

Summary
Aims
It has become increasingly evident that the nigrostriatal degeneration associated with Parkinson's disease initiates at the level of the axonal terminals in the putamen, and this nigrostriatal terminal dystrophy is either caused or exacerbated by the presence of α‐synuclein immunopositive neuronal inclusions. Therefore, strategies aimed at reducing α‐synuclein‐induced early neuronal dystrophy may slow or halt the progression to overt nigrostriatal neurodegeneration. Thus, this study sought to determine if adeno‐associated virus (AAV) mediated overexpression of two molecular chaperone heat shock proteins, namely Hsp27 or Hsp70, in the AAV‐α‐synuclein viral gene transfer rat model of Parkinson's disease could prevent α‐synuclein‐induced early neuronal pathology.
Methods
Male Sprague–Dawley rats were intranigrally coinjected with pathogenic (AAV‐α‐synuclein) and putative therapeutic (AAV‐Hsp27 or AAV‐Hsp70) viral vectors and were sacrificed 18 weeks postviral injection.
Results
Intranigral injection of AAV‐α‐synuclein resulted in significant α‐synuclein accumulation in the substantia nigra and striatal terminals which led to significant dystrophy of nigrostriatal dopaminergic neurons without overt nigrostriatal neurodegeneration. Coinjection of AAV‐Hsp70, but not AAV‐Hsp27, significantly reduced AAV‐α‐synuclein‐induced neuronal dystrophy.
Conclusions
These data confirm that overexpression of Hsp70 holds significant potential as a disease‐modulating therapeutic approach for Parkinson's disease, with protective effects against early‐onset α‐synuclein‐induced pathology demonstrated in the AAV‐α‐synuclein model.
Keywords: Genetic therapy, Heat‐shock proteins, Parkinson's disease
Introduction
The severe and progressive loss of the nigrostriatal dopaminergic pathway is one of the distinguishing features of Parkinson's disease and this is thought to be largely driven by aberrant α‐synuclein expression and aggregation 1, 2, 3, 4, 5, 6. In more recent years, evidence has accumulated to suggest that nigrostriatal neurodegeneration initiates in the axonal terminals in the striatum before progressing in a retrograde degenerative manner to the cell bodies in the substantia nigra 7, 8, 9, 10, 11, 12. This early‐onset terminal pathology is characterized by the presence of morphologically aberrant tyrosine hydroxylase‐immunoreactive axonal fibers (dystrophic neurites) in the putamen of early stage Parkinson's disease patients postmortem, which are associated with α‐synuclein immunopositive neuronal inclusions 7, 8. In support of these clinical findings, several groups have reported that in viral α‐synuclein models of Parkinson's disease [reviewed in 13], α‐synuclein‐induced axonal swellings with truncated, dystrophic profiles appear in the tyrosine hydroxylase‐immunopositive terminals in advance of overt dopaminergic cell loss 8, 9, 14. Therefore, it is conceivable that strategies that can target this early α‐synuclein‐induced neuronal pathology may halt progression to overt nigrostriatal neurodegeneration.
One such approach is to harness the properties of heat shock proteins which, in their role as molecular chaperones, aid in the refolding of misfolded proteins 15, 16. Interestingly, Chu et al. 7 have reported a significant loss of heat shock protein (Hsp73; a member of the Hsp70 family) in the putamen of Parkinson's disease patients compared to age‐matched controls, and this loss was most pronounced in neurons with α‐synuclein inclusions. This suggests that a reduction of heat shock protein expression may actually contribute to the disease pathogenesis by reducing the diseased cells' ability to process α‐synuclein.
Numerous in vitro and in vivo studies suggest that heat shock proteins, particularly Hsp27 and Hsp70, may be capable of decreasing protein aggregation, including α‐synuclein aggregation. In vitro aggregation assays demonstrate that Hsp27 and Hsp70 specifically bind and interfere with α‐synuclein fibrillization kinetics inhibiting fibril assembly 17, 18, 19, 20. In primary neuronal cultures and neuroblastoma cell lines, Hsp27 and Hsp70 protect against α‐synuclein‐induced toxicity 21, 22, 23, 24. Notably, in vivo heat shock protein overexpression has potent protective effects in preclinical models of neurodegenerative diseases where protein aggregates, such as tauopathies and polyglutamine inclusions, are evident 25, 26, 27, 28, 29, 30. While a number of studies have investigated the effect of heat shock protein overexpression in various models of Parkinson's disease 31, 32, 33, 34, 35, 36, 37, 38, to date there remains a paucity of preclinical information to judiciously evaluate the possible therapeutic potential that heat shock proteins may have on early α‐synuclein‐induced neuronal pathology.
Thus, the aim of this study was to determine if adeno‐associated virus (AAV)‐mediated overexpression of Hsp27 and Hsp70 could prevent AAV‐α‐synuclein‐induced pathology, namely those changes associated with early‐stage, predegenerative Parkinson's disease.
Methods
Animals
All procedures were carried out in accordance with European Union Directive 2010/63/EU and S.I. No. 543 of 2012 and were reviewed and approved by The Animal Care and Research Ethics Committee of the National University of Ireland, Galway. Male Sprague–Dawley rats, sourced from Charles Rivers, UK were used in all experiments.
Experimental Designs
Pilot Study
To establish if the viral transgenes employed were expressed in the appropriate neural cell type, that is, tyrosine hydroxylase immunopositive nigrostriatal cell bodies, eighteen male Sprague–Dawley rats (weighing 284 ± 3 g on the day of surgery) received intranigral injections of either control (AAV‐GFP) or putative therapeutic (AAV‐Hsp27 or AAV‐Hsp70) viral vectors (n = 6 rats per group). Rats were sacrificed 2 weeks postviral injection by anesthetic overdose and transcardial perfusion, and brains were processed for immunohistochemical analyses. Overexpression of green fluorescent protein (GFP), Hsp27 or Hsp70 in tyrosine hydroxylase positive cell bodies was assessed by double immunofluorescence.
