

Summary
Background and purpose
The immunosuppressant drug rapamycin was reported to have an antiaging activity, which was attributed to the TORC1 inhibition that inhibits cell proliferation and increases autophagy. However, rapamycin also exhibits a number of harmful adverse effects. Whether rapamycin can be developed into an antiaging agent remains unclear.
Methods and results
We demonstrated that rapamycin at micro‐doses (below the TORC1 inhibiting concentration) exhibits a cell‐protective activity: (1) It protects cultured neurons against neurotoxin MPP + and H2O2. (2) It increases survival time of neuron in culture. (3) It maintains the nonproliferative state of cultured senescent human fibroblasts and prevents cell death induced by telomere dysfunction. (4) In animal models, it decreased the cerebral infarct sizes induced by acute ischemia and dramatically extended the life span of stroke prone spontaneously hypertensive rats (SHR‐SPs).
Conclusion
We propose that rapamycin at micro‐dose can be developed into an antiaging agent with a novel mechanism.
Keywords: Rapamycin, Antiaging, Ischemic infarction, Senescence, Life span
Introduction
Since its discovery as an antifungal agent in 1970s, rapamycin has been developed as an immunosuppressant for organ transplantation in clinical. Based on its antiproliferative activity, its other uses are also explored; for instance, a use in anticancer, autoimmune diseases, and restenosis on coronary artery‐stents 1. Recently, its potential antiaging activity has attracted many attention.
Aging is an important risk factor to many human diseases including degenerative diseases, cardiovascular diseases, cancers, and mellitus diabetes. Antiaging agents could be used for preventing a variety of diseases related to aging. Dietary restriction (DR) is the well‐known practical method to delay aging and prevent these age‐related diseases or disorders 2, 3, 4, 5, 6. Thus, DR‐mimicking drug candidates that target the aging risk could help to control these diseases. Growing evidence suggests that decreased target of rapamycin (TOR) activity is associated with delaying aging in many species, and DR may prolong life span by decreasing mammalian TOR (mTOR) activity 7, 8. Rapamycin, the TOR inhibitor, is well‐known to inhibit the formation of TORC1 complex (TOR‐LST8‐raptor complex) and subsequently inhibit protein translation, slow the progression of cell cycle at G1‐phase, and increase autophagy 1, 9, 10. This mechanism supports its use as an immunosuppressant and antiproliferative agent, as well as a drug candidate to prevent aggregation of abnormal proteins of neurons in such as Huntington disease 11. However, the use of the immunosuppressant rapamycin as an antiaging agent is limited as TORC1 inhibition by rapamycin causes adversary effects such as problems in wound healing and tissue repair, anemia and vulnerable to infection 12. Hyperlipidaemia, nephrotoxicity, and hepatoxicity are also adversary effects that are unfavorable for longevity. Here, we demonstrated that rapamycin at micro‐doses that is below the TORC1 inhibition concentration, exhibits a novel action: prolongs the nonproliferative state and prevent the subsequent cell death. A series of experiments were conducted to test the activity of micro‐dose rapamycin in preventing age‐related diseases and to explore its underline mechanism.
Materials and Methods
Animals
Male SHR‐SPs were provided by the Animal Centre of the Second Military Medical University. Male C57BL/6 mice and Sprague–Dawley (SD) rats were purchased from Sino‐British SIPPR/BK Lab Animals (Shanghai, China). All experiments involving animal subjects were approved by the Institutional Animal Care and Use Committee at the Second Military Medical University.
Drugs and Kits
Rapamycin was purchased from LC laboratories (Woburn, MA, USA), dimethyl sulfoxide (DMSO) and 2‐3‐5‐triphenyltetrazolium chloride (TTC) were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). DAPI and rhodamine phalloidin were purchased from Invitrogen (Carlsbad, CA, USA). 5‐Bromo‐4‐chloro‐3‐indolyl β‐Dgalactopyranoside (X‐gal), 2‐deoxy‐glucose, 1‐methyl‐4‐phenylpyridinium (MPP+), and thiazolyl blue tetrazolium bromide (MTT) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Antibodies were purchased from cell signaling Technology (Danvers, MA, USA). Human primary embryonic lung fibroblast WI‐38 was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were normally cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (from Gemini Bio‐Product, Woodland, CA, USA) at 37°C in a 5% CO2 incubator. WI‐38 cells were split 1:3 upon confluency at early passages and 1:2 at late passages (after passage 29). Cells were replenished with fresh medium every 3 days.
