

Summary
Aims
Sildenafil, a phosphodiesterase type 5 inhibitor, has been found to produce functional recovery in ischemic rats by increasing the cGMP level and triggering neurogenesis. The aim of this study was to investigate further sildenafil mechanisms.
Methods
Male Sprague‐Dawley rats underwent middle cerebral artery occlusion and reperfusion, followed by intraperitoneal or intravenous treatment of sildenafil starting 2 h later. Behavioral tests were performed on day 1 or day 7 after reperfusion, while cerebral infarction, edema, Nissl staining, Fluoro‐Jade B staining, and electron microscopy studies were carried out 24 h poststroke. The cGMP‐dependent Nogo‐66 receptor (Nogo‐R) pathway, synaptophysin, PSD‐95/neuronal nitric oxide synthases (nNOS), brain‐derived neurotrophic factor (BDNF)/tropomyosin‐related kinase B (TrkB), and nerve growth factor (NGF)/tropomyosin‐related kinase A (TrkA) were measured.
Results
Sildenafil enhanced neurological recovery and inhibited infarction, even following delayed administration 4 h after stroke onset. Furthermore, sildenafil reduced the loss of neurons and modulated the expressions of the cGMP‐dependent Nogo‐R pathway. Moreover, sildenafil protected the structure of synapses and mediated the expressions of synaptophysin, PSD‐95/nNOS, BDNF/TrkB, and NGF/TrkA.
Conclusions
Sildenafil produces significant neuroprotective effects on injured neurons in acute stroke, and these are mediated by the cGMP‐dependent Nogo‐R pathway, NGF/TrkA, and BDNF/TrkB.
Keywords: Neuronal network, Neuroprotection, Sildenafil, Stroke
Introduction
Despite stroke being the third most common cause of death 1 and the main cause of permanent disability in adults' worldwide 2, the therapeutic options available remain very limited. Recent lines of experimental evidence have shown that phosphodiesterase type 5 (PDE5) inhibitors have significant therapeutic effects on erectile dysfunction, pulmonary hypertension, and Alzheimer's disease as well as stroke 3, 4, 5, 6. In addition, experimental studies in rodents suggest that PDE5 inhibitors, including sildenafil, tadalafil, ibudilast, and zaprinast, can increase cerebral blood flow and improve functional recovery by increasing the brain level of cGMP, triggering neurogenesis, and reducing neurological deficits after stroke 6, 7, 8, 9, 10. However, this does not fully describe the complicated mechanisms responsible for sildenafil‐induced behavioral recovery.
Neurogenesis, the generation of new neurons, is a strategy that compensates for tissue lost to injury 11. In addition, restructuring the extracellular matrix and enhancing neuronal networks are also necessary for functional recovery after stroke 11. A neuronal network is composed of a group or groups of chemically connected or functionally associated neurons 12. The strengthening synaptic connections or synaptic wiring among neurons leads to enhanced neuronal networks and behavioral recovery 13. Therefore, we hypothesized that the neuroprotection of sildenafil in acute stroke may be due to its protective effect on neuronal networks.
In this study, we investigated the effects of sildenafil on brain injuries in acute cerebral ischemia using rats with a middle cerebral artery occlusion (MCAO). It was found that sildenafil markedly reduced brain edema, infarct volume, neuron damage and improved neurological function. Sildenafil also inhibited neuronal loss by reducing Nogo‐66 receptor (Nogo‐R), RhoA, and p‐PTEN expression and increasing p‐Akt and PI3K levels via a cGMP‐dependent pathway. In addition, sildenafil was able to improve the structure of synapses, increase levels of synaptophysin, brain‐derived neurotrophic factor (BDNF)/tropomyosin‐related kinase B (TrkB), and nerve growth factor (NGF)/tropomyosin‐related kinase A (TrkA), and reduce levels of PSD‐95/neuronal nitric oxide synthases (nNOS). These results suggest that sildenafil may exert significant neuroprotective effects via a cGMP‐dependent Nogo‐R pathway, NGF/TrkA, and BDNF/TrkB.
Methods
Animals MCAO
Male Sprague‐Dawley (SD) rats weighing 270–300 g, supplied by the Experimental Animal Centre of Shenyang Pharmaceutical University, were maintained under standard housing conditions (22 ± 2 °C, 50 ± 10% relative humidity, a 12‐h light/dark cycle, light on at 06:30 a.m.) with food and water available ad libitum. Animals were anesthetized with chloral hydrate (350 mg/kg, intraperitoneal, i.p.) and subjected to 2 h of right MCAO and reperfusion for 1 or 7 days 14. The middle cerebral artery (MCA) was occluded by placement of an embolus at the origin of the MCA. All experiments and procedures were carried out according to the Regulations of Experimental Animal Administration issued by the State Committee of Science and Technology of China.
Drug Preparation and Administration
Sildenafil (sildenafil citrate, purity 98%, kindly supplied by Tianjin Tasly Company Ltd., Tianjin, China) was dissolved in normal saline and administered by the i.p. or intravenous (i.v.) route at doses of 4, 8, 16, or 32 mg/kg. A specific PKG inhibitor DT‐3 (Sigma‐Aldrich, St. Louis, MO, USA, 1 mg/kg, i.p.), a soluble guanylyl cyclase (GC) inhibitor 1H‐[1, 2, 4] Oxadiazolo [4, 3‐a] quinoxalin‐1‐1 (ODQ, Sigma‐Aldrich, 3.5 mg/kg, i.p.), and a PKA inhibitor fragment 5‐24 amide trifluoroacetate salt (IP‐20, Sigma‐Aldrich, 0.34 mg/kg, i.p.) were administered in combination with or without sildenafil (16 mg/kg, i.v.) 15, 16, 17, 18. All animals were randomly assigned to the following experimental treatment groups: a sham‐operated group (natural saline, 10 mL/kg), an MCAO group (rats only suffered from MCAO and given reperfusion and normal saline), MCAO + inhibitor groups (Rats suffered from MCAO and were given reperfusion. The inhibitors were administered immediately after MCAO), MCAO + sildenafil groups (rats suffered from MCAO and were given reperfusion, followed by i.p. or i.v. sildenafil starting 2 h, 4 h, or 6 h later), MCAO + inhibitor + sildenafil groups (Rats suffered from MCAO and were given reperfusion. The inhibitors were administered immediately after MCAO, while sildenafil were administered 2 h later).
