for details.
Beneficial Effects of GDNF on Cognitive Behavior of 3xTg‐AD Mice
In the MWM, control 3xTg‐AD mice (treated with lenti‐GFP) showed severe cognitive loss as expected for this mouse strain 17. Interestingly, the treatment with lenti‐GDNF led to an improvement in both place task acquisition (Figure 1C) and retention of learning (Figure 1D). In the acquisition of learning, repeated‐measures ANOVA showed significant differences in the daily escape latencies between groups (P < 0.05), and Bonferroni test showed differences between control 3xTg‐AD mice and both control NTg mice and 3xTg‐AD treated with lenti‐GDNF (P < 0.01). In the probe trial, column statistics by one sample t‐test showed that all NTg mice and 3xTg‐AD treated with lenti‐GDNF spent more time than expected at random in the platform quadrant (P < 0.05), but not 3xTg‐AD treated with lenti‐GFP. No differences in average swimming speed were detected between groups (not shown). Namely, GDNF overexpression enhanced spatial learning and memory in 3xTg‐AD mice to a level similar to that of NTg mice.
As regards noncognitive behavior, GDNF overexpression in the hippocampus did not induce significant changes in the 3xTgAD phenotype of behavioral and psychological symptoms of BPSD‐like dementia 17. Selected results are shown in Figure S2. As BPSDs are mainly driven by AD pathology in the cortical and basal ganglia areas, these results showed a lack of GDNF effects beyond the hippocampus target area and also refuted the idea that GDNF could generate unwanted effects on general behavior.
Neuroprotective Changes of GDNF in the Hippocampus Tissue
A significant Aβ load in the hippocampus of 3xTg‐AD mice was demonstrated in immunostained pyramidal neurons (Figure 2A) and in Western blots of homogenized tissue (Figure 2B). Treatment with GDNF barely decreased Aβ deposits. With respect to tau pathology, the presence of p‐tau in hippocampus pyramidal neurons (Figure 2C) and in homogenized tissue (Figure 2D) was partially reduced after GDNF overexpression. However, ANOVA showed only a significance of mouse genotype (P < 0.01) and no significant effect of GDNF treatment in both pathologies.
Figure 2.
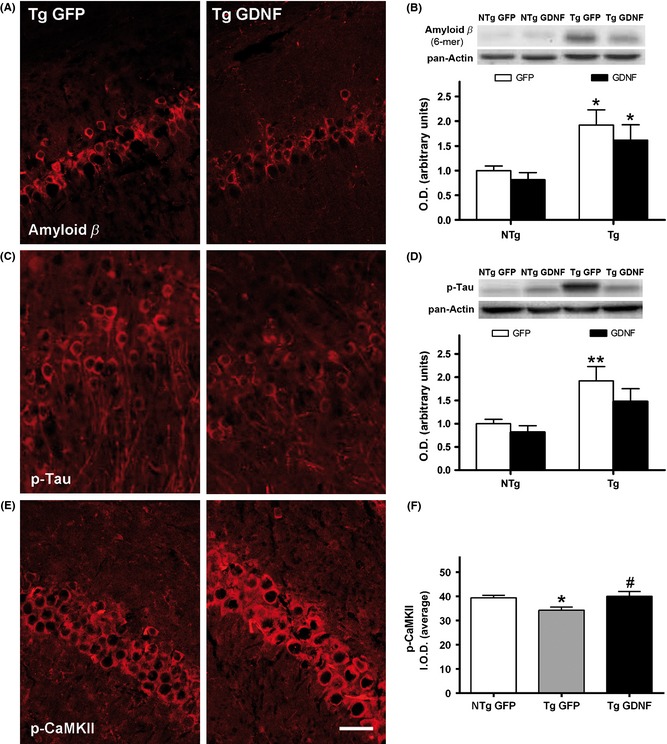
Amyloid and tau pathology and synaptic plasticity status in the hippocampus of 3xTg‐AD mice (Tg) treated with GFP or GDNF. (A) Representative images of immunostaining of intraneuronal amyloid β deposits in the CA1 pyramidal cell layer. (B) Levels of amyloid β oligomers shown by Western blot of the whole hippocampus in Tg and nontransgenic mice (NTg). (C) Representative images of immunostaining of intraneuronal p‐tau deposits in the CA1 pyramidal cell layer. (D) Levels of p‐tau shown by Western blot of the whole hippocampus. (E) Representative images of immunostaining of p‐CaMKII in the CA1 pyramidal neurons. (F) Intensity of p‐CaMKII staining, see text for details. O.D., optical density of Western blots in (B, D); I.O.D., integrated optical density of neurons in (F). Values are the mean ± SEM, n = 8–9. Statistics: *P < 0.05, **P < 0.01 compared to nontransgenic mice (NTg) with the same treatment; #P < 0.05 compared to Tg‐GFP. Scale bar = 50 μm.
Analysis of p‐CaMKII immunostaining showed reduced levels of this signaling enzyme in the CA1 pyramidal neurons of the 3xTg‐AD mice when compared with NTg mice, whereas an upregulation to normal physiological levels was observed in 3xTg‐AD mice treated with lenti‐GDNF (Figure 2E,F) (ANOVA, P < 0.05).