Senescence Extension in WI‐38 Cells by Rapamycin and Other Agents
Cells (passage 29) were incubated with rapamycin or vehicle (DMSO) for 3 days and then incubated in drug‐free medium for 7 days in a 10‐day cycle (3‐day‐on/7‐day‐off cycle), independent of cell splitting. At later passages (30–32), if no cell growth was observed after 10 days, cells were stained with X‐gal to detect cellular β‐galactosidase activity, a hallmark of senescence. If stained positive (blue color), the last split date was defined as day 1 for senescence. If no cell growth was observed after 20 days postsplitting, the last split date was defined as day 1 for senescence.
X‐gal Staining for β‐galactosidase Activity
Cells were briefly fixed with phosphate buffer solution (PBS) buffer containing 2% formaldehyde and 0.2% glutaraldehyde and stained with 1 mg/mL X‐gal (in buffer containing 40 mM citric acid/sodium phosphate, pH 6.0, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, and 2 mM MgCl2) at 37°C for 18 h for cellular β‐galactosidase activity. Cells were viewed under a microscope.
MTT Staining for Viable Cells
In brief, cells were stained with 0.1 mg/mL MTT (Sigma) for 8 h and then dissolved in DMSO. MTT values were measured at 570 nm by a Biorad 3550 microplate reader 13, 14.
Nucleus and Actin Staining
Cells were fixed by PBS containing 2% formaldehyde and 0.2% glutaraldehyde and stained with DAPI (for nuclei) and rhodamine phalloidin that specifically binds to actin (for cell morphology) 15. Cells were viewed under a fluorescent microscopy. More than 100 cells were counted from each treatment.
Measuring Gene Expression Using Quantitative Real‐time Reverse Transcription Polymerase Chain Reaction (RT‐PCR)
Total RNA was isolated with Trizol reagent (Invitrogen, Carlsbad, CA, USA) from treated cells and the RNeasy Mini Kit (Qiagen, Hilden, Germany). About 1–2 μg of total RNA was used for the synthesis of first‐strand cDNA using the SuperScript III First‐Strand Synthesis System (Invitrogen). Real‐time PCR was performed in the 7900HT Fast Real‐Time PCR System (PE Applied Biosystems, Grand Island, NY, USA) with the use of SYBR Green. Results were analyzed with SDS 2.2 software using the—ΔCT method. Primer sets used are as follows: P53, 5′‐GCTGAATGAGGCCTTGGAACTCAA‐3′ (forward primer), 5′‐AGTCAGGCCCTTCTGTCTTGAACA‐3′(reverse primer). P21, 5′‐ACCATGTGGACCTGTCACTGTCTT‐3′ (forward primer), 5′‐AGAAATCTGTCATGCTGGTCTGCC‐3′ (reverse primer). P27, 5′‐AGCAATGCGCAGGAATAAGGAAGC‐3′ (forward primer), 5′‐ TACGTTTGACGTCTTCTGAGGCCA‐3′ (reverse primer).
Middle Cerebral Artery Occlusion (MCAO), Neurological Deficit Scoring and 2, 3, 5‐triphenyltetrazolium Chloride (TTC) Staining
Animals were anesthetized with 2.0% isoflurane. MCAO surgery in mice was performed as described 16, 17, 18. The core temperature was maintained 37.0°C ± 0.5°C during the surgery using a heating pad (Nanjing Xin Xiao Yuan Biotech, Nanjing, China). Focal ischemia was produced by intraluminal occlusion of the left middle cerebral artery (MCA). The silicone rubber–coated nylon monofilament (Beijing Sunbio Biotech Co., Ltd., Beijing, China) was used to occlude the MCA. Cessation of cerebral blood flow (CBF) was verified using a laser Doppler computerized main unit (ML191, ADInstruments, Australia). CBF must be reduced by at least 70% for inclusion in further experiments. Two hours after MCAO, the occluding filament was withdrawn to allow reperfusion. Permanent MCAO in SHR‐SPs was conducted by electric coagulation as reference reported 19.Twenty‐four hours after MCAO, the neurological score was measured as described 20. Then the animals were sacrificed for various examinations.
The coronal slices of brains were incubated in 1% TTC solution at 37°C for 15 min. Then the slices were photographed with a digital camera. The infarct size were traced and quantified with ImageJ software (National Institutes of Health, Bethesda, ML, USA) and expressed as a percentage of the contralateral hemisphere. The possible interference of a brain edema in assessing the infarct size was corrected with a standard method of subtracting the volume of the nonischemic ipsilateral hemisphere from that of the contralateral hemisphere.