Behavioral Testing
Neurological functional deficits were evaluated 24 h after reperfusion using a modified six‐point scoring method applied by an investigator who was blinded as to the experimental treatment groups (n = 10/group). The scale was 0: no neurological deficit; 1: failure to extend the left forepaw fully; 2: circling to the left; 3: falling to the left; 4: no spontaneous walking with a depressed level of consciousness; and 5: dead 19.
Two tests were used to evaluate the behavioral outcome: a beam walking test and a rotarod test after administration of sildenafil (or vehicle) for 7 days consecutively (n = 12/group).
Rotarod Test
Prestroke training was performed three times a week prior to MCAO on a rat rotarod. On testing days, each rat performed the test twice in a row and then the cohort was cycled and the test repeated. The length of time each rat was able to stay on the rotarod, up to 300 s, was recorded 20.
Beam Walking Test
Prestroke training was performed for 3 days and then, on the seventh day after surgery, the test was performed. During training and testing, rats were placed on one end of the beam and they were exposed to noise and light until they entered the goal box. The performance of the rats was evaluated using a modified seven‐point scoring method by an investigator who was blinded as to the experimental treatment groups 20.
Measurement of Cerebral Infarct Volume and Cerebral Edema
Rats were decapitated 24 h after reperfusion, and their brains were quickly removed (n = 10/group). The total wet weight of the brain was measured accurately, and each brain tissue was then sliced into five coronal sections, each of 2 mm thickness and stained with a 2% solution of tetrazolium chloride (Sigma) in saline at 37 °C for 20 min, and then photographic images were taken. Afterward, the brain water content was determined as an indicator of cerebral edema using a wet/dry method as previously described 21. The cross‐sectional areas, with or without infarction, in each brain slice were measured using Image J analysis software (version 1.6 National institutes of Health, Bethesda, MD, USA). The total mean infarct area of each section was examined by the change in coloration.
Histological and Immunohistochemical Assessment
The brains were fixed by transcardial perfusion with saline, followed by perfusion and immersion in 4% paraformaldehyde (n = 4/group). A standard tissue block was obtained from the center of the lesion (bregma −1 mm to +1 mm). A series of 15 μm thick sections were cut from the block. Every 10th coronal section for a total four sections was used for staining. Antibody immunostaining for TrkB, TrkA (1:300, Abcam, Cambridge, UK), and Nogo‐R (1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA) was performed. Control experiments consisted of staining brain coronal tissue sections as outlined above, but nonimmune serum was substituted for the primary antibody. Nissl staining and Fluoro‐Jade B staining were also employed as previously described 22, 23.
TrkA, TrkB, Nogo‐R, and Nissl stained sections were digitized using a 40 × objective (Olympus BX40, Tokyo, Japan) and a 3‐CCD color video camera DP 72 interfaced with an MCID computer imaging analysis system (Image‐Pro 3D Plus Workstation, Media Cybernetics, Inc., Rockville, MD, USA). The number of Nissl‐positive cells and the positive stained areas of TrkA, TrkB, and Nogo‐R were measured in each section. Positive cells or positive areas in four random fields (400 × magnification) across the cortex and striatum were quantified. The data obtained from four sections were averaged for each animal. Data were analyzed in a blind manner and presented as the number of positive cells per field or the percentage of positive areas per field, respectively.
Transmission Electron Microscopy
The brains were fixed by transcardial perfusion with saline, followed by perfusion and immersion in 4% paraformaldehyde and 2.5% glutaraldehyde (n = 4/group). Small blocks from different brain regions, including the striatum and cortex, were processed for electron microscopy and embedded in Epon resin, as previously described 24. The sections were then examined by Transmission Electron Microscopy (TEM) (JEM‐1200EX, Jeol Ltd., Tokyo, Japan) at an accelerating voltage of 80 kV.
Western Blot Analysis
Tissues from the cortex and striatum including the infarct area were homogenized in lysis buffer. Protein extracts were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) (n = 3/group). The following primary antibodies were used: anti‐RhoA (1:500, Abcam), anti‐Akt, anti‐p‐Akt, anti‐PTEN, anti‐p‐PTEN (1:500, Cell Signaling Technology, Bevery, MA, USA), anti‐Nogo‐R, anti‐PI3K, anti‐β‐actin (1:500, Santa Cruz), antisynaptophysin, anti‐PSD‐95 (1:1500, Millipore, Billerica, MA, USA).
Enzyme‐Linked Immunosorbent Assay
The levels of nNOS, BDNF, and NGF in the striatum and cortex were measured 24 h poststroke using ELISA kits according to the manufacturer's instructions (R&D) (n = 8/group).
Statistics
Quantitative data from the experiments are expressed as the mean ± SE. Statistical analysis was carried out using SPSS 13.0 software for Windows (SPSS Inc, Chicago, IL, USA). Statistical significance was determined by one‐way analysis of variance (anova) and least significant difference analysis followed by Dunnett's test. In all cases, differences were considered significant if P < 0.05.