The RNA quantification showed a significant increase of GDNF gene expression in the lenti‐GDNF transduced mouse hippocampi that was paralleled by an increase of BDNF (Figure 3A,B, respectively). The expression of the BDNF receptor TrkB was decreased in these mice following chronic upregulation of BDNF (Figure 3C). Gene expression of the histone deacetylase involved in longevity and neuroprotection SIRT1 was tested for a possible enhancement but, on the contrary, GDNF was found to actually induce a decrease of this deacetylase after chronic overexpression (Figure 3D). ANOVA showed a significance of GDNF treatment (P < 0.01 for GDNF and SIRT1; P < 0.001 for BDNF and TrkB) and no effect of mouse genotype in all analyzed genes.
Figure 3.
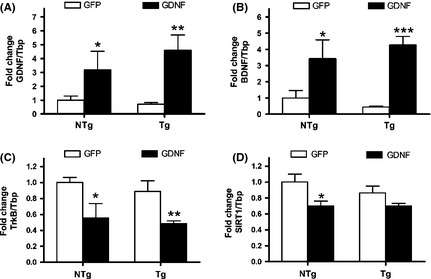
Gene expression induced by GDNF treatment in the hippocampus of 3xTg‐AD mice (Tg) and nontransgenic mice (NTg). RNA levels of GDNF (A), BDNF (B), TrkB (C), and SIRT1 (D) were obtained by quantitative real‐time PCR. GDNF overexpression by lenti‐GDNF induced a chronic increase of BDNF. Values are the mean ± SEM, n = 6–8. Statistics: *P < 0.05, **P < 0.01, and ***P < 0.001 compared to the same strain with a GFP treatment.
Cytoprotective Effects Induced by GDNF in MC65 Cells
The infection of MC65 cultures with lenti‐GFP viral particles demonstrated high efficiency of transduction with 93 ± 2% (n = 6) of cells showing GFP fluorescence (Figure 4A). Incubation of MC65 with lenti‐GDNF induced an elevated secretion of GDNF to the culture media measured by ELISA of 1865 ± 57 pg/mL as compared to that of control cultures of 36 ± 4 pg/mL (n = 6). Gene expression of GDNF in transduced cells is shown in Figure 4B (ANOVA, effect of GDNF treatment P < 0.001). Immunostaining with anti‐APP‐CTF antibody showed an increase in C99 expression after MC65 activation and a tendency toward decreased immunoreactivity after GDNF transduction (Figure 4C).
Figure 4.
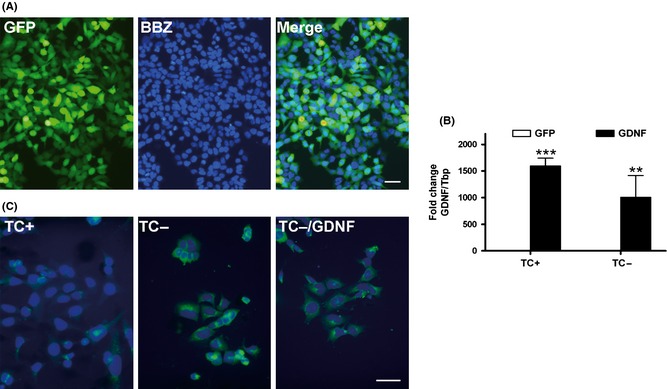
MC65 neuroblastoma characterization after GFP and GDNF transduction. (A) GFP fluorescence after transduction with lenti‐GFP is shown in most cells, as identified by nuclear counterstaining with Hoechst bisbenzimide (BBZ). (B) GDNF gene expression after transduction with lenti‐GDNF, determined by quantitative PCR analysis. (C) Representative immunostaining images of MC65 cells with anti‐APP C‐terminal fragment antibody in nonactivated cell cultures in the presence of tetracycline (TC+), activated for C99 generation in the absence of tetracycline (TC−), and effect of lenti‐GDNF reducing C99 accumulation (TC−/GDNF). Values are the mean ± SEM, n = 4–6. Statistics: **P < 0.01, and ***P < 0.001 compared to GFP. Scale bars = A, 50 μm; B, 20 μm.
Western blot analysis of MC65 cells showed activation of the expression of C99 regardless of the culture medium (DMEM10/TC− or OptiMEM/TC−), with the two antibodies used (anti‐APP‐CTF and anti‐Aβ antibodies; see representative membrane images in Figure 5A,B, respectively). Additional increases of Aβ aggregates, detected with anti‐Aβ‐specific antibody, were only found in cells activated in OptiMEM/TC− (Figure 5B). A nonspecific 37‐kDa band was detected with anti‐Aβ antibody. Therefore, we analyzed the effect of GDNF transduction on a major band corresponding to C99, two upper bands of Aβ oligomers with approximate molecular weights of 18 kDa and 25 kDa, and a lower band probably corresponding to an aggregate of Aβ dimers and cleaved CTFs (Aβ/CTF) previously described 24, 25. Cells transduced with GDNF showed a significant decrease of C99 immunolabelled with anti‐APP‐CTF antibody (Figure 5A, Student's t‐test), whereas the results with anti‐Aβ antibody showed higher variability and the absence of statistical significance for C99. Furthermore, GDNF transduction induced a decrease of Aβ oligomers and Aβ/CTF (Figure 5B). ANOVA indicated an effect of the factor GDNF treatment on the levels of Aβ oligomers (P < 0.001) and CTFs (P < 0.001), and no effect of the factor APP fragment species (Figure 5B).