Isolation of Rat Cerebellar Granule Neurons (CGN) and Neocortical Neurons
CGN cultures were prepared from 7‐day‐old SD rat pups. Briefly, the cerebellum was removed from the brain and placed in a Petri dish containing Basal medium Eagles's medium (BMEM) in 20 mM HEPES buffer (BMEM–HEPES). Cerebellar meninges and blood vessels were discarded to ensure minimal contamination from endothelial cells. Cerebella/cerebral cortices were then minced into fine pieces with dissecting knives and trypsinized at 37°C for 15 min. Then, trypsinization was inhibited by addition of 1 mL of BME containing 0.025% soybean trypsin inhibitor and 0.05% DNase I. The tissue was gently triturated through a fire‐polished Pasteur pipette until it was dispersed into a homogeneous suspension. The suspension was filtered through an ethanol‐sterilized 40‐μm mesh and pelleted by centrifugation. The pellets containing CGNs were resuspended in B27 supplemented neurobasal medium containing 25 mM KCl (Invitrogen). Cells were then seeded into a 24‐well plate (1 plate/cerebellum) and cultured in Neurobasal medium (Invitrogen) supplemented with B27, 20 mM KCl, 0.5 mM Glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin. The procedure of the isolation of neocortical neurons is similar to isolation of CGN. Cell injury assay was established as described by flow cytometric analysis 14, 20.
Immunoblotting
Immunoblotting was performed as previously reported 21, 22, 23. Tissue (from the cerebral cortex) or cells lysate were boiled, subjected to the SDS‐PAGE, and transferred onto nitrocellulose blotting membranes. The membranes were blocked with buffer and incubated with one of the following antibodies overnight at 4°C:
p53, pRB, eEF2‐p, p21cip, p27kip, Hsp27, Hsp70, phospho‐Ser2448, and phospho‐Ser2481 of mTOR (Cell Signaling Technology, Inc., Danvers, MA, USA). The images were captured by the Odyssey infrared imaging system (Li‐Cor Bioscience, Lincoln, NE, USA) and analyzed by ImageJ software (National Institutes of Health). The expression level of each protein was corrected against the loading control of α‐tubulin or GAPDH.
Life Span Experiment
Male SHR‐SPs of 3‐month age were employed. The animal food was purchased from Sino‐British SIPPR/BK Lab. animal LTD., CO. Rapamycin was mixed in the rat chow. Animals were fed with 3‐day rapamycin containing food followed by 4‐day normal food, and this treatment cycle was continued throughout the entire experiment. The survival time was recorded. n = 15 in each group.
Statistics
The investigators were blinded to the procedures when they assessed the infarct size of MCAO animals. The animals were randomly assigned using the random permutations table. Data are expressed as the mean ± SD. We analyzed data with 2‐tailed Student's t test (two groups) or one‐way ANOVA. Survival data were examined using Kaplan–Meier analysis (log‐rank test). P < 0.05 was considered statistically significant.
Results
Micro‐dose Rapamycin Maintains the Senescent State and Prevents Cell Loss in Human Primary Fibroblasts WI‐38
In the first experiment, WI‐38 cells near senescence (passage 29) were intermittently exposed to rapamycin (3‐day exposure for every 10 days) at a concentration (50 pM) far below that is required to inhibit TORC1 activity by 50% (2–5 nM) 24. The cells ceased dividing at passage 31 (Figure S1A) and displayed elevated cellular β‐galactosidase activity, a hallmark of senescence 25, regardless of rapamycin treatment (Figure 1A). Deterioration of senescence, however, was inhibited by micro‐dose rapamycin (25 and 50 pM): cell loss was decreased (Figure 1B,C, and Figure S1A), cell enlargement was inhibited (Figure 1B,D), and the number of the giant multinucleated cells was reduced (from 60% to <30%) (Figure 1D). The β‐galactosidase staining was positive in surviving cells (Figure 1B), suggesting that increased survival in response to rapamycin is due to persistence or prolongation of the senescence, not a result of exiting out of senescence of a mutate cells. Such effects were only observed at the micro‐doses and were in a dose‐dependent manner (10–75 pM; Figure S1B). At ≥100 pM, rapamycin potentiated cell loss (Figure 1C and Figure S1B) and resulted in massive cell loss at a much earlier time point.
Figure 1.