Main Study
A separate cohort of forty male Sprague–Dawley rats (weighing 284 ± 2 g on the day of surgery) were coinjected intranigrally with pathogenic (AAV‐α‐synuclein) and putative therapeutic (AAV‐Hsp27 or AAV‐Hsp70) viral vectors to generate four final groups (n = 10 rats per group) as indicated in Table 1. Rats were sacrificed 18 weeks postviral injection by anesthetic overdose and transcardial perfusion, and brains were processed for immunohistochemical analyses. Eighteen weeks after virus infusion was chosen as a predegenerative time‐point for sacrifice because previous studies in our laboratory have revealed that AAV‐α‐synuclein‐induced motor deficits begin to emerge strongly after this time 39. The effect of Hsp27 and Hsp70 transgene expression on AAV‐α‐synuclein‐induced α‐synuclein expression, nigrostriatal dopaminergic integrity, and neuritic dystrophy was assessed by quantitative immunohistochemistry.
Table 1.
Final experimental groups used in the main experiment. Rats were given unilateral intranigral coinjection of two AAV vectors as indicated. All vectors were based on the AAV2/5 serotypes and the subscripts indicate the promoter used to drive transgene expression (SYN, human synapsin; CMV, cytomegalovirus). Viral titers were as follows: AAV‐GFPSYN and AAV‐α‐synucleinSYN: 1.07 × 1010 drp/μL; AAV‐GFPCMV, AAV‐Hsp27CMV and AAV‐ Hsp70CMV: 8.36 × 108 drp/μL. Coinjection of pathogenic and putative therapeutic viruses yielded a combined viral load of 1.15 × 1010 drp
| Group | Pathogenic vector | Therapeutic vector | n |
|---|---|---|---|
| Intact | AAV‐GFPSYN | AAV‐GFPCMV | 10 |
| Lesion | AAV‐α‐synucleinSYN | AAV‐GFPCMV | 10 |
| Hsp27 | AAV‐α‐synucleinSYN | AAV‐Hsp27CMV | 10 |
| Hsp70 | AAV‐α‐synucleinSYN | AAV‐Hsp70CMV | 10 |
AAV, adeno‐associated virus; GFP, green fluorescent protein.
AAV Preparation
Adeno‐associated virus vectors were prepared as described previously 39, 40. The genes for α‐synuclein (normal human) and GFP were cloned into pTRUF flanked by AAV2 inverted terminal repeats under the human synapsin promoter. The human Hsp27 and Hsp70 genes were subcloned into the pAAV‐MCS vector (Agilent Technologies Inc., Santa Clara, CA, USA), where the transgene is flanked by AAV2 inverted terminal repeats with constitutive expression under the cytomegalovirus promoter. These plasmids and the AAV5 helper plasmid, pDG‐5, were purified for subsequent cell transfection. The AAV2/5 viral vectors were produced by cotransfecting HEK‐293T cells with the relevant transgene plasmid and pDG‐5, as described previously 41, for 48 h, by calcium phosphate precipitation. Viral vectors were purified by the treatment of the transfected cell pellet with a DNA endonuclease to degrade any non‐encapsulated DNA, followed by a single step affinity chromatography purification of the freeze‐thawed cell lysate, using Q‐sepharose columns. Viral titers were established using real‐time PCR and expressed as viral DNAse‐resistant particles (drp) per μL (see Table 1 legend). Viruses were aliquoted into siliconized tubes and stored at −80°C.
Surgery
Unilateral intra‐nigral surgery was conducted under isofluorane anesthesia (5% in O2 for induction and 2% in O2 for maintenance 42) in a stereotaxic frame with the incisor bar set at −2.3 mm. The substantia nigra was targeted with viral infusions delivered at a rate of 1 μL/min for 2 min (with an additional 2 min for diffusion) at stereotaxic coordinates A.P. −5.3 mm, M.L. +2.0 mm from bregma and D.V. −7.2 mm below dura.
Quantitative Immunohistochemistry
Rats were sacrificed by terminal anesthesia (50 mg/kg pentobarbital i.p.) and transcardially perfused with 100 mL heparinized saline followed by 150 mL of 4% paraformaldehyde. Brains were postfixed in 4% paraformaldehyde for 4 h and stored in a 25% sucrose plus 0.1% sodium azide solution. Serial coronal sections (40 μm) were cut using a freezing sledge microtome and a 1:6 series was used for all quantitative immunohistochemistry. Because the viruses were injected unilaterally, the uninfected side served as a control for the immunohistochemical procedures. All quantitative immunohistochemistry was conducted by researchers blinded to the treatment of the rats.
Peroxidase‐based immunohistochemical staining and immunofluorescent staining was completed as described previously 43, 44. In brief, following quenching of endogenous peroxidase activity where appropriate (using a solution of 3% hydrogen peroxide/10% methanol in distilled water) and blocking of nonspecific secondary antibody binding (using 3% normal serum in Tris‐buffered saline (TBS) with 0.2% Triton X‐100 at room temperature for 1 h), sections were incubated overnight at room temperature with the appropriate primary antibody diluted in 1% normal serum in TBS with 0.2% Triton X‐100 (mouse antityrosine hydroxylase, MAB318, 1:1000; Millipore (Billerica, MA, USA); mouse anti‐α‐synuclein, AB36615, 1:1000; Abcam (Cambridge UK); sheep antityrosine hydroxylase, AB113, 1:500; Abcam; rabbit anti‐GFP, A6455, 1:1000; Invitrogen (Carlsbad, CA, USA); mouse anti‐Hsp27, SPA800, 1:1000; Stressgen (Victoria, BC, Canada); mouse anti‐Hsp70, SPA8135, 1:1000; Stressgen; mouse anti‐α‐synuclein, AB36615, 1:1000; Abcam). Sections were then incubated for 3 h at room temperature with the appropriate fluorophore‐labeled secondary antibody (donkey anti‐sheep, A21098, 1:200; Invitrogen; goat anti‐rabbit, A11034, 1:200; Invitrogen; goat anti‐mouse, A11029, 1:200; Invitrogen) or, for peroxidase‐based immunohistochemical staining, in a biotinylated secondary antibody for 3 h (horse anti‐mouse, BA2001, 1:200; Vector, Burlingame, CA, USA), followed by a 2‐h incubation in streptavidin–biotin–horseradish peroxidase solution (Vector). For α‐synuclein immunofluorescent staining, horse anti‐mouse (BA2001, 1:200; Vector) secondary antibody was used followed by a streptavidin–fluorophore conjugate (S11249, 1:1000; Invitrogen). For immunofluorescent staining, sections were then mounted onto gelatine‐coated microscope slides and coverslipped using “Sigma Fluoromount” fluorescence mounting medium (Sigma‐Aldrich, St. Louis, MO, USA). For peroxidase‐based immunohistochemistry, sections were developed in 0.5% solution of diaminobenzidine tetrahydrochloride (DAB; Sigma–Aldrich) in Tris‐buffer containing 0.3 μL/mL of hydrogen peroxide. Sections were mounted on gelatin‐coated microscope slides, dehydrated in ascending concentrations of alcohols, cleared in xylene and cover‐slipped using DPX mountant (BDH chemicals).
