

Abstract
Cancer research relies on model systems, which reflect the biology of actual human tumors only to a certain extent. One important feature of human cancer is its intra-tumor genomic heterogeneity and instability. However, the extent of such genomic instability in cancer models has received limited attention in research. Here we review the state of knowledge about genomic instability of cancer models and discuss its biological origins and implications for basic research and for cancer precision medicine. We discuss strategies to cope with such genomic evolution, and evaluate both the perils and the emerging opportunities associated with it.
Introduction
Cancer is a disease characterized by the genomic instability of somatic cells. Genomic evolution generates genetic and epigenetic diversity, and the resultant cellular heterogeneity constitutes a fertile molecular ground for further evolution. In recent years, largely thanks to the advance of single-cell “omics” and sequencing technologies, clonal heterogeneity and tumor evolution have been studied extensively, and their importance for cancer progression and for the clinical outcome of cancer treatments is now widely appreciated (reviewed in 1, 2).
Any functional interrogation of human cancer cells must rely on patient-derived cancer models, such as patient-derived cell lines (PDCLs), patient-derived organoids (PDOs) and patient-derived xenografts (PDXs). The successful derivation of such models requires that the tumor cells adapt to new environmental conditions, in other words, distinct selection pressures, and their propagation continuously selects for the fittest and most rapidly proliferating cells3–5. Moreover, as cancer cells are often deficient in their ability to properly maintain genome integrity (reviewed in 6), their inherent genomic instability makes them susceptible to rapid acquisition of additional genetic insults throughout propagation. Non-patient-derived cancer models, such as genetically-engineered mouse models (GEMMs), also experience genomic evolution, both at the tumor level and at the host level7. Cancer model evolution is thus emerging as an important aspect of cancer modeling.
In recent years, advances in the development of cancer models have greatly expanded their application in cancer precision medicine. First, large cohorts (also known as “biobanks”) of cancer models have been generated, and extensive genomic and phenotypic characterization of these models performed, in order to uncover genotype-phenotype associations at the patient population level8–31. Second, patient-derived models are increasingly being used as “avatars” of their tumor of origin, in an attempt to predict patient-specific drug response31–35. For both applications, cancer models ought to be faithful representations of the tumors from which they were derived, and remain genomically and phenotypically stable throughout propagation. The proper use of cancer models thus requires critical evaluation of these underlying assumptions in light of the propensity of these models to evolve.
The evolution of cancer models bears potential consequences for another burning issue in cancer research – its reproducibility. The “reproducibility crisis”, that is the inability to replicate results reported in the literature, has drawn much attention recently. Cancer research has been at the focus of this debate, following reports that only 11% to 25% of high-profile cancer studies could be replicated by an industrial lab36, 37. For example, differences between large-scale drug screens of cancer cell lines have been observed and debated in the literature38–40. While many explanations have been suggested to account for, and to some extent reconcile, such discrepancies39–45, the potential contribution of model evolution to observed differences remains underexplored.
In this Opinion, we summarize the emerging evidence for genomic evolution in cancer models, its biological origins and its functional consequences. We then highlight the implications for basic cancer research and for clinical translation, including cancer precision medicine. Finally, we suggest practical ways to mitigate the risks posed by genomic evolution, and propose how to constructively build upon this phenomenon in future research.
Model evolution: proof and prevalence
The factors shaping evolution (Fig. 1) can differ between GEMMs and patient-derived models, and between PDCLs, PDXs, and PDOs (Table 1). The rate of genomic evolution is determined by the genomic heterogeneity within the cell population, and by the genomic stability of the individual cells. Quantitative assessment of these traits can therefore be used to follow genomic evolution and estimate its prevalence (Box 1).
Figure 1: The biological origins of cancer model evolution.
