

Abstract
Fluorescence superquenching is investigated for polyelectrolytes consisting of cyanine dye pendant polylysines ranging in number of polymer repeat units (NPRU) from 1 to 900, both in solution and after adsorption onto silica nanoparticles. As NPRU increases, the absorption and fluorescence evolve from monomer spectra to red-shifted features indicative of molecular J aggregates. In solution, the superquenching sensitivity toward an anionic electron acceptor increases by more than a millionfold over the NPRU range from 1 to 900. The dramatic increase is attributed to enhanced equilibrium constants for binding the quenchers, and the amplified quenching of a delocalized exciton of ≈100 polymer repeat units. The self-assembly of monomer onto silica and clay nanoparticles leads to formation of J aggregates, and surface-activated superquenching enhanced 10,000× over the monomer in solution, indicating the formation of “self-assembled polymers” on the nanoparticle surface. Utilization of these self-assembled polymers as high-sensitivity biosensors is demonstrated.
Recently it has been discovered that certain fluorescent polyelectrolytes—including conjugated polymers and polymers containing pendant cyanine dyes—exhibit very high sensitivity (“superquenching”) to oppositely charged molecular quenchers capable of quenching fluorescence by means of electron or energy transfer (1–5). Superquenching has been used to develop a rapid, homogeneous, and sensitive format for biosensing and bioassay both in solution and with the polyelectrolyte adsorbed onto supports (1, 6–8). We have recently synthesized a series of cyanine pendant poly(l-lysine) derivatives with a number of polymer repeat units (NPRU) ranging from one to nearly 1,000. As anticipated, the superquenching efficiency in solution increases dramatically (roughly one millionfold) over the range NPRU = 1–900. We find that it is possible to mimic the enhancement in superquenching observed for a polymer in solution by using a monomer or small oligomer self-assembled onto the surface of nanometer- to micrometer-sized supports. This “surface-activated” superquenching offers a means of extending the applications of superquenching in biosensing from fluorescent polymers to a range of monomeric and oligomeric dyes. In the present article we report on the remarkable differences in the superquenching observed for the different polymers as a function of the NPRU and the extent of their formation of dye aggregates. We compare the behavior of the individual polymers between solution and supported formats and apply this information to construct a new surface-templated polymeric sensor based on self-assembly of individual oligomeric and monomeric building blocks.
Cyanine Dye Pendant Polymer (CDP) Building Blocks in Solution
The synthesis of the CDP building blocks has been carried out by a two-step synthesis from a starting “scaffold” of commercially available poly(l-lysine) as reported elsewhere (9–11, ¶). The approximate NPRU in the starting poly(l-lysine) used in this study was 5–6, 33, 60, 110, 250, 400, and 900. The monomeric cyanine dye 1
was also studied. As shown in Fig. 1, the absorption spectra of the low molecular weight CDPs in 50:50 vol/vol water/DMSO (similar spectra are obtained in pure water) exhibit broadened transitions in the same region where the monomer absorbs, with an onset of a sharp, red-shifted transition for the polymers having PRUs of 63 and higher. This transition is characteristic of a so-called J aggregate, where the adjacent cyanine dyes on the polymer stack with an offset face-to-face crystalline structure (9, 12–15). For the CDP having higher NPRU, the J aggregate band becomes dominant and shows a gradual sharpening and red-shift consistent with formation of an extended J aggregate structure. All of the CDPs exhibit fluorescence in the same solutions as shown in Fig. 1. Although the absorption spectra do not exhibit J aggregation until the NPRU reaches 63, the fluorescence spectra show predominant J aggregate emission in solution when the NPRU reaches 14, though the band increases notably in sharpness as the NPRU increases. As reported for the CDP with NPRU = 250, simulations suggest that a J aggregate arrangement similar to that obtained in crystalline forms of the monomer cyanine dye 1 may be most readily obtained for the cyanine-pendant poly(l-lysine) when the polypeptide is folded into a β-sheet structure (9).
Figure 1.

Normalized absorption and emission spectra of CDP as a function of NPRU.
The fluorescence of the monomer 1 (C+) can be quenched by the anionic electron acceptor anthraquinone disulfonate (AQS2−; 2) because of the equilibrium association described in Eq. 1.¶
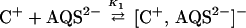 |
1 |
The monotonic decrease in fluorescence intensity (I) with concentration of the quencher is found to give a linear plot (Stern–Volmer relationship) when the intensity of fluorescence with no quencher present (Io) divided by the intensity at any quencher concentration (I) is plotted vs. the concentration of quencher (Q) (Eq. 2).