Image Capture and Analysis
Photomicrographs of immunofluorescently stained sections were captured by an Olympus IX81 with a Hamamatsu C4742‐80 digital camera. Triple immunopositive cell bodies (tyrosine hydroxylase, GFP and α‐synuclein) in the substantia nigra ipsilateral to the viral injection were quantified, and data were expressed as a percentage of the total number of tyrosine hydroxylase immunopositive cell bodies per section.
Photomicrographs of DAB‐stained sections were captured under bright field illumination (using a Nikon DXM1200C digital camera mounted on a Nikon dissecting microscope) and all image analysis was completed using Image J software (ImageJ v1.41o; National Institute of Health, Bethesda, ML, USA). For quantification of AAV‐α‐synuclein‐induced human α‐synuclein expression, the number of α‐synuclein immunopositive cell bodies in the substantia nigra and the optical density of α‐synuclein immunopositive staining in the striatum were quantified, and data were expressed as a percentage of the intact side. For confirmation of the predegenerative status of this AAV‐α‐synuclein model, the impact of α‐synuclein on the number of tyrosine hydroxylase immunopositive cell bodies in the substantia nigra and the optical density of tyrosine hydroxylase immunopositive staining in the striatum were quantified, and data were expressed as a percentage of the intact side. For quantification of AAV‐α‐synuclein‐induced dystrophic neurites (identified as morphologically aberrant (swollen) tyrosine hydroxylase‐immunoreactive axonal fibers), the number of tyrosine hydroxylase immunopositive dystrophic neurites in the injected striatum was quantified, and data were expressed as the number per field examined.
Statistical Analyses
Data were analyzed using a one‐way ANOVA followed by post hoc Fisher's least significant difference test where appropriate. Differences between groups were considered statistically significant when P < 0.05. All data are expressed as mean ± SEM.
Results
Overexpression of Heat Shock Proteins within the Nigral Dopaminergic Neurons
In the initial pilot study, we sought to determine if the transgenes of interest, namely Hsp27 and Hsp70, were successfully overexpressed in the nigral dopaminergic neurons following a stereotaxic nigral injection of either AAV‐Hsp27 or AAV‐Hsp70 (Figure 1). Immunofluorescent staining confirmed that all transgenes of interest, that is, control GFP, Hsp27, and Hsp70, were strongly expressed following AAV delivery. Double immunofluorescent staining for the dopaminergic marker tyrosine hydroxylase revealed that all proteins were successfully colocalized within the dopaminergic neurons.
Figure 1.
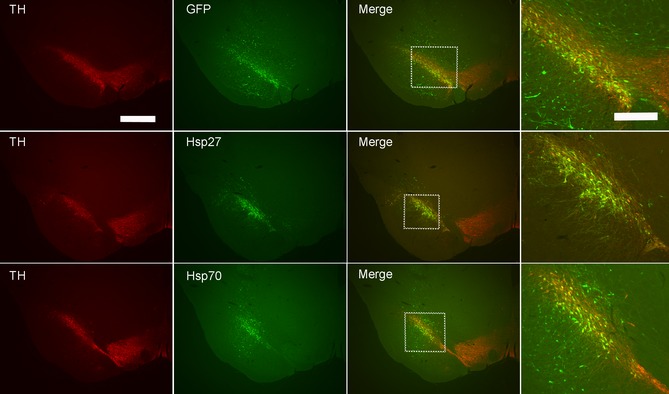
Overexpression of heat shock proteins within the nigral dopaminergic neurons. Representative fluorescent photomicrographs showing tyrosine hydroxylase immunopositive staining in the substantia nigra (left panels – red). Following a stereotaxic injection of AAV‐GFP, AAV‐Hsp27, or AAV‐Hsp70 into the substantia nigra, the transgene protein products were strongly expressed 2 weeks postinjection (center left panels‐green). Colocalization of GFP, Hsp27, and Hsp70 within the tyrosine hydroxylase positive cell bodies in the substantia nigra was confirmed in the merged images (center right and magnified right panels of boxed region). Scale bar left panel = 500 μm, scale bar right panel = 125 μm. AAV, adeno‐associated virus; GFP, green fluorescent protein; TH, tyrosine hydroxylase.
Coexpression of Transgene Protein Products within the Nigral Dopaminergic Neurons
In the main study, we first sought to determine if coinjection of the AAV vectors into the substantia nigra resulted in coexpression of the transgenes in the nigrostriatal dopaminergic neurons using triple immunofluorescence. Results confirmed that when AAV‐α‐synuclein and AAV‐GFP were coinjected into the substantia nigra, both transgenes were successfully coexpressed within nigral tyrosine hydroxylase immunopositive neurons (Figure 2A). Quantitative analysis of the triple immunopositive cells revealed that 32 ± 2% of nigral tyrosine hydroxylase immunopositive cell bodies were successfully transduced by both coinjected viruses (Figure 2B).
Figure 2.

Coexpression of transgene protein products within the nigral dopaminergic neurons. Following unilateral intranigral coinjection of two AAV vectors, triple immunofluorescent staining was used to determine if the different transgenes were coexpressed in the nigral dopaminergic neurons. (A) Representative fluorescent photomicrographs showing tyrosine hydroxylase (red), AAV‐mediated GFP expression (green) and AAV‐mediated human α‐synuclein (blue) expression in the substantia nigra. Colocalization of α‐synuclein and GFP within the tyrosine hydroxylase immunopositive cells in the substantia nigra is shown in the merged image. (B) Quantitative analysis of the triple immunopositive cells revealed that 32 ± 2% of nigral tyrosine hydroxylase immunopositive cell bodies were successfully transduced by both coinjected vectors. Scale bars: Top panels = 500 μm; Bottom panels = 70 μm. AAV, adeno‐associated virus; GFP, green fluorescent protein; TH, tyrosine hydroxylase.