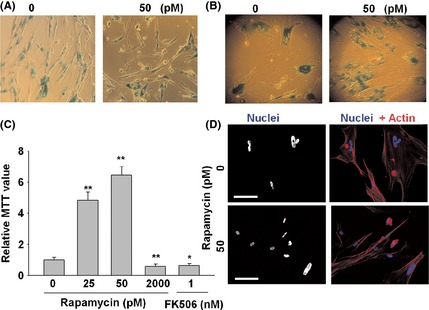
Micro‐dose rapamycin prolongs senescence in human primary fibroblast WI‐38. WI‐38 cells at passage 29 were cultured in DMEM supplemented with 10% fetal bovine serum. Cells were treated with different dose of rapamycin. The β‐galactosidase activity (top panels) was monitored on day 10 (A) or day 45 (B) after the last split (passage 31). (C) MTT assay was performed at day 79 after the last split. (D) Rapamycin reduces the number of multinucleated cells. Eighty‐four days after the last split (senescence), cells were fixed by 2% formaldehyde/0.2% glutaraldehyde solution in PBS and stained with DAPI (for nuclei) and rhodamine phalloidin that specifically binds to actin (for cell morphology). More than 100 cells were counted for the number of nuclei for each sample. Figure shows a representative view. Bars represent 100 μm. The data are expressed as the mean ± SD. *P < 0.05, **P < 0.01 vs. vehicle control (0 pM).
The expression of proteins important for senescence maintenance was increased. As shown in Figure 2A,B, rapamycin (25 and 50 pM) increased the expression of pRB, p53, p21cip, and p27kip at the protein level. Messenger RNA levels of p53, p21, and p27 were also increased (Figure 2C), suggesting a transcriptional regulation. These data support the notion that rapamycin at micro‐dose can maintain the senescent state.
Figure 2.
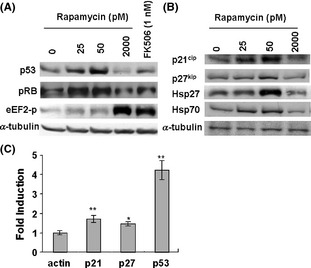
Micro‐dose rapamycin upregulates the senescence pathway components, pRB, p53, p21cip, p27kip, and heat shock protein Hsp70, Hsp27. Senescent WI‐38 cells were treated with rapamycin (25, 50, and 2000 pM) and FK506 (1 nM) for 18 h. (A‐B), Cells were then lyzed and Western blotting was performed using antibodies against p53, pRB, p21cip, p27kip, Hsp70, Hsp27, phospho‐Thr56 of eEF2, and α‐tubulin. (C) Messenger RNA of p21cip, p27kip, and p53 were quantitatively analyzed by real‐time quantitative reverse transcription PCR. The data are expressed as the mean ± SD. *P < 0.05, **P < 0.01 vs actin.
Rapamycin also increased the protein levels of representative heat shock proteins (Hsp27 and Hsp70; Figure 2B). At a higher concentration of 2000 pM, rapamycin, as well as another G1 inhibitor FK506 26, did not increase the levels of these proteins, nor prevented cell loss, but rather inhibited overall protein translation as monitored by an antibody against phospho‐eEF2 Thr56 (Figures 1C and 2A,B). Data were consistent with notion that 2000 pM rapamycin inhibited TORC1 and the subsequent protein translation. These data suggest that G1 inhibition is not responsible for the protection of the senescent cells.
Micro‐dose Rapamycin Prevents Neuron Death Induced by MPP+ and H2O2 and Prolongs the Life Span of Cultured Neurons
We next tested whether micro‐dose rapamycin protected neuron cells against disease‐related stimuli and extend the life span in culture as neuron cells are also nondividing cells. MPP+ is a neurotoxin that inhibits complex I (NADH CoQ1 reductase) in the mitochondrial respiratory chain and is commonly used to induce Parkinson's disease in animal models 27. Exposure to MPP+ (400 μM for 24 h) dramatically decreased cell activity in cultured cortical neurons (Figure 3A). Rapamycin pretreatment (1 pM for 3 h prior to MPP+ exposure) significantly increased the cell activity (Figure 3A).
Figure 3.