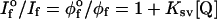 |
2 |
The Stern–Volmer quenching constant, Ksv, in 50:50 (vol/vol) water/DMSO is 630 M−1, a value close to that predicted for a coulombic association constant (K1) between cationic 1 and dianionic 2. Linear Stern–Volmer plots are also obtained for the series of CDPs; however, as discussed below, the interpretation of these plots is not as simple as for the case of the monomer. As shown in Fig. 2, the Ksv values for the series of CDPs show a monotonic increase with increase in NPRU.‖ The data are plotted on a log–log scale for clarity; the Inset shows the same data on a linear scale. The increase in Ksv, though monotonic, shows some interesting trends that delineate the role of J aggregation in amplifying the quenching. For the smallest oligomer (NPRU = 5–6), Ksv is enhanced over the monomer, by ≈103, which can be attributed primarily to an increased coulombic attraction between the more highly charged oligomer and the quencher. For the higher molecular weight CDPs, much of the additional enhancement of Ksv can be attributed to J aggregation. For example, for the polymer with 900 repeat units, Ksv is 5.2 × 108 M−1 in water/DMSO where the polymer is predominantly J aggregate; Ksv for the same polymer in methylene chloride (where no J aggregation is evident from either absorption or fluorescence), is 1.5 × 107 M−1 or a factor of ≈35 lower. In contrast, for the CDP with 33 repeat units, for which minimal J aggregation is observed in either solution, the quenching constants are almost the same, and the same as for the polymer with NPRU = 900 in methylene chloride. As illustrated in Fig. 2, the quenching constants increase rapidly, with saturation at NPRU ≈ 250. Before saturation, the growth in Ksv is superlinear, implying a contribution both from the extended aggregate (expected to give a linear increase in Ksv with NPRU) and also from enhanced coulombic attraction due to the growing net charge on the polymer. Perhaps more informative are the trends in two additional parameters that can be obtained from the quenching data. These are the number of quenchers per polymer molecule (Q/P50) and the number of PRU per quencher at 50% quenching (PRU/Q50). These are shown in Figs. 2 and 3. The value of Q/P50 initially decreases as the size (NPRU) of the polymer increases and reaches a minimum value of 2.5 near PRU ≈250. The second parameter, PRU/Q50 increases until a strong J aggregate is formed and saturates at ≈100.
Figure 2.

Fluorescence quenching of CDP as a function of NPRU. Left axis, Ksv; right axis, PRU/Q50. Solid blue line (▴) is for silica-supported CDP, dashed red line (□) is for solution phase polymers. In the main figure, the data are plotted for clarity on a log–log scale. (Inset) The same data on a linear scale.
Figure 3.

Q/P50 for CDP as a function of NPRU. Solid blue line (▴) is for silica-supported CDP, dashed red line (□) is for solution-phase polymers.
The results obtained for quenching of the fluorescence of the CDPs in 50:50 (vol/vol) water/DMSO solution (very similar Ksv values are obtained in pure water; the oligomers and polymers generally exhibit greater solubility and stability toward adsorption in aqueous DMSO compared with pure water) show that both polymer size and aggregation contribute to the extent of superquenching. The quenching constants observed in this study are, to our knowledge, the highest Stern–Volmer quenching constants reported. The fact that the Ksv values level off for CDPs having more than 250 PRUs suggests that there may be a practical limit or optimum size polymer for solution-phase superquenching based on fluorescent polyelectrolytes.
CDP Building Blocks on Silica Microspheres and Laponite Clay.