Results
Sildenafil Improved Behavioral Outcome and Reduced Cerebral Infarct Volume
Ischemia leads to severe behavioral disturbance and histological changes in rats. The results of the behavioral studies showed that sildenafil‐treated rats obtained higher beam walking scores and took longer to fall from a rotating rod (P < 0.05, Figure S1). Furthermore, compared with the sham‐operated group, the neurological deficit, cerebral infarct volume, and cerebral edema were significantly higher in the MCAO group and could be significantly reversed by sildenafil, even after a 4‐h delay in administration poststroke (P < 0.05, Figure 1). In addition, it was found that sildenafil (8–32 mg/kg) could significantly reduce the cerebral infarct volume in a dose‐dependent manner with an ED50 of 15.92 mg/kg. These results suggest that sildenafil was able to significantly improve behavioral outcome and reduce cerebral infarct volume after stroke.
Figure 1.
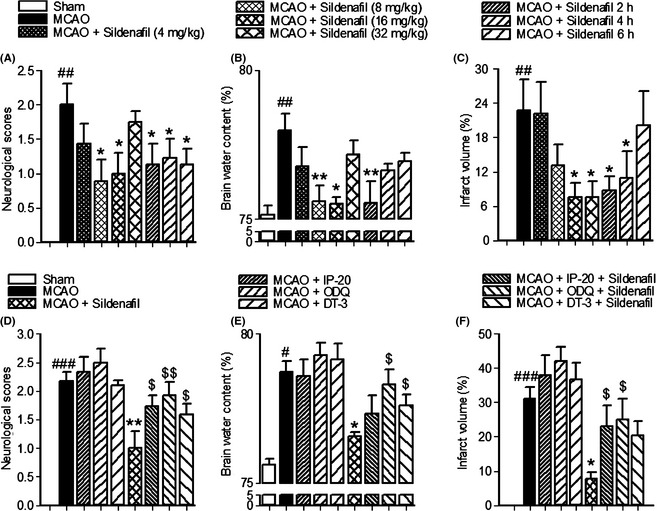
The results showed that the neurological deficit (A), cerebral infarct volume (C), and cerebral edema (B) were significantly higher in the middle cerebral artery occlusion (MCAO) group compared with the sham‐operated animals, and these could be significantly reversed by sildenafil, even after a 4‐h delay in administration after MCAO. The reductions in neurological deficit scores (D), brain water content (E), and infarct volume (F) induced by sildenafil were significantly reversed by ODQ, IP‐20, or DT‐3, respectively. # P < 0.05, ## P < 0.01, ### P < 0.001 compared with the sham‐operated group, *P < 0.05, **P < 0.01 compared with the MCAO group. $ P < 0.05, $$ P < 0.01 compared with the MCAO + sildenafil group.
Sildenafil Reduced the Loss of Neurons through a cGMP‐Dependent Nogo‐R Pathway
The number of surviving neurons is a reliable indicator of cerebral injury. After cerebral ischemia, the number of surviving neurons was reduced in both the cortex and the striatum (P < 0.001, Figure 2A,E). When examined by TEM, a number of dying neurons in MCAO rats displayed features of apoptosis, including nuclear breakdown and chromatin condensation (Figure 2D). Our results suggest that sildenafil could significantly reduce the number of degenerated neurons, prevent neuronal damage, and increase the number of surviving neurons after stroke.
Figure 2.
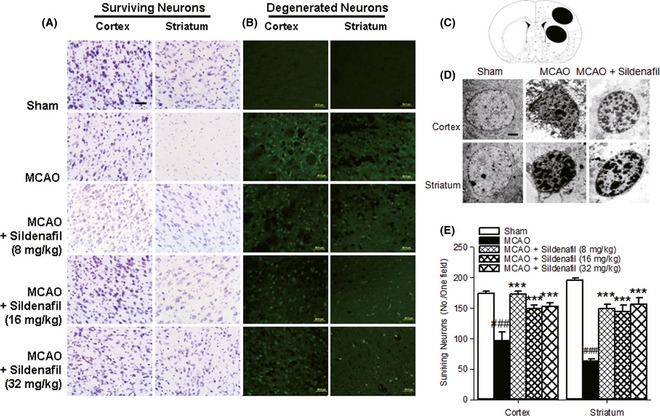
Sildenafil could significantly reduce the number of degenerated neurons (B) and increase the number of surviving neurons (A, E) after stroke. Scale bar = 50 μm. (C) The two black ovals indicate the regions selected for immunohistochemistry. The TEM study showed that sildenafil could significantly protect against neuronal damage (D). Scale bar = 2 μm. ### P < 0.001 compared with the sham‐operated group, ***P < 0.001 compared with the middle cerebral artery occlusion (MCAO) group.
DT‐3 is a PKG inhibitor 17, 25, 26. ODQ could reduce cGMP expression by inhibiting nitric oxide‐sensitive GC. IP‐20 could bind to the catalytic subunit of PKA, mimicking the protein substrate 15, 16, 17, 18. These inhibitors could reduce the expression of cGMP or inhibit the binding affinity of PKA/PKG to the downstream substrate, respectively. To explore whether a cGMP‐dependent pathway plays a role in sildenafil‐induced neuroprotection, rats were simultaneously treated with sildenafil (16 mg/kg) and ODQ, IP‐20, or DT‐3. The reductions in neurological deficit scores, brain water content and infarct volume, and the increase in surviving neurons induced by sildenafil were significantly reversed by ODQ, IP‐20, or DT‐3, respectively (P < 0.05, Figures 1, 3). Meanwhile, the neurological deficit scores, brain water content, and cerebral infarct volume were not significantly increased in the rats of MCAO + inhibitor groups compared with that of the MCAO group (Figure 1). These data show that the inhibitors alone could not influence the MCAO/reperfusion‐induced injury. These results show that cGMP/PKG/PKA inhibitors were able to inhibit the protective response of sildenafil.
Figure 3.