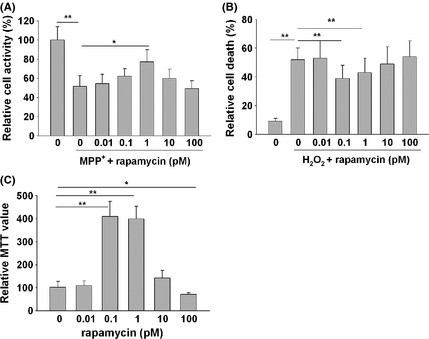
Micro‐dose rapamycin prevents cortical neuron death induced by MPP+ and H2O2 and extends the life span of cultured cerebellar granule neurons (CGN). (A‐B). Micro‐dose rapamycin prevents neuron death induced by MPP+ and H2O2. The cortical neurons from SD rats were pretreated with rapamycin for 3 h. MPP+ (200 μM) or H2O2 (50 μM) were then added into the culture and continued to incubate for 24 hours. Surviving cells after MPP+ treatment were monitored by measuring the optical density. The death cells induced by H2O2 were detected by flow cytometric analysis. All experiments were repeated three times. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01. (C) Isolated CGNs from 7‐day‐old rat pups were cultured in neurobasal medium supplemented with B27, 20 mM KCl, 0.5 mM Glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin. Rapamycin was added 7 days after the seeding. Medium was replenished every 3 days. Thirty‐one days later, surviving CGNs were determined using a MTT assay. All experiments were conducted in triplicate. Data were analyzed by one‐way analysis of variance (ANOVA) followed by LSD post hoc testing.
As shown in Figure 3B, oxidative free radical agent H2O2 (50 μM for 24 h) greatly increased cell death of cultured cortical neurons as evaluated by flow cytometric analysis 14. Rapamycin pretreatment (0.1 and 1 pM for 3 h) prevented this increase.
To monitor life span of cultured neurons, primary culture of CGN was prepared from 7‐day‐old rat pups. The majority of the cells were dead after 31 days in culture (Figure 3C; MTT assay). Rapamycin treatment (0.1 and 1 pM, starting from the 7th day of the culture) prevented this loss. In contrast, higher doses of rapamycin (100 pM) expedited neuron loss. These results suggest a protective action of rapamycin on cultured neurons.
Micro‐dose Rapamycin Limits Acute Cerebral Ischemic Infarction and Extends Life Span of SHR‐SPs
We next tested whether the neuron‐protective activities of micro‐dose rapamycin could be translated into disease prevention in animal models. Acute cerebral ischemic infarction (an aged‐related disease that causes neuron death and brain damage 28, 29) was employed. In a C57BL/6 mice model of acute ischemic cerebral infarction induced by transient MCAO, the animals were divided into six groups (n = 10 in each group) and pretreated by different dose of rapamycin (0.1, 1, 10, 100, 1000 μg/kg/day, intraperitoneal (i.p.) injection) for 3 days. The rate of death after the MCAO experiments was 10% and did not differ between the vehicle control group and the groups receiving rapamycin. CBF reduction was <70% in 8% of the animal subjects (not included in the treatment assignment). At micro‐dose (from 0.1 to 10 μg/kg/day), rapamycin significantly reduced the cerebral infarct size and neurologic deficit score (Figure 4A,B). Higher doses of rapamycin (0.1 and 1 mg/kg) did not reduce the infarct size or neurologic deficit score.
Figure 4.
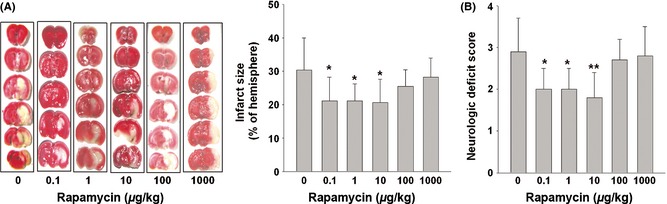
Micro‐dose rapamycin decreased cerebral ischemic infarct size in the C57BL/6 mice MCAO model. Male C57BL/6 mice were randomly divided into six groups (n = 10 in each group) and received daily rapamycin treatment at the indicated doses for 3 days prior to MCAO using a silicone rubber–coated nylon monofilament. (A) The ischemic infarct size (white) was measured by TTC staining of the six coronal brain sections 1 day after MCAO. (B) The neurologic deficit scores of different groups. Data are expressed as mean ± SD and analyzed by one‐way analysis of variance (ANOVA) followed by Dunnett's t‐test. *P < 0.05, **P < 0.01 vs. control (0 μg/kg).