Figure 5.
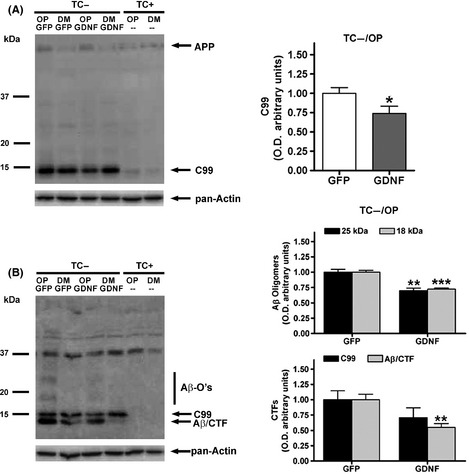
Western blot analyses of C99 and amyloid β levels in MC65 cells after GFP and GDNF transduction. Lysates from cells cultured in OptiMEM (OP), activated for C99 generation in the absence of tetracycline (TC−) and transduced with lenti‐GFP or lenti‐GDNF were analyzed with an anti‐APP‐C‐terminal antibody (A) and an anti‐amyloid β‐specific antibody (clone 6E10) (B). Lysates from cells cultured in growth medium DMEM10 (DM) in the absence of tetracycline (TC−) and also lysates from nonactivated cells grown in the presence of tetracycline (TC+) were added as reference. The levels of amyloid β oligomers (Aβ‐O's) and aggregates of amyloid β dimers and C99 shorter fragments (Aβ/CTF) were increased in OP/TC− medium and decreased by GDNF as compared to GFP. GDNF also decreased C99 levels. Values are the mean ± SEM, n = 4–6. Statistics: *P < 0.05, **P < 0.01 and ***P < 0.001 compared to the GFP treatment in activated TC− cells.
The activation of the neuroblastoma MC65 cells to generate the C99 induced an increase of hydroperoxide and cell death, as previously described 19. GDNF transduction suppressed both oxidative stress and cytotoxicity (Figure 6A,B, respectively). ANOVA indicated an effect of GDNF treatment factor in both DCF and LDH results (P < 0.05), and of cell activation factor (P < 0.001) and interaction between both factors (P < 0.001) in LDH.
Figure 6.
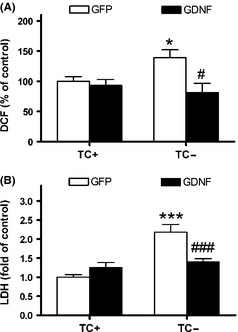
Cytoprotective effects of GDNF in MC65 cells. (A) GDNF treatment protected from oxidative stress measured by the dichlorofluorescein (DCF) generation method. (B) Cell death measured by LDH leakage was protected by GDNF. Values are the mean ± SEM, n = 4–6. Statistics: *P < 0.05 and ***P < 0.001 compared to the nonactivated state with the same treatment (control, TC+; activated for generation of C99, TC−); #P < 0.05 and ###P < 0.001 compared to the GFP treatment in activated TC− cells.
Gene expression of several genes tested to further explore GDNF mechanisms did not give significant changes. Namely, the relevant α‐secretase ADAM10, the autophagy regulator beclin 1, and neuritin which is involved in neurite outgrowth (not shown).
Discussion
Gene expression of GDNF mediated by lentiviral vectors was able to induce neuroprotective effects in both, in vivo and in vitro experimental models of AD. In vivo, the chronic overexpression of GDNF in astrocytes from the dorsal CA1 hippocampus protected against cognitive loss in 3xTg‐AD mice, as demonstrated by their good performance in the MWM. Spatial cognitive abilities require the dorsal hippocampal function to be intact, with the target CA1 area being particularly crucial for spatial learning 26 and memory 27. The sustained secretion of GDNF by transduced astrocytes exerted neuroprotective effects on the hippocampal neurons. In vitro, GDNF overexpression induced a potent cytoprotective action in MC65 neuroblastoma. There was a recovery from cell death induced by the intracellular generation of C99 in this AD cell model.