The cationic CDPs can be adsorbed from aqueous solution onto silica microspheres or Laponite RDS clay nanoparticles (7–9). Both the monomer 1 and the CDPs incorporating the chromophore of 1 give evidence of J aggregation on both silica microspheres and clay nanoparticles. J aggregates of cyanine dyes assembled on silver halide particles have been widely applied in color photographic film. Many cyanine dyes are known to form aggregates, but the molecular mechanisms governing aggregate formation are still not well understood. Although J aggregation on supports such as Laponite and other clays and in assemblies has been observed for several specific cyanine dye chromophores, it is not observed for other structurally similar cyanines and the structural factors that control J aggregation on these supports are also not well-understood (ref. 9 and W. Herkstroeter, S. Farid, L.L., D.W.M., and D.G.W., unpublished studies). For the larger CDPs that form J aggregates in water, the transfer from solution to the adsorbed phase is accompanied by a broadening and weakening of the J aggregate absorption (8). The fluorescence from the adsorbed polymer on the particles is in the same spectral region and slightly broadened compared with the solution J aggregate. For the smaller CDPs, for which the J aggregate is not the predominant species in solution, the absorption spectra of the adsorbed species are quite broad, yet the fluorescence of the adsorbed species is always predominantly that from a J aggregate. In previous studies with the CDP (NPRU = 250) we found that, depending on the loading and the nature of the oppositely charged support, either “normal” superquenching by oppositely charged energy or electron acceptors could occur, or “charge reversal” superquenching by like-charged quenchers could be observed (7, 8). In the present study using the series of CDP polymers on silica microspheres, we observe “normal” superquenching by oppositely charged AQS2−. This appears reasonable based on the fact that the microspheres at neutral pH have a low net negative charge such that at moderate loading the CDP-coated spheres should have an overall positive charge and thus still attract anionic quenchers. As shown in Fig. 2, the variation of Ksv with NPRU for the silica-supported CDPs is remarkably different from that for the same series of polymers in solution. Thus although there is again a monotonic increase in Ksv with NPRU, the quenching constants for the monomer 1 and the low molecular weight CDPs are much higher than for solution, whereas the Ksv values for the three highest CDPs are slightly lower than in solution. For the supported CDP Ksv varies by only a factor of 15 over the range from monomer to NPRU = 900. The most noteworthy change is the increase of the quenching constant for monomer 1 by a factor of almost 20,000 on adsorption into a J aggregate on the silica microspheres.
The striking increase in Ksv for the lower molecular weight CDPs and monomer adsorbed onto the silica microspheres (surface-activated superquenching) seems most consistent with effective collection of smaller units together on the microsphere surface such that enhanced superquenching occurs. Some of this effect can be ascribed to the greater effective charge of the “collected polymer” on the spheres compared with individual oligomer “units” in solution. However a second, and conceivably more interesting effect is the potential of exciton delocalization extending across aggregates consisting of more than one oligomer. To gain some insights into the relative importance of these two effects it is useful to examine the parameters Q/P50 and PRU/Q50 for the supported molecules.
Not surprisingly, the factors Q/P50 and PRU/Q50 are quite different for the microsphere-supported CDPs compared with the solution-phase polymers. The values for Q/P50 for the monomer and small oligomers are quite small (<1), and much lower than the corresponding values for the same compounds in solution. The values for PRU/Q50 are higher for monomer and the low molecular weight oligomers, but somewhat smaller for the higher molecular weight polymers. As will be discussed below, the value of PRU/Q50 should provide a rough estimate of the effective exciton size for a given oligomer or polymer. The values for the supported oligomers having NPRU = 1, 6 and 33 are much larger than for solutions, but still very modest compared with other systems in which extended J aggregates are evident. On the other hand, the values for Q/P50 less than 1 for these three compounds indicate that some cooperativity exists among nearby molecules.
The Role of J Aggregation in CDP Superquenching.
When we compare the quenching of the monomer and CDP polymers with AQS2− it is clear that the largest PRU/Q50 values are obtained for the solutions of the polymers with 250, 400, and 900 PRUs. The saturation of PRU/Q50 at about 100 PRU per quencher can be compared with quenching of other J aggregated systems in some quite different formats. Previous studies of a cyanine J aggregate quenching by energy acceptors in a solid (Langmuir–Blodgett) film indicate that incorporation of an imbedded energy acceptor at a level of 1 quencher per 12,000 monomers in the aggregate results in 50% quenching (13). However, for incorporation of the same energy acceptor in an adjacent layer, a more than 40-fold reduction in the number of monomers quenched (to ≈300) is observed (13). Interestingly, a study of quenching of a similar cyanine J aggregate Langmuir–Blodgett film by an electron acceptor in an adjacent layer (amphiphilic viologen) indicated 50% quenching of the J aggregate fluorescence occurs at a level of 1 quencher per 44 molecules of the cyanine, a value about half that found in this study (16). Studies with electron acceptors in Langmuir–Blodgett films of squaraine dyes suggest quenching occurs at similar levels (1 “trap” per 25 aggregated molecules) of aggregate component molecule/electron acceptor (17). These lower numbers are also consistent with the “exciton migration distance” of ≈50 nm estimated from near-field scanning optical microscope (NSOM) studies of pseudoisocyanine aggregates grown in poly(vinyl sulfate) thin films (18). Other optical studies of J aggregate excitons suggest that the exciton domain may be small, even at low temperatures (19–22). These previous studies were all carried out in rigid solid media and it is not clear to what extent the solution-phase polymer J aggregates can be compared with them. It would seem possible that in fluid media the conformations of these polymers may be mobile and more random than in a crystal or solid and that imperfections might limit the size of J aggregate domains and the effective exciton range. However, studies of liposomes containing J aggregate cyanine dyes give quenching results similar to those in the solid phase and it is reasonable to expect that a liposome in water might be somewhat comparable to a semiorganized J aggregate polymer (23). The observation that PRU/Q50 is lower for the larger polymers when they are supported on the silica microspheres is consistent with the broadening or “denaturation” of the J aggregate absorption when the polymers are adsorbed onto the silica. Thus in these cases instead of having a more crystalline-like J aggregate absorption, it is clear that there must be a mixture of environments with patches of “J” aggregate serving as traps from which the predominant emission occurs.