AAV‐Mediated Overexpression of Human α‐Synuclein in Nigrostriatal Neurons
Quantitative immunohistochemical analysis for α‐synuclein in the ventral midbrain and striatum was completed in order to confirm that the transgene was successfully delivered to the nigrostriatal neurons and expressed in both the cell bodies and terminals. In line with previous studies in our laboratory 39, 40, injection of AAV‐α‐synuclein caused significant accumulation of the human α‐synuclein protein in the ipsilateral nigrostriatal cell bodies (Figure 3Ai,Bi; Group, F 3,36 = 7.52, P < 0.01) along with a significant increase in the density of human α‐synuclein immunopositive staining in the striatal terminals (Figure 3Aii,Bii; Group, F 3,36 = 9.40, P < 0.001). Although there was a tendency for reduction in α‐synuclein levels at the terminal level, neither coexpression of Hsp27 nor Hsp70 had any significant impact on the overall levels of human α‐synuclein expression in either the ventral mesencephalon (Figure 3Ai,Bi) or the striatal terminals (Figure 3Aii,Bii). It should also be noted that the α‐synuclein antibody used also cross‐reacted with endogenous rat α‐synuclein as α‐synuclein immunoreactivity was also detected in the nigrostriatal cell bodies in the Intact group (Figure 3Ai,Bi).
Figure 3.

Adeno‐associated virus (AAV)‐mediated overexpression of human α‐synuclein in nigrostriatal neurons. Immunohistochemical staining for α‐synuclein revealed significant human α‐synuclein accumulation in the substantia nigra (Ai) and in the striatal terminals (Aii) in the lesion group compared to those animals which received control viral injections. A similar pattern of human α‐synuclein staining was also evident in animals that received both AAV‐α‐synuclein and AAV‐Hsp27 or AAV‐Hsp70 indicating that overall human α‐synuclein expression was unaffected by coexpression of Hsp27 or Hsp70. Quantification of immunopositive staining revealed that injection of AAV‐α‐synuclein resulted in a significant increase in human α‐synuclein expression in the nigral cell bodies (Bi) and in the striatal terminals (Bii). This level of human α‐synuclein expression was not significantly affected by coexpression of Hsp27 or Hsp70. Scale bars: Left panels = 500 μm; center panels = 100 μm; right panels = 3 mm. **P < 0.01 and ***P < 0.001 versus intact group by one‐way ANOVA with post hoc Fisher's least significant difference test. All data are expressed as mean ± SEM AAV, adeno‐associated virus; VM, ventral mesencephalon.
Confirmation of the Predegenerative Status of this AAV‐α‐Synuclein Model
As indicated above, previous studies using the AAV‐α‐synuclein model in our laboratory have revealed that α‐synuclein‐induced motor dysfunction and neurodegeneration begins to emerge approximately 18 weeks after viral infusion 39. Therefore, this time‐point was chosen for sacrifice in the present study to ensure a presymptomatic, predegenerative AAV model. In line with this previous report, we did not observe any motor dysfunction in these animals up to this time‐point (data not shown). To confirm the predegenerative status of this AAV‐α‐synuclein model, the impact of α‐synuclein on tyrosine hydroxylase immunopositive cell bodies in the substantia nigra and terminals in the striatum was assessed. Injection of AAV‐α‐synuclein did not cause any loss of tyrosine hydroxylase immunopositive cell bodies from the substantia nigra (Figure 4Ai,Bi; Group, F 3,36 = 2.33, P = 0.09, ns) or tyrosine hydroxylase immunopositive terminals from the striatum (Figure 4Aii,Bii; Group, F 3,36 = 1.45, P = 0.24, ns). In the combined AAV‐α‐synuclein and AAV‐Hsp27 or AAV‐Hsp70 groups, there was no effect of coinjection of both viruses on nigrostriatal integrity at the level of the cell bodies (Figure 4Ai,Bi) or terminals (Figure 4Aii,Bii).
Figure 4.
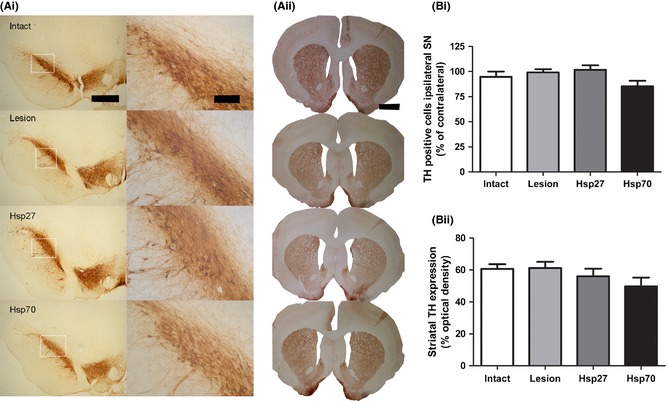
Confirmation of the predegenerative status of this AAV‐α‐synuclein model. Representative photomicrographs of immunohistochemical staining for tyrosine hydroxylase revealed that AAV‐α‐synuclein did not induce any loss of dopaminergic neurons from the substantia nigra (Ai) or striatal terminals (Aii). Coadministration of AAV‐Hsp27 or AAV‐Hsp70 with pathogenic AAV‐α‐synuclein also had no effect on nigrostriatal integrity. Quantification of tyrosine hydroxylase immunopositive cell bodies (Bi) and striatal terminals (Bii) confirmed that injection of AAV‐α‐synuclein alone or coinjection with putative therapeutic viruses AAV‐Hsp27 or Hsp70 had no effect on nigrostriatal integrity. Scale bars: Left panels = 500 μm; center panels = 100 μm, right panels = 3 mm. All data are expressed as mean ± SEM AAV, adeno‐associated virus; SN, substantia nigra; TH, tyrosine hydroxylase.