The brain neuroprotection driven by lenti‐GDNF transduced astrocytes confirmed previous results obtained in cognitively deficient aged rats 6. It also supports the mediation of GDNF secreted by astrocytes in the neuroprotection afforded by several anti‐AD drugs 10, 11. There was a small and nonsignificant reduction of amyloid and tau pathologies in vivo, whereas activation of BDNF gene expression was substantial. BDNF has already been demonstrated to induce a broad neuroprotection in several models of AD, including reversion of synapse loss and restoration of learning and memory in APP‐Tg mice and prevention of death induced by Aβ in primary neuron cultures 28. In APP‐Tg mice, BDNF had no effect on amyloid plaque load and its neuroprotection was reported as mediated through amyloid‐independent mechanisms 28. The decreased levels of BDNF reported in the hippocampus and cortex of AD patients are suggested to contribute to the atrophy and cognitive dysfunctions 29. Similarly, a reduction of p‐CaMKII in the hippocampus and cortex of AD patients is believed to contribute to memory impairment 30. Activation of the α‐subunit of CaMKII by autophosphorylation at Thr286 is essential for the induction of long‐term potentiation (LTP) and for consolidating learning through synaptic plasticity changes 31. Furthermore, cognitive deficiencies in APP/PS1‐Tg mice 32, in SAMP8 mice 33, and in diabetic rats 34 have been associated with p‐CaMKII reduction. Therefore, recovery of p‐CaMKII levels after GDNF treatment may contribute to reversing the synaptic plasticity impairment of pyramidal neurons in the hippocampus of 3xTg‐AD mice. Upregulation of BDNF by GDNF could be at least partially mediated through the signaling pathway CaMKII—cyclic AMP‐response element binding protein cAMP (CREB)—BDNF, in which p‐CaMKII phosphorylates CREB which, in turn, binds to the promoter of BDNF. A mediator of GDNF‐induced BDNF expression, as recently described in nigrostriatal dopaminergic neurons, is the transcription factor pituitary homeobox 3 (Pitx3) 35. Both GDNF and BDNF are increased in the hippocampus of mice by a known plasticity enhancer therapy such as physical exercise 36, 37, 38, although the sequence of gene activation has not been analyzed. Furthermore, similar enhancement of both neurotrophic factors have been described in astrocyte cultures treated with the protective polyphenol resveratrol 39. In addition to the neuroprotection attained through BDNF upregulation, GDNF itself is a potent neurotrophic factor. Therefore, both factors could have worked in concert to improve synaptic plasticity and hippocampal circuitry in the 3xTg‐AD injected with lenti‐GDNF. Neurotrophic effects of GDNF secreted by astrocytes enhanced neurotransmitter functionality, as previously demonstrated by local increases in the synthesis of the neurotransmitters acetylcholine, dopamine, and serotonin in aged rats similarly transduced with lenti‐GDNF 6. An in vitro study with septal cholinergic neurons has shown that GDNF increases the expression of choline acetyl transferase, in addition to protecting against the cytotoxicity induced by oligomeric Aβ42 8. GDNF is also a potent inductor of cell growth pathways and progenitor cell differentiation throughout brain embryonic development and adulthood. GDNF‐induced neurogenesis has been described as being present in the dentate gyrus of adult rats 40, whereas in vitro studies have shown GDNF facilitation of astrogliogenesis from hippocampal neural progenitors 41. Although hippocampal neural progenitors are based in the gyrus dentate zone, we cannot rule out the contribution of GDNF overexpression in dorsal CA1 to the recovery of the impaired adult neurogenesis, as reported in the 3xTg‐AD mice 42. Therefore, we can speculate that GDNF‐mediated hippocampal neurogenesis and/or astrogliogenesis contributed to the rescue of the cognitive impairment in these mice.
GDNF protected from the oxidative stress induced by C99‐derived fragment in activated MC65 cells. Similarly, it has been reported that GDNF neuroprotection involves antioxidant mechanisms in both in vivo and in vitro models of PD 43, 44, 45, 46 and of other pathologies 47, 48. GDNF has been demonstrated to induce the activity of the antioxidant enzymes glutathione peroxidase, superoxide dismutase, and catalase in brain tissue 49 and neuronal cultures 46, 50. In this sense, the antioxidant action of GDNF may be a significant mechanism of neuroprotection also against AD. The presence of oxidative damage is well documented in the AD brain, and oxidative stress is considered an early event that plays an important role in the pathogenesis of the disease 51, 52, 53. The basis of this oxidative damage appears to derive from mitochondrial dysfunction, overaccumulation of iron, defective proteolysis, and other interacting mechanisms that generate ROS in vulnerable neuronal populations, such as those of the hippocampus CA1 region 54, 55, 56. Previous studies had reported that Aβ induces ROS generation in neurons 57, whereas oxidative stress, in turn, activates the production of pathological levels of Aβ through a positive feedback relationship between γ‐ and β‐secretase activities 58. Furthermore, brain tissue and cultured neurons from the 3xTg‐AD mouse model have showed early ROS damage and disturbances in the antioxidant defense 17, 59. Also, C99‐induced cell death in the MC65 cell model of AD has been demonstrated to involve oxidative stress pathways 19. It is known that CTFs appear in neuritic plaques 60 and accumulate intracellularly in patients with hereditary AD 61. Furthermore, C99 and derived fragments have proven to be highly neurotoxic in several experimental systems in vivo and in vitro, through mechanisms that include generation of ROS 62. Regarding tau pathology, several oxidative stress‐induced mechanisms have been demonstrated to exacerbate hyperphosphorylation of tau 63, 64. The neuroprotection conferred to both AD models by overexpressing GDNF in this study is at least partially mediated by antioxidant mechanisms, as demonstrated by the fact that the inhibition of ROS generation in MC65 cultures was paralleled by the recovery of neuronal survival. An enhanced functionality of the proteasome system in the degradation and processing of toxic proteins could also be involved in the GDNF neuroprotection, as suggested by the decrease of overexpressed CTFs in MC65 cells.