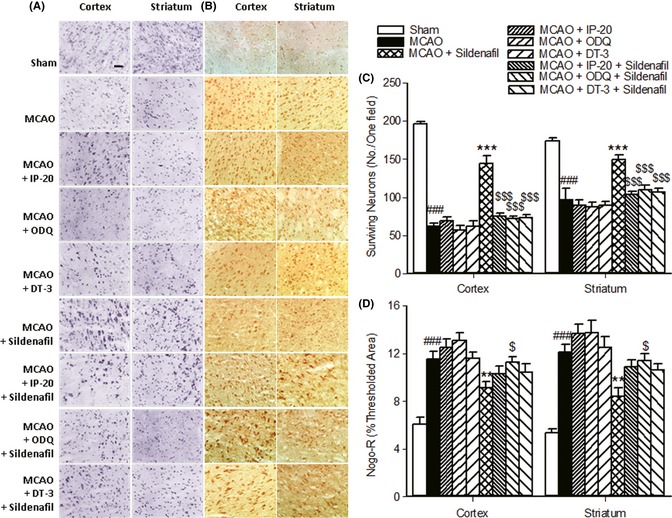
Sildenafil protected neurons from injury via a cGMP‐dependent Nogo‐R pathway. The increase in surviving neurons (A, C) and the decrease in Nogo‐R expression (B, D) induced by sildenafil were significantly reversed by ODQ, IP‐20, or DT‐3. Scale bar = 50 μm. ### P < 0.001 compared with the sham‐operated group. **P < 0.01, ***P < 0.001 compared with the middle cerebral artery occlusion (MCAO) group. $ P < 0.05, $$$P < 0.001 compared with the MCAO + sildenafil group.
To further investigate the molecular mechanism of sildenafil‐induced neuroprotection, the expressions of Nogo‐R, RhoA, p‐Akt/Akt, p‐PTEN/PTEN, and PI3K were further examined. It was found that Nogo‐R, RhoA, and p‐PTEN/PTEN expressions were significantly increased, while p‐Akt/Akt and PI3K expressions were markedly reduced in the cortex and striatum after stroke. Sildenafil reduced the protein expressions of Nogo‐R, RhoA, and p‐PTEN, and increased p‐Akt and PI3K levels (Figure 4A). When rats were treated with sildenafil and cGMP/PKG/PKA inhibitors, the reduction in Nogo‐R expression was reversed significantly (Figure 3B,D). These results indicate that sildenafil reduced the loss of neurons through a cGMP‐dependent Nogo‐R pathway after cerebral ischemia.
Figure 4.
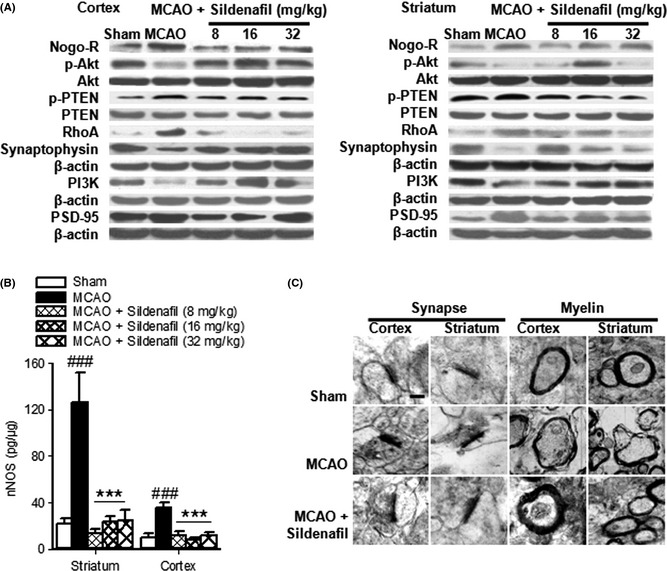
Sildenafil increased neuron survival by downregulation of the Nogo‐R pathway (A). Ischemia often causes low levels of synaptophysin (A) and high levels of uncoupling PSD‐95 (A) and nNOS (B), which could be blocked by sildenafil. ### P < 0.001 compared with the sham‐operated group, ***P < 0.001 compared with the middle cerebral artery occlusion (MCAO) group. Sildenafil could prevent synapse structure degeneration and demyelination in acute stroke (C). Scale bar = 500 nm.
Sildenafil Alleviated Synapse Damage
Further morphological diversity was determined by TEM in synapses which displayed structure degeneration, including a reduction in synaptic vesicles and disappearance of the synaptic cleft, fracture, or fusion. Local damage, swelling, and demyelination were evident in many of the axonal processes in ischemic stroke and these could be reversed by sildenafil (Figure 4C), suggesting that it had a protective effect on the structure of synapses.
Synaptophysin is a marker for quantification of synapses 27. PSD‐95 and nNOS are coupled to the postsynaptic membrane. To elucidate the mechanism of sildenafil‐induced synaptic protection, the expressions of synaptophysin, PSD‐95, and nNOS were measured. Importantly, ischemia often causes a reduction in the level of synaptophysin and an increase in the level of uncoupling PSD‐95 and nNOS, which could be blocked by sildenafil (cortex: P < 0.001. striatum: P < 0.001, Figure 4A,B). These data suggest that sildenafil significantly alleviated synaptic damage in the ischemic brain. Furthermore, sildenafil (16 mg/kg) significantly increased TrkB protein expression in both the cortex and striatum (P < 0.001, Figure 5A,C), and increased BDNF expression (cortex: P < 0.05. striatum: P < 0.001, Figure 5D) compared with the MCAO group. In addition, compared with the MCAO group, sildenafil (16, 32 mg/kg) increased TrkA protein expression in the cortex (P < 0.01, Figure 5B,C), but not in the striatum (P > 0.05, Figure 5B), while sildenafil (32 mg/kg) also increased NGF protein expression in the striatum (P < 0.05, Figure 5E), but not in the cortex (P > 0.05, Figure 5E). These data suggest that sildenafil significantly enhanced neuronal function in the ischemic brain by modulating the expressions of BDNF/TrkB and NGF/TrkA.