Overexpression of Hsp70 Reduces AAV‐α‐Synuclein‐Induced Neuritic Dystrophy
In this predegenerative AAV model, tyrosine hydroxylase immunohistochemical staining revealed that injection of AAV‐α‐synuclein caused a significant increase in the numbers of dystrophic axon terminals present in the striatum (Figure 5A,B; Group, F 3,36 = 6.42, P < 0.01). Quantification of the numbers of distorted axon terminals revealed that injection of AAV‐Hsp27 did not significantly reduce the number of dystrophic neurites in the striatum. However, coinjection of AAV‐α‐synuclein and AAV‐Hsp70 resulted in a statistically significant decrease in the number of dystrophic neurites present in the ipsilateral striatum (Figure 5A,B; post hoc Fisher's least significant difference confirmed Lesion ≈ Hsp27 > Hsp70 > Intact).
Figure 5.

Overexpression of Hsp70 reduces AAV‐α‐synuclein‐induced neuritic dystrophy. (A) Representative photomicrographs of immunohistochemical staining for tyrosine hydroxylase revealed that AAV‐α‐synuclein caused nigrostriatal terminal pathology evidenced by morphologically aberrant, swollen axonal terminal in the striatum (arrows and inset). (B) Quantitative analysis confirmed that AAV‐α‐synuclein resulted in a significant increase in the expression of dystrophic neurites compared to intact controls. The levels of dystrophic neurites were unaffected by Hsp27 expression, however, Hsp70 expression resulted in a significant decrease in the levels of dystrophic neurites compared to the lesioned animals. Dystrophic neurites indicated by arrows. Scale bar = 50 μm. *P < 0.05; **P < 0.01 and ***P < 0.001 versus intact group, + P < 0.05 versus lesion group by one‐way ANOVA with post hoc Fisher's least significant difference test. All data are expressed as mean ± SEM. AAV, adeno‐associated virus.
Discussion
The present experiment sought to determine if virally mediated heat shock protein overexpression could modulate α‐synuclein‐induced pathology in a predegenerative, early‐stage model of Parkinson's disease, thereby providing a potential disease‐modulating therapeutic approach for the human condition. To address this, we used the AAV human wild‐type α‐synuclein overexpression rat model of Parkinson's disease 14 and investigated the effect of AAV‐mediated overexpression of Hsp27 and Hsp70 on α‐synuclein‐induced nigrostriatal pathology. This study found that viral overexpression of Hsp70, but not Hsp27, significantly reduced AAV‐α‐synuclein‐induced axonal aberrations in the striatum. This finding supports the continued preclinical evaluation of heat shock protein based therapeutics to modulate α‐synuclein‐induced neuronal pathology.
For almost two decades, interest in α‐synuclein as a potential disease‐modulating therapeutic target in Parkinson's disease has increased steadily. This interest has led to the development of reliable animal models where genetic or viral overexpression of the wild‐type or mutant protein can recapitulate many of the key features of the disease [reviewed in 13, 45]. In this study, following an intra‐nigral AAV‐α‐synuclein injection, significant overexpression of the human α‐synuclein protein was evident both in the nigrostriatal cell bodies as well as the striatal terminals, thereby confirming successful transduction of the target neurons 46. This increased α‐synuclein expression induced formation of dystrophic neurites, a well‐established characteristic of the AAV α‐synucleinopathy model 9, 14. These dystrophic neurites are also present in the human condition, and are evident postmortem in the putamen of early stage Parkinson's disease patients before overt dopaminergic cell loss in the substantia nigra 8. Moreover, a recent postmortem report of Parkinson's disease patients highlights that loss of dopaminergic markers of the axonal terminals in the dorsal putamen occurs rapidly and is virtually complete by 4 years postdiagnosis 47. This indicates that there is a therapeutic “window‐of‐opportunity” for early neuroprotective intervention in Parkinson's disease, and it is becoming increasingly evident that the stage at which neuroprotective therapies are applied in Parkinson's disease patients will have significant influence on achieving satisfactory clinical outcome measures 48. This emphasizes the importance of defining patients at the early disease phase, with premotor and prodromal Parkinson's disease, where enrollment in neuroprotective studies is more likely to preserve the nigrostriatal dopaminergic pathway. Given that we saw dystrophic neurites without overt dopaminergic cell loss in our preclinical model, this provided the possibility of evaluating putative therapeutic heat shock proteins in early‐stage disease pathology when the nigrostriatal dopaminergic pathway is intact but dystrophic.
Given the expanse of in vitro and in vivo data supporting the potential of heat shock proteins to modulate misfolded proteins 49, we used viral vectors to overexpress Hsp27 and Hsp70 in this predegenerative AAV‐α‐synuclein animal model. Significant overexpression of the putative therapeutic proteins in the targeted dopaminergic cells was achieved using the AAV vectors. Overexpression of the putative therapeutic proteins reduced the appearance of dystrophic neurites in the striatum, with coinjection of AAV‐α‐synuclein and AAV‐Hsp70, but not AAV‐Hsp27, leading to a significant decrease in the number of dystrophic terminals in the striatum indicating that Hsp70 is protective against AAV‐α‐synuclein‐induced pathology in the nigrostriatal terminals. Moreover, because only 32% of tyrosine immunopositive nigral cells were cotransduced by the disease‐causing and therapeutic viruses, it is possible that a greater therapeutic effect would have been observed if greater expression had been achieved.
The positive effects of Hsp70 overexpression on α‐synuclein‐induced pathology seen in our study are in line with previously reported studies on the protective effects of Hsp70 in models of Parkinson's disease. In a seminal study, Auluck et al. 31 demonstrated that directed coexpression of Hsp70 protected against dopaminergic neuronal loss associated with α‐synuclein in Drosophila melanogaster. Similarly, it was demonstrated that cross breeding α‐synuclein transgenic mice with Hsp70 transgenic mice led to a significant reduction in both the high molecular weight and detergent‐insoluble α‐synuclein species 33. Given the numerous protective roles of Hsp70, it is difficult to definitively postulate as to the mechanism through which Hsp70 may act as a neuroprotectant in these α‐synuclein overexpression animal models. One theory is based on a large body of literature highlighting that Hsp70 is capable of altering α‐synuclein fibrillization kinetics, thus modulating the toxicity of α‐synuclein. For example, Dedmon et al. 18 demonstrated that Hsp70 strongly inhibited α‐synuclein fibril formation via preferential binding to prefibrillar species. Similarly, Huang et al. 19 demonstrated in vitro that Hsp70 is able to change the properties of fibril formation of α‐synuclein by interactions with diverse intermediates of α‐synuclein formed during fibrillization. Luk et al. 20 found that in vitro assembly of α‐synuclein was efficiently inhibited by substoichiometric concentrations of purified Hsp70 in the absence of cofactors. In this same study, experiments using α‐synuclein deletion mutants indicated that interactions between the Hsp70 substrate binding domain and the α‐synuclein core hydrophobic region underlie assembly inhibition. Thus, there is substantial preclinical evidence that Hsp70 can reduce the neurotoxicity of a‐synuclein by preventing the aggregation of this protein. However, it is important to note that it is not yet known if this molecular chaperone can assist with the clearance of pre‐formed Lewy bodies which may be an important determinant of clinical efficacy in Parkinson's disease.