Multiple pathways are in involved GDNF signaling and survival pathways 6, 7, 65, 66, 67, 68, in some of which there is an evident neuroprotective role of astrocytes. For instance, inflammatory mediators have been shown to induce the release of GDNF from astrocytes, leading to neuron survival 69 and recovery from cognitive impairment 70. Furthermore, it has been reported that GDNF induces an upregulation of the astrocyte glutamate transporter GLAST‐1 against cell death mediated through excitotoxic processes 71. There was also an upregulation of GDNF secretion by astrocytes after ischemic brain injury 48. Furthermore, it has been proposed that injured dopaminergic neurons signal astrocytes trigger GDNF upregulation 72. Therefore, in addition to GDNF direct survival effects as seen in MC65 cells, this neurotrophic factor appears to be involved in a neuron‐astrocyte cross talk to generate astrocyte‐mediated neuroprotection. This work further demonstrates the feasibility of using astrocytes as engineered minipumps that stably oversecrete GDNF at neuroprotective doses 6, 73, 74.
Conclusion
GDNF demonstrated to be neuroprotective in AD‐like pathological alterations through astrocyte transduction with a lentiviral vector in hippocampus of 3xTg‐AD mice, with a preservation of spatial learning and memory. The most significant molecular change was an upregulation of BDNF. Overexpression of GDNF by lentivirus transduction of neuroblastoma MC65 cells protected against oxidative stress and cell death mediated by C99. Engineered lenti‐GDNF showed high effectiveness and safety, which might contribute to the advancement of gene‐therapy‐based approaches in AD experimental and clinical research for this promising neutrophic agent.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Figure S1. Area of transgene expression of GFP driven by a lentiviral vector in the hippocampus of 3xTgAD mice.
Figure S2. Behavior of 3xTgAD mice (Tg) and non‐transgenic mice (NTg) treated with GFP or GDNF.
Acknowledgments
MC65 cells were kindly given by Bryce Sopher and George M Martin (University of Washington, Seattle, WA). This study was supported by Grants: SAF2009‐13093, SAF2012‐39852, and CSD2010‐45 from the Spanish MINECO; 2009/SGR/214 from the Generalitat and 062931 from the Fundació La Marató de TV3, of Catalonia; and the European Regional Development Fund (ERDF).
References
- 1. Lee CK, Weindruch R, Prolla TA. Gene‐expression profile of the ageing brain in mice. Nat Genet 2000;25:294–297. [DOI] [PubMed] [Google Scholar]
- 2. Miyazaki H, Okuma Y, Nomura J, Nagashima K, Nomura Y. Age‐related alterations in the expression of glial cell line‐derived neurotrophic factor in the senescence‐accelerated mouse brain. J Pharmacol Sci 2003;92:28–34. [DOI] [PubMed] [Google Scholar]
- 3. Ray S, Britschgi M, Herbert C, et al. Classification and prediction of clinical Alzheimer's diagnosis based on plasma signaling proteins. Nat Med 2007;13:1359–1362. [DOI] [PubMed] [Google Scholar]
- 4. Siegel GJ, Chauhan NB. Neurotrophic factors in Alzheimer's and Parkinson's disease brain. Brain Res Brain Res Rev 2000;33:199–227. [DOI] [PubMed] [Google Scholar]
- 5. Airavaara M, Pletnikova O, Doyle ME, Zhang YE, Troncoso JC, Liu QR. Identification of novel GDNF isoforms and cis‐antisense GDNFOS gene and their regulation in human middle temporal gyrus of Alzheimer disease. J Biol Chem 2011;2011:45093–45102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pertusa M, García‐Matas S, Mammeri H, et al. Expression of GDNF transgene in astrocytes improves cognitive deficits in aged rats. Neurobiol Aging 2008;29:1366–1379. [DOI] [PubMed] [Google Scholar]
- 7. Ghribi O, Herman MM, Pramoonjago P, Spaulding NK, Savory J. GDNF regulates the Aβ‐induced endoplasmic reticulum stress response in rabbit hippocampus by inhibiting the activation of gadd 153 and the JNK and ERK kinases. Neurobiol Dis 2004;16:417–427. [DOI] [PubMed] [Google Scholar]
- 8. Kitiyanant N, Kitiyanant Y, Svendsen CN, Thangnipon W. BDNF‐, IGF‐1‐ and GDNF‐secreting human neural progenitor cells rescue amyloid β‐induced toxicity in cultured rat septal neurons. Neurochem Res 2012;37:143–152. [DOI] [PubMed] [Google Scholar]
- 9. Martorana A, Esposito Z, Koch G. Beyond the cholinergic hypothesis: do current drugs work in Alzheimer's disease? CNS Neurosci Ther 2010;16:235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maruyama W, Nitta A, Shamoto‐Nagai M, et al. N‐Propargyl‐1 (R)‐aminoindan, rasagiline, increases glial cell line‐derived neurotrophic factor (GDNF) in neuroblastoma SH‐SY5Y cells through activation of NF‐κB transcription factor. Neurochem Int 2004;44:393–400. [DOI] [PubMed] [Google Scholar]
- 11. Caumont AS, Octave JN, Hermans E. Amantadine and memantine induce the expression of the glial cell line‐derived neurotrophic factor in C6 glioma cells. Neurosci Lett 2006;394:196–201. [DOI] [PubMed] [Google Scholar]