We then used SHR‐SPs with MCAO to investigate the cerebral protection of rapamycin. Fifty SHR‐SPs were used in this study and divided randomly into five groups (n = 10 in each group). According to the mice data, the dose of rapamycin was selected only at lower micro‐doses (0.3, 1, 3, and 10 μg/kg). Permanent MCAO model by electric coagulation of MCA was employed. This mild treatment yielded smaller infarct size and no death. Consistent with the mice result, micro‐dose rapamycin at 1, 3, and 10 μg/kg/day also decreased infarct size and neurologic deficit score induced by MCAO (Figure 5A,C).
Figure 5.
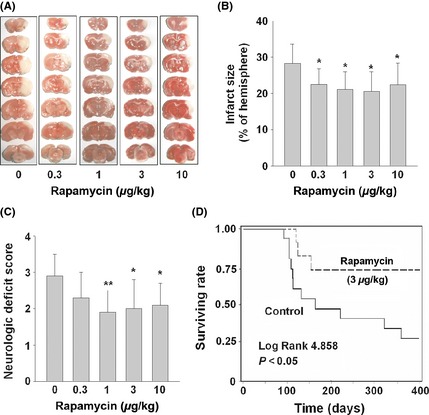
Micro‐dose rapamycin decreased cerebral ischemic infarct size of SHR‐SPs and prolonged their life span. SHR‐SPs were randomly divided into five groups (n = 10 in each group) and administrated with indicated doses of rapamycin once a day. MCA was occluded by electric coagulation after the drug administration for 3 days. (A and B) The ischemic infarct size was measured by TTC staining of the seven coronal brain sections 1 day after MCAO. (C) The neurologic deficit scores of different groups. Data are expressed as mean ± SD and analyzed by one‐way analysis of variance (ANOVA) followed by Dunnett's t‐test. *P < 0.05, **P < 0.01. (D) SHR‐SPs (aged 90 days) were randomly divided into rapamycin and control groups (n = 15 per group). Animals in the rapamycin group received intermittent rapamycin treatment (in a 7‐day cycle) throughout the 310‐day experiment. Rapamycin was delivered at 3 μg/kg/day for 3 days, followed by 4 days of normal food. Survival data were plotted against time and examined using Kaplan–Meier analysis (log‐rank test). P < 0.05 was considered statistically significant.
We next selected one dose of rapamycin (3 μg/kg/day) to test whether micro‐dose rapamycin could prolong the life span of SHR‐SPs. In the control group of SHR‐SPs receiving no intervention, the mortality increased sharply starting at 3 month of age (Figure 5D). Rapamycin treatment for 3 days in every 7 days via food started from 90 days of age attenuated the increase in mortality (Figure 5D). Compared to control group, rapamycin treatment increased survival rate from 60% to 83% at 120 days of age, from 52% to 75% at 150 days of age (P < 0.05). Remarkably, no rats receiving rapamycin treatment died between 150 and 400 days of age, while the survivals in control group continued to decrease.
Micro‐dose Rapamycin Alters Phosphorylation Status of TOR
To determine whether the aforementioned effects of micro‐dose rapamycin are mediated by targeting at mammalian TOR (mTOR), we examined the phosphorylation of mTOR at Ser2448 (a measure of mTOR or TORC1 activation in response to insulin) 30 and Ser2481 (a measure of autophosphorylation by mTOR kinase) 31. SHR‐SPs were used (n = 3 in each group) and treated by two doses of rapamycin (1 and 1000 μg/kg, i.p.) or vehicle (DMSO) for 5 h. As shown in Figure 6A, Micro‐dose rapamycin at 1 μg/kg (5 h treatment) significantly increased phospho‐Ser2448 of mTOR in isolated cortical tissue. However, rapamycin at 1 mg/kg reversely reduced phospho‐Ser2448 signal. Both doses of rapamycin did not have much effect on phospho‐Ser2481 of TOR (Figure 6B), indicating no effect on the auto‐kinase activity of mTOR.
Figure 6.
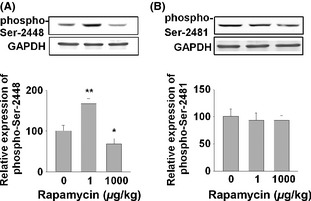
Rapamycin at 1 μg/kg increases the phospho‐Ser2448 of mTOR, but not phospho‐Ser2481. Male SHR‐SPs (n = 3 per group) received rapamycin at two doses (1 and 1000 μg/kg). The rats were sacrificed at 5 h after the treatment. The cerebral cortex cell lysates were extracted and subjected to Western blotting analysis using antibodies against phospho‐Ser2448 (A) and phospho‐Ser2481 of mTOR (B). Data were analyzed by one‐way analysis of variance (ANOVA) followed by Dunnett's t‐test. *P < 0.05, **P < 0.01.