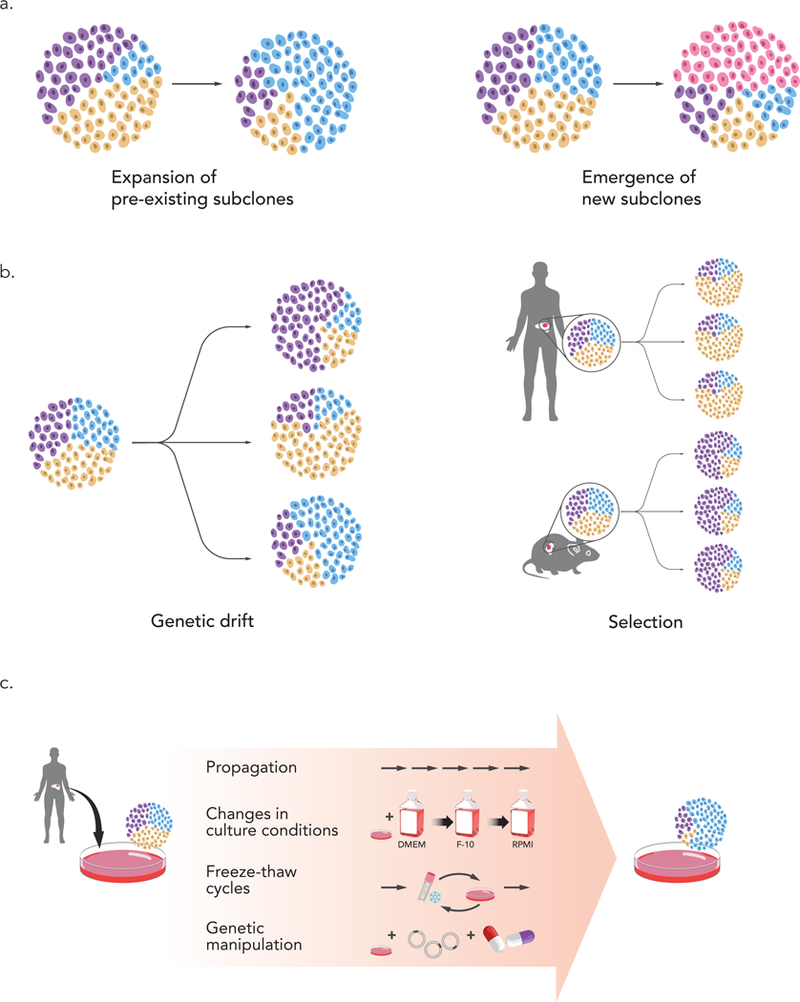
(a) Genomic evolution could be the outcome of clonal dynamics that lead to the expansion of pre-existing subclones (left), or the outcome of the emergence of new subclones during the derivation or the propagation of the model (right). (b) In both cases, such evolution could result from a genetic drift, which would lead to stochastic changes (left), or from clonal section, which would lead to reproducible changes (right). Selection pressures are different between the natural tumor environment in the patient’s body and the new environment of the model (e.g., mouse in the case of PDXs). (c) Bottlenecks associated with model propagation can promote genomic evolution. In ECLs, the main bottlenecks are extensive propagation, changes in culture conditions, multiple freeze-thaw cycles, and genetic manipulations that involve viral infection and/or antibiotic selection.
Table 1:
Determinants of genomic evolution in cancer models.
| Model type | Major advantages for research | Factors impacting genomic evolution | |
|---|---|---|---|
| Genetically-engineered mouse models |
* De novo tumorigenesis in vivo * Interactions with the microenvironment and with other cell types |
* Somatic evolution of the tumor as an integral part of tumorigenesis * Germline evolution of the host throughout colony propagation |
|
| Patient-derived models | New cancer cell lines | * Short time and few cell divisions from primary tumors to functional assays | * Physical constraints (2D) * Variations in culture conditions (media, passaging practices, etc.) * Continuous selection for rapidly proliferating cells |
| Established cancer cell lines | * Widely accessible and easy to work with * Ample genomic data available |
* Numerous cell divisions * Variations in culture conditions * Deficient mechanisms of genome maintenance inherited from tumor of origin |
|
| Xenografts | * No growth on plastic * Functional investigation of human tumors in vivo |
* Differences in physiology and metabolism between species * Immune-deficient environment * Site of transplantation * Multiple cell divisions within each passage |
|
| Organoids | * Complex cellular interactions * Culture conditions mimic in vivo conditions better than 2D culture * 3D environment * Matched normal controls |
* Variations in culture conditions * Deficient mechanisms of genome maintenance inherited from tumor of origin * Immune-deficient environment |
|
Box 1: Measuring genomic evolution in cancer models.