Particularly interesting, and highlighting a potential reason as to why we observed a beneficial effect on α‐synuclein‐induced pathology with Hsp70 but not Hsp27, is the ability of Hsp70 to chaperone α‐synuclein extracellularly. Danzer et al. 21 reported that α‐synuclein is present in its oligomeric form in extracellular space, from where it taken up by neighboring cells and mediates its toxic effects (contributing to theory of prion mediated transfer of α‐synuclein disease pathology 50, 51). In their study, Hsp70 chaperoned α‐synuclein in the extracellular space and prevented extracellular oligomer formation. The authors show that Hsp70's rescue of oligomer‐induced toxicity is accompanied by a concomitant modification of the α‐synuclein oligomeric species in the extracellular space. Thus, it is intriguing to speculate that this extracellular chaperone role of Hsp70 52, a phenomenon not yet reported for Hsp27, could have been partly responsible for the beneficial effects seen in this study.
In toxin models of Parkinson's disease, Hsp70 has also shown to have positive effects on pathology. In an MPTP Parkinson's disease model, AAV‐Hsp70 gene transfer significantly protected the mouse dopaminergic system against MPTP‐induced dopamine neuron loss and the associated decline in striatal dopamine levels and tyrosine hydroxylase‐positive fibers 32. Systemic application of cell‐permeable Hsp70 has also been shown to be protective for dopamine neurons of the substantia nigra against subacute toxicity of MPTP 37. The mechanisms through which Hsp70 appears to be neuroprotectant in these models may reflect Hsp70s ability to regulate the apoptotic pathway. Hsp70 is capable of binding the caspase‐recruitment domain of Apaf‐1 thereby preventing recruitment of procaspase‐9 to the apoptosome complex and blocking the assembly of a functional apoptosome 53, 54. Hsp70 can modulate apoptosis inducing factor activity 55, 56, 57, interfere with the Bid‐dependent apoptotic pathway via inhibition of JNK 58 and inhibit the death‐associated permeabilization of lysosomes 59. Taken together, a number of in vivo studies indicate the significant potential of Hsp70 as a potential disease modifying, neuroprotective target for Parkinson's disease, while its precise mechanism of action in these models remains to be fully elucidated.
In this study, Hsp27 overexpression did not reduce dystrophic neurites in the striatum compared to the AAV‐α‐synuclein lesion group. To our knowledge, no in vivo studies have been completed determining the effect of Hsp27 on α‐synuclein aggregation making it difficult to compare our results in an in vivo context. Overexpression of Hsp27 in a chronic model of Huntington's disease, the R2/6 transgenic mouse, where intraneuronal polyglutamine protein aggregations are characteristic, did not prevent the formation of aggregates or the progression of the disease 60. However, in other in vitro and in vivo models of Huntington's disease, overexpression of Hsp27 has been shown to decrease cell death without reducing the formation of aggregations 61, 62. In our experimental paradigm, it is conceivable that the levels of expression of Hsp27 that we achieved were not sufficient to have an appreciable protective effect. Thus, while a number of studies suggest that Hsp27 has protective effects against α‐synuclein in vitro 17, 22, our limited in vivo effect suggests that continued preclinical studies are warranted to establish its potential.
Conclusion
As dystrophic terminals with degenerating bulbs are thought to precede neuronal loss in Parkinson's disease 9, 63, efforts to delay axonal dysfunction may preserve nigrostriatal connectivity thereby allowing afflicted patients to maintain an improved quality of life. In this experiment, Hsp70 significantly reduced the number of dystrophic neurites in the striatum in an early‐stage, predegenerative model of Parkinson's disease. These data suggest that overexpression of Hsp70 holds significant potential as a disease‐modulating therapeutic approach for Parkinson's disease.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
We thank Dr. Sinéad Walsh for assistance with surgery. This project was funded by Science Foundation Ireland under the Research Frontiers Program (07/RFP/BIMF463).
The first two authors contributed equally to this work.