Figure 5.
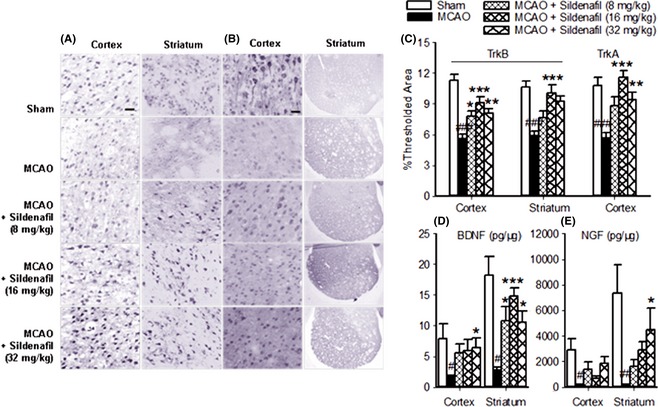
Sildenafil improved neuronal function through upregulation of BDNF/TrkB (A, C, D), NGF/TrkA (B, C, E) in the ischemic brain. Scale bar = 50 μm for TrkB (cortex and striatum), TrkA (cortex). Scale bar = 500 μm for TrkA (striatum). # P < 0.05, ## P < 0.01, ### P < 0.001 compared with the sham‐operated group, *P < 0.05, **P < 0.01, ***P < 0.001 compared with the middle cerebral artery occlusion (MCAO) group.
Discussion
The lack of functional recovery following acute injury to the central nervous system (CNS), such as motor recovery, can mainly be attributed to the restriction of restructuring extracellular matrix and modifying neuronal networks. Recent experimental studies in rodents suggest that treatment with different PDE5 inhibitors not only increases cerebral blood flow but also improves functional recovery after stroke 8, 9, 10. However, the extent and the underlying mechanisms for these inhibitors are still not totally understood. In this study, we examined the beneficial effects and possible mechanism of sildenafil on the structures and function of the neuronal networks.
First of all, sildenafil had a beneficial effect on behavioral outcome. Moreover, sildenafil (8–32 mg/kg) significantly reduced cerebral infarct volume in a dose‐dependent manner with an ED50 of 15.92 mg/kg. This is the first and most direct preclinical study to report the inhibitory effect of sildenafil on acute stroke‐induced infarct volume. In actual fact, a prior study reported that sildenafil failed to decrease the infarct volume when administered 24 h after the onset of ischemia 7. There are two possible explanations for these contradictory results. Firstly, the strains and the age of rats used by the two labs were not identical. Male Wistar rats weighing 320–380 g (age unknown) were used in the previous report 7, while, in the current study, male SD rats weighing 270–300 g (age 8 weeks approximately) were used. It has been reported that there are substantial differences in the temporal evolution of ischemic lesions and the putative penumbra between Wistar and SD rats 28. Slight differences in gene expression between Wistar and SD rats might also influence the outcome involving neuroprotection. Secondly, the administration method and the dosage of sildenafil were different. In the earlier report, sildenafil was administered orally starting 2 or 24 h after stroke onset at doses of 2 or 5 mg/kg 7, while, in the present study, sildenafil was administered intravenously at doses of 16 or 32 mg/kg, which significantly reduced the infarct volume, even with a 4‐h delay in administration post‐MCAO.
Phosphodiesterase type 5 is the main cGMP‐specific enzyme that promotes degradation of cGMP 7. Binding of cGMP to allosteric sites is essential for regulation of PKG or PKA 29. PKG could phosphorylate various effectors that are important in cellular survival, proliferation, and relaxation of vascular smooth muscle 15. The cGMP‐PKG cascade could prevent activation of a proapoptotic pathway to promote neural cell survival and enhance the levels of cAMP response element binding protein, which regulates the expression and survival of immature neurons in the adult hippocampus 30, 31. DT‐3 is a specific PKG inhibitor 17, 25, 26. Recent experimental studies in rodents suggest that administration of DT‐3 diminishes the enhanced blood flow recovery by vardenafil 18.
It is reported that activation of PKA plays a number of neuroprotective roles such as inhibiting the Ca2+ release from the endoplasmic reticulum 32, activating the high‐affinity uptake of glutamate 33, and suppressing the cytokine‐induced expression of adhesion molecules 34, 35. IP‐20 is a specific PKA inhibitor. The amino acid sequence of IP‐20 is Thr – Thr – Tyr – Ala – Asp – Phe – Ile – Ala – Ser – Gly – Arg – Thr – Gly – Arg – Arg – Asn – Ala – Ile – His – Asp – NH2. IP‐20 could bind to the catalytic subunit of PKA, mimicking the protein substrate 18.
In the present study, the observed changes could not distinguish the relative contributions of PKA and PKG for neuronal survival, the Nogo‐R expression, and the neurological deficits. But some changes in brain water content and infarct volume could distinguish the relative contributions of PKA and PKG. The reduction in brain water content induced by sildenafil was significantly reversed by DT‐3 (P < 0.05, Figure 1E). However, the brain water content was not significantly increased in rats of MCAO + IP‐20 + sildenafil group, as compared to MCAO + sildenafil group. These results show that sildenafil was able to reduce the brain water content mainly by cGMP/PKG pathway. More, the reduction in infarct volume induced by sildenafil was significantly reversed by IP‐20 (P < 0.05, Figure 1F). However, the infarct volume was not significantly increased in rats of MCAO + DT‐3 + sildenafil group compared with that of the MCAO + sildenafil group. These results show that sildenafil was able to reduce the infarct volume mainly by cGMP/PKA pathway.