The ability to follow the genomic evolution of cancer models has considerably advanced in recent years. First, the drop in the costs of DNA and RNA sequencing has increased standard sequencing depths121, enabling to detect genetic alterations that are rare within the bulk tumor population, and to characterize the genomic composition of cancer models at multiple time points throughout their propagation52, 81, 83. Second, single-cell sequencing technologies now enable cellular heterogeneity to be studied at the resolution of individual cells83, 108, 109, and improvements in the isolation and expansion of clones from single tumor cells enables their functional interrogation115. Third, analytical tools have been developed that use genomic data to infer the clonal structures of cell populations107, 122, 123, and additional tools make use of genomic data to characterize and quantify signatures of genomic instability124–126. Together, these methods now enable to measure both the heterogeneity and the instability of cancer models in much greater detail than before. It is important to note that genomic heterogeneity and genomic instability, although conceptually related and quantitatively correlated81, 107, are not synonymous terms – heterogeneity refers to the genetic variation within the cell population, whereas instability refers to the stability of that genetic composition over time. Measuring both of these traits in cancer models is required to understand how these models evolve over time.
Genetically-engineered mouse models
De novo tumor evolution
GEMMs are a powerful tool to study tumor heterogeneity and follow tumor evolution. They have been used extensively for these purposes, especially with the advance of technologies to edit the genome and to molecularly profile tumors (reviewed in 7, 46, 47). As GEMMs are primarily generated by manipulating a single gene, or a handful of genes, genomic evolution of the manipulated tissue must occur in order for tumors to form. Numerous studies have shown that diverse routes of genomic evolution could lead to the formation of molecularly distinct tumors, within the same mouse model48, 49. Nonetheless, genomic evolution is not stochastic; we and others have shown that specific transgenes induce specific secondary genetic events, thus identifying driver-specific evolutionary trajectories of tumorigenesis50–52. Tumor formation in GEMMs is thus inherently associated with genomic evolution. Despite differences in the tumor genomic landscapes and in the relatively short time for tumor formation required in mice compared with humans46, this type of evolution is clearly a desired trait of GEMMs, as it largely mimics tumor evolution in human patients49, 52, 53.
Genetic instability in mouse colonies
Applying GEMMs for research involves their continuous breeding for the purpose of colony expansion and maintenance, which generates a risk for genomic diversification throughout the generations. Two populations of the same laboratory mouse strain will ultimately evolve in different directions, if maintained and propagated separately. Based on spontaneous mutation rates, 0.96 deleterious germline mutations are expected to arise in wildtype laboratory mice each generation, and this number is much higher in genomically unstable mice54. As colony maintenance relies on inbred breeding, there is ~25% likelihood for a new mutation to become homozygous, and thus fixed, in the population.
Indeed, different substrains of common mouse strains are acknowledged in the literature. For example, the commonly used C57BL/6 strain has evolved into multiple substrains, with the two major ones – C57BL/6J and C57BL/6N – differing in 34 single-nucleotide variants, 2 indels and 15 structural variants in coding genes55. This genetic variation is associated with marked phenotypic variation55–58, and can severely mislead the interpretation of experimental results59–61. Therefore, the US Institute for Laboratory Animal Research (ILAR) assigns unique identifiers that designate the laboratory in which mice were maintained (see Related Links), and strain diversification is thus reflected in proper mouse strain nomenclature.