References
- 1. Chartier‐Harlin MC, Kachergus J, Roumier C, et al. Alpha‐synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 2004;364:1167–1169. [DOI] [PubMed] [Google Scholar]
- 2. Farrer M, Kachergus J, Forno L, et al. Comparison of kindreds with parkinsonism and alpha‐synuclein genomic multiplications. Ann Neurol 2004;55:174–179. [DOI] [PubMed] [Google Scholar]
- 3. Kruger R, Kuhn W, Muller T, et al. Ala30Pro mutation in the gene encoding alpha‐synuclein in Parkinson's disease. Nat Genet 1998;18:106–108. [DOI] [PubMed] [Google Scholar]
- 4. Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha‐synuclein gene identified in families with Parkinson's disease. Science 1997;276:2045–2047. [DOI] [PubMed] [Google Scholar]
- 5. Singleton AB, Farrer M, Johnson J, et al. Alpha‐Synuclein locus triplication causes Parkinson's disease. Science 2003;302:841. [DOI] [PubMed] [Google Scholar]
- 6. Zarranz JJ, Alegre J, Gomez‐Esteban JC, et al. The new mutation, E46K, of alpha‐synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004;55:164–173. [DOI] [PubMed] [Google Scholar]
- 7. Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH. Alterations in lysosomal and proteasomal markers in Parkinson's disease: Relationship to alpha‐synuclein inclusions. Neurobiol Dis 2009;35:385–398. [DOI] [PubMed] [Google Scholar]
- 8. Chu Y, Morfini GA, Langhamer LB, He Y, Brady ST, Kordower JH. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson's disease. Brain 2012;135:2058–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha‐synucleinopathy. J Neurosci 2009;29:3365–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morfini GA, Burns M, Binder LI, et al. Axonal transport defects in neurodegenerative diseases. J Neurosci 2009;29:12776–12786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Raff MC, Whitmore AV, Finn JT. Axonal self‐destruction and neurodegeneration. Science 2002;296:868–871. [DOI] [PubMed] [Google Scholar]
- 12. Roy S, Zhang B, Lee VM, Trojanowski JQ. Axonal transport defects: A common theme in neurodegenerative diseases. Acta Neuropathol 2005;109:5–13. [DOI] [PubMed] [Google Scholar]
- 13. Low K, Aebischer P. Use of viral vectors to create animal models for Parkinson's disease. Neurobiol Dis 2012;48:189–201. [DOI] [PubMed] [Google Scholar]
- 14. Kirik D, Rosenblad C, Burger C, et al. Parkinson‐like neurodegeneration induced by targeted overexpression of alpha‐synuclein in the nigrostriatal system. J Neurosci 2002;22:2780–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luo GR, Le WD. Collective roles of molecular chaperones in protein degradation pathways associated with neurodegenerative diseases. Curr Pharm Biotechnol 2010;11:180–187. [DOI] [PubMed] [Google Scholar]
- 16. Stetler RA, Gan Y, Zhang W, et al. Heat shock proteins: Cellular and molecular mechanisms in the central nervous system. Prog Neurobiol 2010;92:184–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bruinsma IB, Bruggink KA, Kinast K, et al. Inhibition of alpha‐synuclein aggregation by small heat shock proteins. Proteins 2011;79:2956–2967. [DOI] [PubMed] [Google Scholar]
- 18. Dedmon MM, Christodoulou J, Wilson MR, Dobson CM. Heat shock protein 70 inhibits alpha‐synuclein fibril formation via preferential binding to prefibrillar species. J Biol Chem 2005;280:14733–14740. [DOI] [PubMed] [Google Scholar]
- 19. Huang C, Cheng H, Hao S, et al. Heat shock protein 70 inhibits alpha‐synuclein fibril formation via interactions with diverse intermediates. J Mol Biol 2006;364:323–336. [DOI] [PubMed] [Google Scholar]
- 20. Luk KC, Mills IP, Trojanowski JQ, Lee VM. Interactions between Hsp70 and the hydrophobic core of alpha‐synuclein inhibit fibril assembly. Biochemistry 2008;47:12614–12625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Danzer KM, Ruf WP, Putcha P, et al. Heat‐shock protein 70 modulates toxic extracellular alpha‐synuclein oligomers and rescues trans‐synaptic toxicity. FASEB J 2011;25:326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Outeiro TF, Klucken J, Strathearn KE, et al. Small heat shock proteins protect against alpha‐synuclein‐induced toxicity and aggregation. Biochem Biophys Res Commun 2006;351:631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Outeiro TF, Putcha P, Tetzlaff JE, et al. Formation of toxic oligomeric alpha‐synuclein species in living cells. PLoS ONE 2008;3:e1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zourlidou A. Payne Smith MD, Latchman DS. HSP27 but not HSP70 has a potent protective effect against alpha‐synuclein‐induced cell death in mammalian neuronal cells. J Neurochem 2004;88:1439–1448. [DOI] [PubMed] [Google Scholar]
- 25. Abisambra JF, Jinwal UK, Jones JR, Blair LJ, Koren J 3rd, Dickey CA. Exploiting the diversity of the heat‐shock protein family for primary and secondary tauopathy therapeutics. Curr Neuropharmacol 2011;9:623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Howarth JL, Glover CP, Uney JB. HSP70 interacting protein prevents the accumulation of inclusions in polyglutamine disease. J Neurochem 2009;108:945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malik B, Nirmalananthan N, Gray AL, La Spada AR, Hanna MG, Greensmith L. Co‐induction of the heat shock response ameliorates disease progression in a mouse model of human spinal and bulbar muscular atrophy: Implications for therapy. Brain 2013;136:926–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miyata Y, Li X, Lee HF, et al. Synthesis and initial evaluation of YM‐08, a blood‐brain barrier permeable derivative of the heat shock protein 70 (Hsp70) inhibitor MKT‐077, which reduces tau levels. ACS Chem Neurosci 2013;4:930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Patterson KR, Ward SM, Combs B, et al. Heat shock protein 70 prevents both tau aggregation and the inhibitory effects of preexisting tau aggregates on fast axonal transport. Biochemistry 2011;50:10300–10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Toth ME, Szegedi V, Varga E, et al. Overexpression of Hsp27 ameliorates symptoms of Alzheimer's disease in APP/PS1 mice. Cell Stress Chaperones 2013;18:759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha‐synuclein toxicity in a Drosophila model for Parkinson's disease. Science 2002;295:865–868. [DOI] [PubMed] [Google Scholar]
- 32. Dong Z, Wolfer DP, Lipp HP, Bueler H. Hsp70 gene transfer by adeno‐associated virus inhibits MPTP‐induced nigrostriatal degeneration in the mouse model of Parkinson disease. Mol Ther 2005;11:80–88. [DOI] [PubMed] [Google Scholar]
- 33. Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ. Hsp70 reduces alpha‐synuclein aggregation and toxicity. J Biol Chem 2004;279:25497–25502. [DOI] [PubMed] [Google Scholar]