Discussion
We demonstrated that rapamycin at micro‐doses (<1% of normal dose for TORC1 inhibition), exhibited cell protection activity in nondividing cells (senescent fibroblasts and neurons). This activity may be not related to the TORC1 inhibition, for the following reasons: (1) The concentration is below the TORC1 inhibition that leads to G1 slowing down (as shown by Figure S1A, Figures 1C and 2A,B), (2) Rapamycin at micro‐dose increases phosphorylation of mTOR at Ser2448 (Figure 6A), an indication of increased TORC1 formation; (3) The G1 inhibitor, such as higher dose of rapamycin or FK506, did not exhibit cell protective activity (Figures 1C and 2A,B). We propose that micro‐dose rapamycin regulates a novel low‐affinity complex of mTOR (TORCLA).
We also demonstrated that rapamycin at micro‐dose prevented stroke in animal models, possibly through its cell protection activity to limit neuron death. Rapamycin at the TORC1‐inhibition concentration did not protect cell nor prevent acute cerebral infarction. Interestingly, re‐entry of cell cycle has been found to be associated with neuron death 32.
We also demonstrated that rapamycin at micro‐dose prolonged senescence. Senescence has been proposed to be a barrier of cancer, as it arrests oncogene‐activated cells from proliferation. Maintaining of senescent state thus is a natural cancer prevention mechanism.
Rapamycin at micro‐dose can prolong senescent state in fibroblasts, protect neurons from death, and prevent stroke. These effects suggest a common mechanism for them and for age‐related diseases. It is possible that maintenance of the nondividing state (or G0 state) in most somatic cells, that is responsible for preventing abnormal proliferation or cell death, is an important antiaging mechanism for preventing cancer, stroke, and other aging diseases related to the exit of the nondividing state or to cell death. This mechanism can explain why DR can prevent various diseases that seem to have different mechanisms.
Although rapamycin at the TORC1‐inhibition doses can be used to treat‐specific diseases, such as abnormal growth in cancer and abnormal protein aggregation in Parkinson's and Huntington's diseases 11, 33, 34, the adverse effects of TORC1 inhibition may minimize its antiaging use. Our results suggest that rapamycin at micro‐dose could be developed to a safe antiaging agent to prevent various age‐related diseases.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Figure S1. The effect of rapamycin on doubling time, replication potential and senescence maintenance in human primary fibroblast WI‐38.
Acknowledgments
This study was supported by National Natural Science Foundation of China (81273505, 81230083).
The first two authors contributed equally to this work.
References
- 1. Tsang CK, Qi H, Liu LF, Zheng XF. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discovery Today 2007;12:112–124. [DOI] [PubMed] [Google Scholar]
- 2. Maswood N, Young J, Tilmont E, et al. Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson's disease. Proc Natl Acad Sci USA 2004;101:18171–18176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hursting SD, Dunlap SM, Ford NA, Hursting MJ, Lashinger LM. Calorie restriction and cancer prevention: A mechanistic perspective. Cancer Metab 2013;1:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sohal RS, Forster MJ. Caloric restriction and the aging process: A critique. Free Radic Biol Med 2014;73C:366–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zang P, Dong J, Song XR, Zhang LL, Liu AJ. Involvement of fibroblast growth factor in the restoration of arterial baroreflex by dietary restriction. CNS Neurosci Ther 2013;19:367–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu AJ, Guo JM, Liu W, et al. Involvement of arterial baroreflex in the protective effect of dietary restriction against stroke. J Cereb Blood Flow Metab 2013;33:906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009;460:392–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kaeberlein M, Powers RW 3rd, Steffen KK, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005;310:1193–1196. [DOI] [PubMed] [Google Scholar]
- 9. Abraham RT. Identification of TOR signaling complexes: More TORC for the cell growth engine. Cell 2002;111:9–12. [DOI] [PubMed] [Google Scholar]
- 10. Wei K, Wang P, Miao CY. A double‐edged sword with therapeutic potential: An updated role of autophagy in ischemic cerebral injury. CNS Neurosci Ther 2012;18:879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ravikumar B, Duden R, Rubinsztein DC. Aggregate‐prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 2002;11:1107–1117. [DOI] [PubMed] [Google Scholar]
- 12. Kuypers DR. Benefit‐risk assessment of sirolimus in renal transplantation. Drug Saf 2005;28:153–181. [DOI] [PubMed] [Google Scholar]