- 12. Wu HM, Tzeng NS, Qian L, et al. Novel neuroprotective mechanisms of memantine: increase in neurotrophic factor release from astroglia and anti‐inflammation by preventing microglial activation. Neuropsychopharmacology 2009;34:2344–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nicole O, Ali C, Docagne F, et al. Neuroprotection mediated by glial cell line‐derived neurotrophic factor: involvement of a reduction of NMDA‐induced calcium influx by the mitogen‐activated protein kinase pathway. J Neurosci 2001;21:3024–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marco S, Canudas AM, Canals JM, Gavaldà N, Pérez‐Navarro E, Alberch J. Excitatory amino acids differentially regulate the expression of GDNF, neurturin, and their receptors in the adult rat striatum. Exp Neurol 2002;174:243–252. [DOI] [PubMed] [Google Scholar]
- 15. Liberto CM, Albrecht PJ, Herx LM, Yong VW, Levison SW. Pro‐regenerative properties of cytokine‐activated astrocytes. J Neurochem 2004;89:1092–1100. [DOI] [PubMed] [Google Scholar]
- 16. Oddo S, Caccamo A, Shepherd JD, et al. Triple‐transgenic model of Alzheimer's disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron 2003;39:409–421. [DOI] [PubMed] [Google Scholar]
- 17. García‐Mesa Y, López‐Ramos JC, Giménez‐Llort L, et al. Physical exercise protects against Alzheimer's disease in 3xTg‐AD mice. J Alzheimers Dis 2011;24:421–454. [DOI] [PubMed] [Google Scholar]
- 18. Sopher BL, Fukuchi K, Smith AC, Leppig KA, Furlong CE, Martin GM. Cytotoxicity mediated by conditional expression of a carboxylterminal derivative of the β‐amyloid precursor protein. Brain Res Mol Brain Res 1994;26:207–217. [DOI] [PubMed] [Google Scholar]
- 19. Sebastià J, Pertusa M, Vílchez D, et al. Carboxyl‐terminal fragment of amyloid precursor protein and hydrogen peroxide induce neuronal cell death through different pathways. J Neural Transm 2006;113:1837–1845. [DOI] [PubMed] [Google Scholar]
- 20. Kutner RH, Zhang XY, Reiser J. Production, concentration and titration of pseudotyped HIV‐1‐based lentiviral vectors. Nat Protoc 2009;4:495–505. [DOI] [PubMed] [Google Scholar]
- 21. Jin LW, Hua DH, Shie FS, Maezawa I, Sopher B, Martin GM. Novel tricyclic pyrone compounds prevent intracellular APP C99‐induced cell death. J Mol Neurosci 2002;19:57–61. [DOI] [PubMed] [Google Scholar]
- 22. Copenhaver PF, Anekonda TS, Musashe D, et al. A translational continuum of model systems for evaluating treatment strategies in Alzheimer's disease: isradipine as a candidate drug. Dis Model Mech 2011;4:634–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Biju K, Zhou Q, Li G, et al. Macrophage‐mediated GDNF delivery protects against dopaminergic neurodegeneration: a therapeutic strategy for Parkinson's disease. Mol Ther 2010;18:1536–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maezawa I, Hong HS, Wu HC, et al. A novel tricyclic pyrone compound ameliorates cell death associated with intracellular amyloid‐β oligomeric complexes. J Neurochem 2006;98:57–67. [DOI] [PubMed] [Google Scholar]
- 25. Hong HS, Maezawa I, Budamagunta M, et al. Candidate anti‐A beta fluorene compounds selected from analogs of amyloid imaging agents. Neurobiol Aging 2010;31:1690–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Naghdi N, Majlessi N, Bozorgmehr T. The effects of anisomycin (a protein synthesis inhibitor) on spatial learning and memory in CA1 region of rats hippocampus. Behav Brain Res 2003;139:69–73. [DOI] [PubMed] [Google Scholar]
- 27. Volpe BT, Davis HP, Towle A, Dunlap WP. Loss of hippocampal CA1 pyramidal neurons correlates with memory impairment in rats with ischemic or neurotoxin lesions. Behav Neurosci 1992;106:457–464. [DOI] [PubMed] [Google Scholar]
- 28. Nagahara AH, Merrill DA, Coppola G, et al. Neuroprotective effects of brain‐derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med 2009;15:331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Connor B, Young D, Yan Q, Faull RL, Synek B, Dragunow M. Brain‐derived neurotrophic factor is reduced in Alzheimer's disease. Brain Res Mol Brain Res 1997;49:71–81. [DOI] [PubMed] [Google Scholar]
- 30. Amada N, Aihara K, Ravid R, Horie M. Reduction of NR1 and phosphorylated Ca2+/calmodulin‐dependent protein kinase II levels in Alzheimer's disease. NeuroReport 2005;16:1809–1813. [DOI] [PubMed] [Google Scholar]
- 31. Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long‐term potentiation. Nat Rev Neurosci 2012;13:169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang DM, Yang YJ, Zhang L, Zhang X, Guan FF, Zhang LF. Naringin enhances CaMKII activity and improves long‐term memory in a mouse model of Alzheimer's disease. Int J Mol Sci 2013;14:5576–5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin N, Pan XD, Chen AQ, et al. Tripchlorolide improves age‐associated cognitive deficits by reversing hippocampal synaptic plasticity impairment and NMDA receptor dysfunction in SAMP8 mice. Behav Brain Res 2014;258:8–18. [DOI] [PubMed] [Google Scholar]