From our study of the present series of CDP polymers, it appears that J aggregation can provide an enhancement of quenching (in terms of Ksv) of up to two orders of magnitude for electron transfer quenchers. The results of previous investigations suggest that the use of energy transfer quenchers (13, 15, 16) might lead to somewhat larger exciton harvesting and hence even greater quenching constants. It is reasonable that the largest factor controlling superquenching for both the polymers in solution and the adsorbed monomer and oligomers on silica microspheres is the enhanced coulombic and hydrophobic attraction. What is remarkably demonstrated by both the solution and supported format investigations is the ability to “tune” the superquenching by control of the size and environment of the polymers or smaller building blocks.
Surface-Activated Superquenching: Biosensing Using “Self-Assembled Polymers”
Surface-activated superquenching offers a route to design biosensors from a potentially wide range of dyes that form supramolecular assemblies. We have demonstrated a biosensor using surface-activated superquenching with a quencher-tether-ligand (QTL) conjugate, followed by reversal on addition of a receptor for the conjugate. For this demonstration we used the cyanine dye monomer 1 assembled into a J aggregate on Laponite clay nanoparticles. As observed for the silica microspheres, addition of Laponite clay to an aqueous solution (1.2% methanol) of 1 results in its adsorption onto the clay concurrent with the formation of a very sharp J aggregate spectrum (Fig. 4). The adsorbed J aggregate of 1 can be quenched by the cationic viologen-biotin conjugate (V-B+) but not by the anionic anthraquinone-biotin conjugate (AQ-B−).
Figure 4.

Absorption spectra showing adsorption of 1 (17.2 μM/1.2% methanol in water) onto Laponite clay nanoparticles: black curve is starting mixture of monomeric 1 and J aggregate microcrystals. Successive curves (follow arrows) show spectral changes occurring on successive addition of microgram portions of Laponite nanoparticles. (Inset) Quench–unquench experiments of Laponite-supported 1 J aggregate in water. Black curve is starting emission from nanoparticles in water; red curve is emission quenched by the addition of 100 nM V-B+; blue and green curves show recovery on successive additions of 25 nM avidin.
The quenching charge reversal (quenching by a cationic electron acceptor) is similar to that reported earlier for CDP coated onto clay (8), and reflects the higher charge density of the anionic clay, and that coated clay retains a net negative charge. The quenching constant Ksv ≈ 7 × 106 M−1 is similar to that of 1 adsorbed onto silica and quenched by the anionic AQS2−. As shown in Fig. 4 Inset, addition of dilute avidin solutions causes a quantitative unquenching of the J aggregate fluorescence of adsorbed 1. Only a slight stoichiometric excess of the protein is needed (each avidin has four biotin-binding sites) to accomplish complete unquenching and very little “over-recovery” (indicative of nonspecific association of the protein with the adsorbed J aggregate) is detected on further addition of avidin. The results of this study indicate that superquenching can be extended from polymers to small oligomers and even monomers by collecting the smaller molecules into self-assembled polymers. The self-assembled polymers thus obtained by adsorption onto charged supports can be tuned in both their quenching and quench-recovery behavior to widen the possibilities for sensing and other applications.
Scheme 1.

Scheme 2.

Acknowledgments
We thank Ileana Place, Tom Penner, Sriram Kumaraswamy, and Fred Wudl for technical assistance and helpful discussions. This work was supported by the Defense Advanced Research Projects Agency under Contract MDA972-00-C-006.