RhoA is a PKA substrate 36. Accumulating evidence indicates that Nogo, MAG, and OMgp are present on the surface of oligodendrocytes giving them inhibitory activity via Nogo‐R to activate RhoA. RhoA further activates ROCK to phosphorylate several substrates that are involved in the regulation of cell shape and motility as well as in survival pathways. The downstream target of ROCK is PTEN, which negatively regulates Akt signaling by antagonizing PIP3. Akt signaling finally leads to an increase in general translation with an increase in protein synthesis and cell simultaneous modulation of neurite outgrowth and survival 37, 38, 39. As Nogo‐R plays a key role in the regeneration of axons in the CNS, inhibiting the expression of Nogo‐R could support axon sprouting after neuron injury 40. Our results indicate that sildenafil could reduce the expressions of Nogo‐R, RhoA, and p‐PTEN, but increase the expressions of PI3K and p‐Akt. When the rats were treated with sildenafil and cGMP/PKG/PKA inhibitors, the increase in surviving neurons and the decrease in Nogo‐R expression were markedly reversed. Recent studies have shown that Nogo‐R expression is significantly increased to inhibit neurite growth after ischemia 41, 42. Akt could promote growth factor‐mediated cell survival 43. Accordingly, a novel finding was that sildenafil reduced the loss of neurons through a cGMP‐dependent Nogo‐R pathway (Figure 6).
Figure 6.
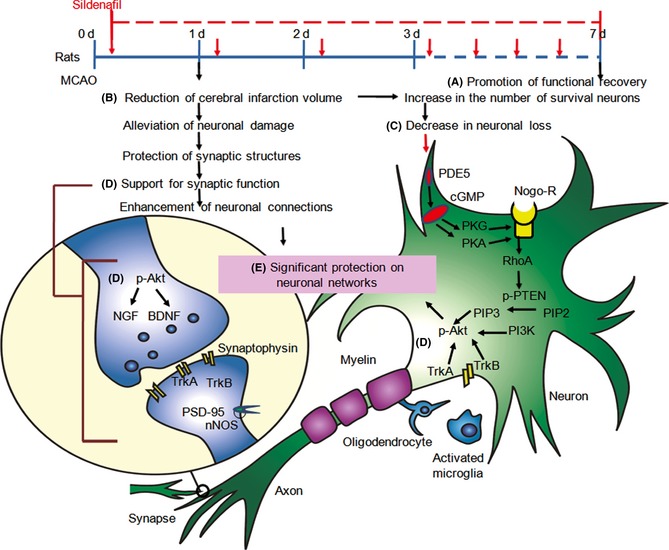
Sildenafil improved the behavioral outcome at day 1 or day 7 poststroke (A), and reduced cerebral infarct volume 24 h after stroke (B). Infarction, the main pathophysiological outcome of cerebral ischemia, involves neuronal degeneration and necrosis. Accordingly, sildenafil could reduce the loss of neurons through a cGMP‐dependent Nogo‐R pathway to activate RhoA. RhoA further activates PTEN, which negatively regulates Akt signaling by antagonizing PIP3 (C). Meanwhile, Akt signaling could promote NGF/TrkA and BDNF/TrkB to modulate neuronal survival and control synaptic function (D). Overall, sildenafil improves neuronal networks (E) by reducing the loss of neurons and boosting neuronal function, which are all mediated by a cGMP‐dependent Nogo‐R pathway, NGF/TrkA, and BDNF/TrkB.
Evidence from animal models suggests that a time‐limited window opens following a stroke, during which the greatest gains in recovery occur 11. The challenge for improving recovery is to understand how to optimally engage and enhance the neuronal networks in this time‐limited window and to provide new response pathways that compensate for the tissue lost to injury 11. Growing evidence supports the association between synaptic connections and neuronal function in stroke 5, 11. Reductions in blood flow to the brain of sufficient duration and extent lead to ischemia, which results in loss of neurons, synaptic connections, and damage to neuronal networks 11. In the present study, sildenafil was found to increase the expression of synaptophysin and reduce the expressions of uncoupling PSD‐95 and nNOS. These data suggest that sildenafil significantly reduced synaptic damage and protected the synapse structure in the ischemic brain. Furthermore, NGF/TrkA, which has an important role in regulating growth cones, motility, and biosynthesis of enzymes for neurotransmitters, could prevent neuronal cell death after stroke 44. In the mature nervous system, BDNF/TrkB modulates neuronal survival by regulating neuronal migration, morphological and biochemical differentiation, and the control of synaptic function and synaptic plasticity 40. Moreover, sildenafil is able to increase the levels of BDNF/TrkB and NGF/TrkA. Thus, we initially assumed that sildenafil enhanced neuronal function by protecting the structure of synapses possibly via upregulation of BDNF/TrkB and NGF/TrkA in acute stroke (Figure 6).
Overall, the present study found that sildenafil had a significant protective effect on neuronal networks during stroke by reducing the loss of neurons and enhancing neuronal function. A recent study has reported that sildenafil produces an immediate and long‐lasting improvement in synaptic function and memory in an Alzheimer's disease mouse model 5. Our previous study demonstrated that sildenafil could inhibit microglial activation, showing its possible benefits in neuroinflammation‐mediated neurological disorders 45. Thus, it is assumed that sildenafil possesses a significant neuroprotective effect by restructuring the extracellular matrix, reducing the loss of neurons and increasing the function of neuronal networks.
Summary
Sildenafil has a significant protective effect on neuronal networks during stroke by reducing the loss of neurons and enhancing the function of neuronal networks, and this is mediated by cGMP‐dependent Nogo‐R, BDNF/TrkB, and NGF/TrkA pathways.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Figure S1. The results of the behavioral studies showed that sildenafil‐treated rats obtained higher beam walking scores and took longer to fall from a rotating rod.