- 34. Liao MH, Xiang YC, Huang JY, et al. The disturbance of hippocampal CaMKII/PKA/PKC phosphorylation in early experimental diabetes mellitus. CNS Neurosci Ther 2013;19:329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peng C, Aron L, Klein R, et al. Pitx3 is a critical mediator of GDNF‐induced BDNF expression in nigrostriatal dopaminergic neurons. J Neurosci 2011;31:12802–12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Revilla S, Suñol C, García‐Mesa Y, Giménez‐Llort L, Sanfeliu C, Cristòfol R. Physical exercise improves synaptic dysfunction and recovers the loss of survival factors in 3xTg‐AD mouse brain. Neuropharmacology 2014;81:55–63. [DOI] [PubMed] [Google Scholar]
- 37. García‐Mesa Y, Pareja‐Galeano H, Bonet‐Costa V, et al. Physical exercise neuroprotects ovariectomized 3xTg‐AD mice through BDNF mechanisms. Psychoneuroendocrinology 2014;45:154–166. [DOI] [PubMed] [Google Scholar]
- 38. Aguiar AS Jr, Stragier E, da Luz Scheffer D, et al. Effects of exercise on mitochondrial function, neuroplasticity and anxio‐depressive behavior of mice. Neuroscience 2014;271:56–63. [DOI] [PubMed] [Google Scholar]
- 39. Zhang F, Lu YF, Wu Q, Liu J, Shi JS. Resveratrol promotes neurotrophic factor release from astroglia. Exp Biol Med (Maywood) 2012;237:943–948. [DOI] [PubMed] [Google Scholar]
- 40. Chen Y, Ai Y, Slevin JR, Maley BE, Gash DM. Progenitor proliferation in the adult hippocampus and substantia nigra induced by glial cell line‐derived neurotrophic factor. Exp Neurol 2005;196:87–95. [DOI] [PubMed] [Google Scholar]
- 41. Boku S, Nakagawa S, Takamura N, et al. GDNF facilitates differentiation of the adult dentate gyrus‐derived neural precursor cells into astrocytes via STAT3. Biochem Biophys Res Commun 2013;434:779–784. [DOI] [PubMed] [Google Scholar]
- 42. Rodríguez JJ, Jones VC, Tabuchi M, et al. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer's disease. PLoS ONE 2008;3:e2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Smith MP, Cass WA. GDNF reduces oxidative stress in a 6‐hydroxydopamine model of Parkinson's disease. Neurosci Lett 2007;412:259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sandhu JK, Gardaneh M, Iwasiow R, et al. Astrocyte‐secreted GDNF and glutathione antioxidant system protect neurons against 6OHDA cytotoxicity. Neurobiol Dis 2009;33:405–414. [DOI] [PubMed] [Google Scholar]
- 45. Littrell OM, Granholm AC, Gerhardt GA, Boger HA. Glial cell‐line derived neurotrophic factor (GDNF) replacement attenuates motor impairments and nigrostriatal dopamine deficits in 12‐month‐old mice with a partial deletion of GDNF. Pharmacol Biochem Behav 2013;104:10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gharib E, Gardaneh M, Shojaei S. Upregulation of glutathione peroxidase‐1 expression and activity by glial cell line‐derived neurotrophic factor promotes high‐level protection of PC12 cells against 6‐hydroxydopamine and hydrogen peroxide toxicities. Rejuvenation Res 2013;16:185–199. [DOI] [PubMed] [Google Scholar]
- 47. Dong A, Shen J, Krause M, Hackett SF, Campochiaro PA. Increased expression of glial cell line‐derived neurotrophic factor protects against oxidative damage‐induced retinal degeneration. J Neurochem 2007;103:1041–1052. [DOI] [PubMed] [Google Scholar]
- 48. Ikeda T, Xia XY, Xia YX, Ikenoue T, Han B, Choi BH. Glial cell line‐derived neurotrophic factor protects against ischemia/hypoxia‐induced brain injury in neonatal rat. Acta Neuropathol 2000;100:161–167. [DOI] [PubMed] [Google Scholar]
- 49. Chao CC, Lee EH. Neuroprotective mechanism of glial cell line‐derived neurotrophic factor on dopamine neurons: role of antioxidation. Neuropharmacology 1999;38:913–916. [DOI] [PubMed] [Google Scholar]
- 50. Korsak K, Dolatshad NF, Silva AT, Saffrey MJ. Ageing of enteric neurons: oxidative stress, neurotrophic factors and antioxidant enzymes. Chem Cent J 2012;6:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nunomura A, Perry G, Aliev G, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 2001;60:759–767. [DOI] [PubMed] [Google Scholar]
- 52. Praticò D, Zhukareva V, Yao Y, et al. 12/15‐Lipoxygenase is increased in Alzheimer's disease: possible involvement in brain oxidative stress. Am J Pathol 2004;164:1655–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sultana R, Butterfield DA. Role of oxidative stress in the progression of Alzheimer's disease. J Alzheimers Dis 2010;19:341–353. [DOI] [PubMed] [Google Scholar]