- 13. Fu X, Wan S, Lyu YL, Liu LF, Qi H. Etoposide induces ATM‐dependent mitochondrial biogenesis through AMPK activation.PLoS ONE 2008;3:e2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luan P, Zhou HH, Zhang B, et al. Basic fibroblast growth factor protects C17.2 cells from radiation‐induced injury through ERK1/2. CNS Neurosci Ther 2012;18:767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Du CP, Tan R, Hou XY. Fyn kinases play a critical role in neuronal apoptosis induced by oxygen and glucose deprivation or amyloid‐beta peptide treatment.CNS Neurosci Ther 2012;18:754–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun Y, Gui H, Li Q, et al. MicroRNA‐124 protects neurons against apoptosis in cerebral ischemic stroke. CNS Neurosci Ther 2013;19:813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gui H, Guo YF, Liu X, et al. Effects of combination therapy with levamlodipine and bisoprolol on stroke in rats. CNS Neurosci Ther 2013;19:178–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen CH, Jiang Z, Yan JH, et al. The involvement of programmed cell death 5 (PDCD5) in the regulation of apoptosis in cerebral ischemia/reperfusion injury. CNS Neurosci Ther 2013;19:566–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ma XJ, Cheng JW, Zhang J, et al. E‐selectin deficiency attenuates brain ischemia in mice. CNS Neurosci Ther 2012;18:903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu AJ, Zang P, Guo JM, et al. Involvement of acetylcholine‐alpha7nAChR in the protective effects of arterial baroreflex against ischemic stroke. CNS Neurosci Ther 2012;18:918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wei N, Yu SP, Gu XH, et al. The involvement of autophagy pathway in exaggerated ischemic brain damage in diabetic mice. CNS Neurosci Ther 2013;19:753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peng T, Britton GL, Kim H, et al. Therapeutic time window and dose dependence of xenon delivered via echogenic liposomes for neuroprotection in stroke. CNS Neurosci Ther 2013;19:773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zheng YJ, Wang XR, Chen HZ, et al. Protective effects of the delta opioid peptide [D‐Ala2, D‐Leu5]enkephalin in an ex vivo model of ischemia/reperfusion in brain slices. CNS Neurosci Ther 2012;18:762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Galarneau A, Primeau M, Trudeau LE, Michnick SW. Beta‐lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein protein interactions. Nat Biotechnol 2002;20:619–622. [DOI] [PubMed] [Google Scholar]
- 25. Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo . Proc Natl Acad Sci USA 1995;92:9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Karashima T, Hachisuka H, Sasai Y. FK506 and cyclosporin A inhibit growth factor‐stimulated human keratinocyte proliferation by blocking cells in the G0/G1 phases of the cell cycle. J Dermatol Sci 1996;12:246–254. [DOI] [PubMed] [Google Scholar]
- 27. Chun HS, Gibson GE, DeGiorgio LA, et al. Dopaminergic cell death induced by MPP(+), oxidant and specific neurotoxicants shares the common molecular mechanism. J Neurochem 2001;76:1010–1021. [DOI] [PubMed] [Google Scholar]
- 28. Pompili M, Venturini P, Campi S, et al. Do stroke patients have an increased risk of developing suicidal ideation or dying by suicide? An overview of the current literature CNS Neurosci Ther 2012;18:711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deng YX, Wang YL, Gao BQ, et al. Age differences in clinical characteristics, health care, and outcomes after ischemic stroke in China. CNS Neurosci Ther 2012;18:819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: Identification of a convergence point for opposing effects of insulin and amino‐acid deficiency on protein translation. Biochem J 1999;344(Pt 2):427–431. [PMC free article] [PubMed] [Google Scholar]
- 31. Peterson RT, Beal PA, Comb MJ, Schreiber SL. FKBP12‐rapamycin‐associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J Biol Chem 2000;275:7416–7423. [DOI] [PubMed] [Google Scholar]
- 32. Bhaskar K, Miller M, Chludzinski A, et al. The PI3K‐Akt‐mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener 2009;4:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Malagelada C, Jin ZH, Jackson‐Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's disease. J Neurosci 2010;30:1166–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morris LG, Veeriah S, Chan TA. Genetic determinants at the interface of cancer and neurodegenerative disease. Oncogene 2010;29:3453–3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The effect of rapamycin on doubling time, replication potential and senescence maintenance in human primary fibroblast WI‐38.