{kind=link}
Figure S2. The statistical results of western blot analysis showed that sildenafil could increase neuron survival by downregulation of the Nogo‐R pathway. Ischemia could cause low levels of synaptophysin and high levels of uncoupling PSD‐95, which could be blocked by sildenafil.
{kind=link}
Acknowledgments
This research was partially supported by the National Natural Science Foundation of China (81102455) and the Research Fund for the Doctoral Program of Higher Education of China (20122134110007).
References
- 1. Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 2003;4:399–415. [DOI] [PubMed] [Google Scholar]
- 2. Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet 2008;371:1612–1623. [DOI] [PubMed] [Google Scholar]
- 3. Bivalacqua TJ, Champion HC, Hellstrom WJ, Kadowitz PJ. Pharmacotherapy for erectile dysfunction. Trends Pharmacol Sci 2000;21:484–489. [DOI] [PubMed] [Google Scholar]
- 4. Weimann J, Ullrich R, Hromi J, et al. Sildenafil is a pulmonary vasodilator in awake lambs with acute pulmonary hypertension. Anesthesiology 2000;92:1702–1712. [DOI] [PubMed] [Google Scholar]
- 5. Puzzo D, Staniszewski A, Deng SX, et al. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid‐β load in an Alzheimer's disease mouse model. J Neurosci 2009;29:8075–8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang L, Zhang RL, Wang Y, et al. Functional recovery in aged and young rats after embolic stroke: treatment with a phosphodiesterase type 5 inhibitor. Stroke 2005;36:847–852. [DOI] [PubMed] [Google Scholar]
- 7. Zhang R, Wang Y, Zhang L, et al. Sildenafil (Viagra) induces neurogenesis and promotes functional recovery after stroke in rats. Stroke 2002;33:2675–2680. [DOI] [PubMed] [Google Scholar]
- 8. Royl G, Balkaya M, Lehmann S, et al. Effects of the PDE5‐inhibitor vardenafil in a mouse stroke model. Brain Res 2009;1265:148–157. [DOI] [PubMed] [Google Scholar]
- 9. Wakita H, Tomimoto H, Akiguchi I, et al. Ibudilast, a phosphodiesterase inhibitor, protects against white matter damage under chronic cerebral hypoperfusion in the rat. Brain Res 2003;992:53–59. [DOI] [PubMed] [Google Scholar]
- 10. Zhang L, Zhang Z, Zhang RL, et al. Tadalafil, a long‐acting type 5 phosphodiesterase isoenzyme inhibitor, improves neurological functional recovery in a rat model of embolic stroke. Brain Res 2006;1118:192–198. [DOI] [PubMed] [Google Scholar]
- 11. Murphy TH, Corbett D. Plasticity during stroke recovery: from synapse to behavior. Nat Rev Neurosci 2009;10:861–872. [DOI] [PubMed] [Google Scholar]
- 12. Hopfield JJ. Neural networks and physical systems with emergent collective computational abilities. Proc Natl Acad Sci U S A 1982;79:2554–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rich MM, Wenner P. Sensing and expressing homeostatic synaptic plasticity. Trends Neurosci 2007;30:119–125. [DOI] [PubMed] [Google Scholar]
- 14. Chen J, Cui X, Zacharek A, et al. Niaspan increases angiogenesis and improves functional recovery after cerebral ischemia. Ann Neurol 2007;62:49–58. [DOI] [PubMed] [Google Scholar]
- 15. Chau VQ, Salloum FN, Hoke NN, Abbate A, Kukreja RC. Mitigation of the progression of heart failure with sildenafil involves inhibition of RhoA/Rho‐kinase pathway. Am J Physiol Heart Circ Physiol 2011;300:H2272–H2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. García‐Herrera J, Marca MC, Brot‐Laroche E, et al. Protein kinases, TNF‐a, and proteasome contribute in the inhibition of fructose intestinal transport by sepsis in vivo. Am J Physiol Gastrointest Liver Physiol 2008;294:G155–G164. [DOI] [PubMed] [Google Scholar]
- 17. Chen LW, Hwang YC, Chen CJ, Wang JS, Chen JS, Hsu CM. Burn‐induced lung damage in rat is mediated by a nitric oxide/cGMP system. Shock 2003;20:369–374. [DOI] [PubMed] [Google Scholar]
- 18. Sahara M, Sata M, Morita T, Nakajima T, Hirata Y, Nagai R. A phosphodiesterase‐5 inhibitor vardenafil enhances angiogenesis through a protein kinase G‐dependent hypoxia‐inducible factor‐1/vascular endothelial growth factor pathway. Arterioscler Thromb Vasc Biol 2010;30:1315–1324. [DOI] [PubMed] [Google Scholar]
- 19. Minematsu K, Li L, Sotak CH, Davis MA, Fisher M. Reversible focal ischemic injury demonstrated by diffusion‐weighted magnetic resonance imaging in rats. Stroke 1992;23:1304–1310. [DOI] [PubMed] [Google Scholar]
- 20. Guzman R, De Los Angeles A, Cheshier S, et al. Intracarotid injection of fluorescence activated cell‐sorted CD49d‐positive neuronal stem cells improves targeted cell delivery and behavior after cerebral ischemia in a mouse cerebral ischemia model. Stroke 2008;39:1300–6. [DOI] [PubMed] [Google Scholar]
- 21. Jiang W, Zhang S, Fu F, Zhu H, Hou J. Inhibition of nuclear factor‐κB by 6‐O‐acetyl shanzhiside methyl ester protects brain against injury in a rat model of ischemia and reperfusion. J Neuroinflammation 2010;7:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suh SW, Aoyama K, Chen Y, et al. Hypoglycemic neuronal death and cognitive impairment are prevented by poly(ADP‐ribose) polymerase inhibitors administered after hypoglycemia. J Neurosci 2003;23:10681–10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Han J, Pollak J, Yang T, et al. Delayed administration of a small molecule tropomyosin‐related Kinase B ligand promotes recovery after hypoxic‐ischemic stroke. Stroke 2012;43:1918–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alfonso‐Loeches S, Pascual M, Gómez‐Pinedo U, Pascual‐Lucas M, Renau‐Piqueras J, Guerri C. Toll‐like receptor 4 participates in the myelin disruptions associated with chronic alcohol abuse. Glia 2012;60:948–964. [DOI] [PubMed] [Google Scholar]