- 54. Zhu X, Su B, Wang X, Smith MA, Perry G. Causes of oxidative stress in Alzheimer disease. Cell Mol Life Sci 2007;64:2202–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. García‐Matas S, de Vera N, Aznar AO, et al. In vitro and in vivo activation of astrocytes by amyloid‐β is potentiated by pro‐oxidant agents. J Alzheimers Dis 2010;20:229–245. [DOI] [PubMed] [Google Scholar]
- 56. Gandhi S, Abramov AY. Mechanism of oxidative stress in neurodegeneration. Oxid Med Cell Longev 2012;2012:428010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell 1994;77:817–827. [DOI] [PubMed] [Google Scholar]
- 58. Tamagno E, Guglielmotto M, Aragno M, et al. Oxidative stress activates a positive feedback between the γ‐ and β‐secretase cleavages of the β‐amyloid precursor protein. J Neurochem 2008;104:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ghosh D, LeVault KR, Barnett AJ, Brewer GJ. A reversible early oxidized redox state that precedes macromolecular ROS damage in aging nontransgenic and 3xTg‐AD mouse neurons. J Neurosci 2012;32:5821–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Selkoe DJ, Podlisny MB, Joachim CL, et al. β‐Amyloid precursor protein of Alzheimer disease occurs as 110‐ to 135‐kilodalton membrane‐associated proteins in neural and nonneural tissues. Proc Natl Acad Sci USA 1988;85:7341–7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McPhie DL, Lee RK, Eckman CB, et al. Neuronal expression of β‐amyloid precursor protein Alzheimer mutations causes intracellular accumulation of a C‐terminal fragment containing both the amyloid β and cytoplasmic domains. J Biol Chem 1997;272:24743–24746. [DOI] [PubMed] [Google Scholar]
- 62. Chang KA, Suh YH. Pathophysiological roles of amyloidogenic carboxy‐terminal fragments of the β‐amyloid precursor protein in Alzheimer's disease. J Pharmacol Sci 2005;97:461–471. [DOI] [PubMed] [Google Scholar]
- 63. Zhu X, Rottkamp CA, Boux H, Takeda A, Perry G, Smith MA. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle‐related events in Alzheimer disease. J Neuropathol Exp Neurol 2000;59:880–888. [DOI] [PubMed] [Google Scholar]
- 64. Melov S, Adlard PA, Morten K, et al. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS ONE 2007;2:e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li F, Wang M, Zhu S, Li L, Xiong Y, Gao DS. The potential neuroprotection mechanism of GDNF in the 6‐OHDA‐induced cellular models of Parkinson's disease. Cell Mol Neurobiol 2013;33:907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang L, Deng QQ, Wu XH, Yu J, Yang XL, Zhong YM. Upregulation of glutamate‐aspartate transporter by glial cell line‐derived neurotrophic factor ameliorates cell apoptosis in neural retina in streptozotocin‐induced diabetic rats. CNS Neurosci Ther 2013;19:945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Duarte EP, Curcio M, Canzoniero LM, Duarte CB. Neuroprotection by GDNF in the ischemic brain. Growth Factors 2012;30:242–257. [DOI] [PubMed] [Google Scholar]
- 68. Ghribi O, Herman MM, Forbes MS, DeWitt DA, Savory J. GDNF protects against aluminum‐induced apoptosis in rabbits by upregulating Bcl‐2 and Bcl‐XL and inhibiting mitochondrial Bax translocation. Neurobiol Dis 2001;8:764–773. [DOI] [PubMed] [Google Scholar]
- 69. Iravani MM, Sadeghian M, Leung CC, Jenner P, Rose S. Lipopolysaccharide‐induced nigral inflammation leads to increased IL‐1β tissue content and expression of astrocytic glial cell line‐derived neurotrophic factor. Neurosci Lett 2012;510:138–142. [DOI] [PubMed] [Google Scholar]
- 70. Bian Y, Zhao X, Li M, Zeng S, Zhao B. Various roles of astrocytes during recovery from repeated exposure to different doses of lipopolysaccharide. Behav Brain Res 2013;253:253–261. [DOI] [PubMed] [Google Scholar]
- 71. Koeberle PD, Bähr M. The upregulation of GLAST‐1 is an indirect antiapoptotic mechanism of GDNF and neurturin in the adult CNS. Cell Death Differ 2008;15:471–483. [DOI] [PubMed] [Google Scholar]
- 72. Saavedra A, Baltazar G, Santos P, Carvalho CM, Duarte EP. Selective injury to dopaminergic neurons up‐regulates GDNF in substantia nigra postnatal cell cultures: role of neuron‐glia crosstalk. Neurobiol Dis 2006;23:533–542. [DOI] [PubMed] [Google Scholar]
- 73. Ericson C, Wictorin K, Lundberg C. Ex vivo and in vitro studies of transgene expression in rat astrocytes transduced with lentiviral vectors. Exp Neurol 2002;173:22–30. [DOI] [PubMed] [Google Scholar]
- 74. Safi R, Gardaneh M, Panahi Y, Maghsoudi N, Zaefizadeh M, Gharib E. Optimized quantities of GDNF overexpressed by engineered astrocytes are critical for protection of neuroblastoma cells against 6‐OHDA toxicity. J Mol Neurosci 2012;46:654–665. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Area of transgene expression of GFP driven by a lentiviral vector in the hippocampus of 3xTgAD mice.
Figure S2. Behavior of 3xTgAD mice (Tg) and non‐transgenic mice (NTg) treated with GFP or GDNF.