- 34. Lo Bianco C, Shorter J, Regulier E, et al. Hsp104 antagonizes alpha‐synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J Clin Invest 2008;118:3087–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marber MS, Mestril R, Chi SH, Sayen MR, Yellon DM, Dillmann WH. Overexpression of the rat inducible 70‐kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J Clin Invest 1995;95:1446–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Masliah E, Rockenstein E, Veinbergs I, et al. Dopaminergic loss and inclusion body formation in alpha‐synuclein mice: Implications for neurodegenerative disorders. Science 2000;287:1265–1269. [DOI] [PubMed] [Google Scholar]
- 37. Nagel F, Falkenburger BH, Tonges L, et al. Tat‐Hsp70 protects dopaminergic neurons in midbrain cultures and in the substantia nigra in models of Parkinson's disease. J Neurochem 2008;105:853–864. [DOI] [PubMed] [Google Scholar]
- 38. Shimshek DR, Mueller M, Wiessner C, Schweizer T, van der Putten PH. The HSP70 molecular chaperone is not beneficial in a mouse model of alpha‐synucleinopathy. PLoS ONE 2010;5:e10014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mulcahy P, O'Doherty A, Paucard A, O'Brien T, Kirik D, Dowd E. Development and characterisation of a novel rat model of Parkinson's disease induced by sequential intranigral administration of AAV‐alpha‐synuclein and the pesticide, rotenone. Neuroscience 2012;203:170–179. [DOI] [PubMed] [Google Scholar]
- 40. Mulcahy P, O'Doherty A, Paucard A, O'Brien T, Kirik D, Dowd E. The behavioural and neuropathological impact of intranigral AAV‐alpha‐synuclein is exacerbated by systemic infusion of the Parkinson's disease‐associated pesticide, rotenone, in rats. Behav Brain Res 2013;243:6–15. [DOI] [PubMed] [Google Scholar]
- 41. Grimm D, Kern A, Rittner K, Kleinschmidt JA. Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum Gene Ther 1998;9:2745–2760. [DOI] [PubMed] [Google Scholar]
- 42. Smith JC, Bolon B. Isoflurane leakage from non‐rebreathing rodent anaesthesia circuits: Comparison of emissions from conventional and modified ports. Lab Anim 2006;40:200–209. [DOI] [PubMed] [Google Scholar]
- 43. Moloney TC, Rooney GE, Barry FP, Howard L, Dowd E. Potential of rat bone marrow‐derived mesenchymal stem cells as vehicles for delivery of neurotrophins to the Parkinsonian rat brain. Brain Res 2010;1359:33–43. [DOI] [PubMed] [Google Scholar]
- 44. Moloney TC, Dockery P, Windebank AJ, Barry FP, Howard L, Dowd E. Survival and immunogenicity of mesenchymal stem cells from the green fluorescent protein transgenic rat in the adult rat brain. Neurorehabil Neural Repair 2010;24:645–656. [DOI] [PubMed] [Google Scholar]
- 45. Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson's disease. Neuron 2010;66:646–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bjorklund A, Kirik D, Rosenblad C, Georgievska B, Lundberg C, Mandel RJ. Towards a neuroprotective gene therapy for Parkinson's disease: Use of adenovirus, AAV and lentivirus vectors for gene transfer of GDNF to the nigrostriatal system in the rat Parkinson model. Brain Res 2000;886:82–98. [DOI] [PubMed] [Google Scholar]
- 47. Kordower JH, Olanow CW, Dodiya HB, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain 2013;136:2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marks WJ Jr, Bartus RT, Siffert J, et al. Gene delivery of AAV2‐neurturin for Parkinson's disease: A double‐blind, randomised, controlled trial. Lancet Neurol 2010;9:1164–1172. [DOI] [PubMed] [Google Scholar]
- 49. Arawaka S, Machiya Y, Kato T. Heat shock proteins as suppressors of accumulation of toxic prefibrillar intermediates and misfolded proteins in neurodegenerative diseases. Curr Pharm Biotechnol 2010;11:158–166. [DOI] [PubMed] [Google Scholar]
- 50. Olanow CW, Brundin P. Parkinson's disease and alpha synuclein: Is Parkinson's disease a prion‐like disorder? Mov Disord 2013;28:31–40. [DOI] [PubMed] [Google Scholar]
- 51. Steiner JA, Angot E, Brundin P. A deadly spread: Cellular mechanisms of alpha‐synuclein transfer. Cell Death Differ 2011;18:1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tytell M. Release of heat shock proteins (Hsps) and the effects of extracellular Hsps on neural cells and tissues. Int J Hyperthermia 2005;21:445–455. [DOI] [PubMed] [Google Scholar]
- 53. Beere HM, Wolf BB, Cain K, et al. Heat‐shock protein 70 inhibits apoptosis by preventing recruitment of procaspase‐9 to the Apaf‐1 apoptosome. Nat Cell Biol 2000;2:469–475. [DOI] [PubMed] [Google Scholar]
- 54. Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES. Negative regulation of the Apaf‐1 apoptosome by Hsp70. Nat Cell Biol 2000;2:476–483. [DOI] [PubMed] [Google Scholar]
- 55. Gurbuxani S, Schmitt E, Cande C, et al. Heat shock protein 70 binding inhibits the nuclear import of apoptosis‐inducing factor. Oncogene 2003;22:6669–6678. [DOI] [PubMed] [Google Scholar]
- 56. Ravagnan L, Gurbuxani S, Susin SA, et al. Heat‐shock protein 70 antagonizes apoptosis‐inducing factor. Nat Cell Biol 2001;3:839–843. [DOI] [PubMed] [Google Scholar]
- 57. Ruchalski K, Mao H, Singh SK, et al. HSP72 inhibits apoptosis‐inducing factor release in ATP‐depleted renal epithelial cells. Am J Physiol Cell Physiol 2003;285:C1483–C1493. [DOI] [PubMed] [Google Scholar]
- 58. Gabai VL, Mabuchi K, Mosser DD, Sherman MY. Hsp72 and stress kinase c‐jun N‐terminal kinase regulate the bid‐dependent pathway in tumor necrosis factor‐induced apoptosis. Mol Cell Biol 2002;22:3415–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nylandsted J, Gyrd‐Hansen M, Danielewicz A, et al. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med 2004;200:425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zourlidou A, Gidalevitz T, Kristiansen M, et al. Hsp27 overexpression in the R6/2 mouse model of Huntington's disease: Chronic neurodegeneration does not induce Hsp27 activation. Hum Mol Genet 2007;16:1078–1090. [DOI] [PubMed] [Google Scholar]
- 61. Perrin V, Regulier E, Abbas‐Terki T, et al. Neuroprotection by Hsp104 and Hsp27 in lentiviral‐based rat models of Huntington's disease. Mol Ther 2007;15:903–911. [DOI] [PubMed] [Google Scholar]
- 62. Wyttenbach A, Sauvageot O, Carmichael J, Diaz‐Latoud C, Arrigo AP, Rubinsztein DC. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet 2002;11:1137–1151. [DOI] [PubMed] [Google Scholar]
- 63. Lundblad M, Decressac M, Mattsson B, Bjorklund A. Impaired neurotransmission caused by overexpression of alpha‐synuclein in nigral dopamine neurons. Proc Natl Acad Sci U S A 2012;109:3213–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]