- 25. Ramchandran R, Pilipenko E, Bach L, Raghavan A, Reddy SP, Raj JU. Hypoxic regulation of pulmonary vascular smooth muscle cyclic guanosine monophosphate – dependent kinase by the ubiquitin conjugating system. Am J Respir Cell Mol Biol 2012;46:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krieg T, Philipp S, Cui L, Dostmann WR, Downey JM, Cohen MV. Peptide blockers of PKG inhibit ROS generation by acetylcholine and bradykinin in cardiomyocytes but fail to block protection in the whole heart. Am J Physiol Heart Circ Physiol 2005;288:H1976–H1981. [DOI] [PubMed] [Google Scholar]
- 27. Calhoun ME, Jucker M, Martin LJ, Thinakaran G, Price DL, Mouton PR. Comparative evaluation of synaptophysin‐based methods for quantification of synapses. J Neurocytol 1996;25:821–828. [DOI] [PubMed] [Google Scholar]
- 28. Bardutzky J, Shen Q, Henninger N, Bouley J, Duong TQ, Fisher M. Differences in ischemic lesion evolution in different rat strains using diffusion and perfusion imaging. Stroke 2005;36:2000–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murthy KS. Activation of phosphodiesterase 5 and inhibition of guanylate cyclase by cGMP‐dependent protein kinase in smooth muscle. Biochem J 2001;360:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakagawa S, Kim JE, Lee R, et al. Localization of phosphorylated cAMP response element‐binding protein in immature neurons of adult hippocampus. J Neurosci 2002;22:9868–9876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fiscus RR. Involvement of cyclic GMP and protein kinase G in the regulation of apoptosis and survival in neural cells. Neurosignals 2002;11:175–190. [DOI] [PubMed] [Google Scholar]
- 32. Supattapone S, Danoff SK, Theibert A, Joseph SK, Steiner J, Snyder SH. Cyclic AMP‐dependent phosphorylation of a brain inositol trisphosphate receptor decreases its release of calcium. Proc Natl Acad Sci U S A 1988;85:8747–8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pisano P, Samuel D, Nieoullon A, Kerkerian‐Le Goff L. Activation of the adenylate cyclase‐dependent protein kinase pathway increases high affinity glutamate uptake into rat striatal synaptosomes. Neuropharmacology 1996;35:541–7. [DOI] [PubMed] [Google Scholar]
- 34. Ballestas ME, Benveniste EN. Elevation of cyclic AMP levels in astrocytes antagonizes cytokine‐induced adhesion molecule expression. J Neurochem 1997;69:1438–1448. [DOI] [PubMed] [Google Scholar]
- 35. Tanaka K, Ito D, Suzuki S, Dembo T, Kosakai A, Fukuuchi Y. A novel voltage‐sensitive Na (+) and Ca (2+) channel blocker, NS‐7, prevents suppression of cyclic AMP‐dependent protein kinase and reduces infarct area in the acute phase of cerebral ischemia in rat. Brain Res 2002;924:98–108. [DOI] [PubMed] [Google Scholar]
- 36. Tkachenko E, Sabouri‐Ghomi M, Pertz O, et al. Protein kinase A governs a RhoA‐RhoGDI protrusion‐retraction pacemaker in migrating cells. Nat Cell Biol 2011;13:660–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tönges L, Koch JC, Bähr M, Lingor P. ROCKing regeneration: Rho kinase inhibition as molecular target for neurorestoration. Front Mol Neurosci 2011;4:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shin HK, Salomone S, Potts EM, et al. Rho‐kinase inhibition acutely augments blood flow in focal cerebral ischemia via endothelial mechanisms. J Cereb Blood Flow Metab 2007;27:998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rikitake Y, Kim HH, Huang Z, et al. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke 2005;36:2251–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cui X, Chopp M, Zacharek A, et al. Niacin treatment of stroke increases synaptic plasticity and axon growth in rats. Stroke 2010;41:2044–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Deguchi K, Miyazaki K, Tian F, et al. Modifying neurorepair and neuroregenerative factors with tPA and edaravone after transient middle cerebral artery occlusion in rat brain. Brain Res 2012;1436:168–177. [DOI] [PubMed] [Google Scholar]
- 42. Wang H, Yao Y, Jiang X, Chen D, Xiong Y, Mu D. Expression of Nogo‐A and NgR in the developing rat brain after hypoxia‐ ischemia. Brain Res 2006;1114:212–220. [DOI] [PubMed] [Google Scholar]
- 43. Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med 2005;9:59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jang SW, Okada M, Sayeed I, et al. Gambogic amide, a selective agonist for TrkA receptor that possesses robust neurotrophic activity, prevents neuronal cell death. Proc Natl Acad Sci U S A 2007;104:16329–16334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhao S, Zhang L, Lian G, et al. Sildenafil attenuates LPS‐induced pro‐inflammatory responses through down‐regulation of intracellular ROS‐related MAPK/NF‐κB signaling pathways in N9 microglia. Int Immunopharmacol 2011;11:468–474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The results of the behavioral studies showed that sildenafil‐treated rats obtained higher beam walking scores and took longer to fall from a rotating rod.
Figure S2. The statistical results of western blot analysis showed that sildenafil could increase neuron survival by downregulation of the Nogo‐R pathway. Ischemia could cause low levels of synaptophysin and high levels of uncoupling PSD‐95, which could be blocked by sildenafil.