Abbreviations
- NPRU
number of polymer repeat units
- CDP
cyanine dye pendant polylysine
- PRU/Q50
number of polymer repeat units per quencher at 50% quenching
- Q/P50
quenchers per polymer at 50% quenching
- AQS2−
anthraquinone disulfonate
Footnotes
The counterion for the polymer as synthesized
is predominantly the tosylate
(C7H7SO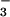 ). Although we do not
have any indication of the degree of association in water or aqueous
dimethyl sulfoxide (the media used for solution studies), it is
reasonable that anion exchange should occur readily with
AQS2−.
). Although we do not
have any indication of the degree of association in water or aqueous
dimethyl sulfoxide (the media used for solution studies), it is
reasonable that anion exchange should occur readily with
AQS2−.
There is generally a variation of 5–10% in multiple determinations of Ksv; R2 values are 0.95 or greater.
References
- 1.Chen L, McBranch D W, Wang H-L, Helgeson R, Wudl F, Whitten D G. Proc Natl Acad Sci USA. 1999;96:12287–12292. doi: 10.1073/pnas.96.22.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen L, McBranch D, Wang R, Whitten D. Chem Phys Lett. 2000;330:27–33. [Google Scholar]
- 3.Chen L, Xu S, McBranch D, Whitten D. J Am Chem Soc. 2000;122:9302–9303. [Google Scholar]
- 4.Wang J, Wang D, Miller E K, Moses D, Bazan G C, Heeger A J. Macromolecules. 2000;33:5153–5158. [Google Scholar]
- 5.Harrison B S, Ramey M B, Reynolds J R, Schanze K S. J Am Chem Soc. 2000;122:8561–8562. [Google Scholar]
- 6.Whitten D, Chen L, Jones R, Bergstedt T, Heeger P, McBranch D. In: Molecular and Supramolecular Photochemistry. Schanze K S, Ramamurthy V, editors. Vol. 7. New York: Dekker; 2001. , Ch. 4, pp. 189–208. [Google Scholar]
- 7.Jones R M, Bergstedt T S, Buscher C T, McBranch D, Whitten D. Langmuir. 2001;17:2568–2571. [Google Scholar]
- 8.Jones R M, Bergstedt T A, McBranch D, Whitten D. J Am Chem Soc. 2001;123:6726–6727. doi: 10.1021/ja0157797. [DOI] [PubMed] [Google Scholar]
- 9.Place I, Perlstein J, Penner T L, Whitten D G. Langmuir. 2000;16:9042–9048. [Google Scholar]
- 10.Roberts M R, Coltrain B C, Melpolder S M, Wake R W. Ceramic Trans. 1991;19:287–293. [Google Scholar]
- 11.Lu, L., Helgeson, R., Jones, R. M., McBranch, D. & Whitten, D. G. (2002) J. Am. Chem. Soc. 124, in press. [DOI] [PubMed]
- 12.Scheibe G, Schoentag A, Katheder F. Naturwissenschaften. 1939;29:499–501. [Google Scholar]
- 13.Kuhn H, Kuhn C. In: J Aggregates. Kobayashi T, editor. Singapore: World Scientific; 1996. pp. 1–40. [Google Scholar]
- 14.Moebius D, Kuhn H. J Appl Phys. 1988;64:5138–5144. [Google Scholar]
- 15.Kuhn H, Foersterling H-D. Principles of Physical Chemistry. Chichester, UK: Wiley; 2000. [Google Scholar]
- 16.Penner T L, Moebius D. J Am Chem Soc. 1982;104:7407–7413. [Google Scholar]
- 17.Liang K, Law K-Y, Whitten D G. J Phys Chem. 1995;99:16704–16708. [Google Scholar]
- 18.Higgins D A, Barbara P F. J Phys Chem. 1995;99:3–7. [Google Scholar]
- 19.Tani T, Suzimoto T, Kemnitz K, Yoshihara K. J Phys Chem. 1992;96:2778–2783. [Google Scholar]
- 20.Moll J, Daehne S, Durrant J R, Wiersma D A. J Chem Phys. 1995;102:6362–6370. [Google Scholar]
- 21.De Boer S, Wiersma D A. Chem Phys Lett. 1990;165:45–53. [Google Scholar]
- 22.Fidder H, Knoester J, Wiersma D A. Chem Phys Lett. 1990;171:529–536. [Google Scholar]
- 23.Sato T, Kurahasi M, Yonezawa Y. Langmuir. 1993;97:3395–3401. [Google Scholar]