Patient-derived models
Established cancer cell lines
The diversification of established cell lines (ECLs) in culture has been appreciated for decades, and some of the most commonly used cancer ECLs, such as HeLa and MCF7, have become notorious for it62–67. In fact, similar to the mouse nomenclature, it has become common practice to designate the exact strain of HeLa used in one’s study (e.g., HeLa-CCL2, HeLa-S3 or HeLa-Kyoto). Nonetheless, ECLs are often considered to be clonal and stable for most applications, as evident by efforts to provide definitive characterization of their genomic landscapes and cellular dependencies8–14, as well as by lack of routine documentation of culture history (e.g., passage number is rarely tracked, reported or controlled for).
Recent molecular analyses comprehensively characterized the genomic variation that exists between strains of ECLs, revealing rather extensive heterogeneity and evolution. Comparing multiple strains of commonly used cancer and non-cancer ECLs, we found evidence for extensive genetic variation at all genetic levels – point mutations, rearrangements and copy number alterations – and affecting many cancer-related genes. A comparison between over 100 ECLs cultured independently in two laboratories (in the U.S.A and the U.K.), found a ~20% chance that a mutation would be detected in only one of the two compared strains68. Variation in gene expression mirrored genetic variation, and specific genetic alterations were often associated with a transcriptional signature of the genetically perturbed pathway68. Preliminary observations from 14 strains of HeLa revealed similar magnitudes of genetic and transcriptional variation, which translated into corresponding proteomic variation as well69. In both of these studies, routine maintenance of cells under standard culture conditions resulted in rapid genomic diversification within a handful of passages. In agreement with these findings, a recent analysis of RNAseq data from eight cell lines found genetic variability across labs, which was associated with gene expression variability70. Therefore, the genetic, transcriptional and proteomic landscape of cancer ECLs keeps evolving in culture, leading to genomic differences across separate cultures of the same ECL.
In addition to genetic variation, cell line diversification could be induced by epigenetic variation. Given their more transient nature, epigenetic marks would likely be more sensitive to culture conditions than DNA sequences, making cell lines susceptible to epigenetic instability. Indeed, instability of DNA methylation and erosion of X-chromosome inactivation have been documented throughout the culture of human pluripotent stem cell lines71, 72. Similarly, continuous propagation of human mammary epithelial cells is associated with silencing of the tumor suppressor p16INK4A by hypermethylation of its transcription start site73, demonstrating that epigenetic instability can alter the tumorigenicity of cultured cell lines. Systematic analysis of epigenomic variation across strains of cell line cultures is yet to be performed.
New patient-derived cell lines
As described above, cancer ECLs, which were derived from patients many years ago, experience ongoing genomic evolution. To what extent would such evolution also affect recently derived early-passage PDCLs? Recently, systematic attempts to generate PDCLs have been initiated, in order to increase representation of certain genetic alterations and tumor lineages available for cancer research74–76. It has been hoped that these freshly derived PDCLs would resemble their tumors of origin better than ECLs that have been cultured, and have evolved, over decades.
While PDCLs were reported to largely retain the genomic features of primary tumors, differences between these models and their parental tumors were observed77–80. We recently analyzed copy number alterations (CNAs) in 38 samples of PDCL models from 5 cancer types, and determined how the genomic landscapes of these models evolved throughout their derivation (p0/1) and early propagation (through p20)81. We found evidence for continuous genomic evolution throughout passaging, with an average of ~20% of the genome differentially affected by CNAs between early and later passages81. Interestingly, CNA landscapes of PDCLs evolved more rapidly during the first few passages (<p5), compared to the later time points (>p10). This suggests that the derivation of the models is associated with genomic evolution, and that the models eventually become more stable as they adapt to their new environment.
Patient-derived xenografts
PDXs are considered to be more physiologically relevant than cell lines, and to mimic the human disease more accurately, as their generation and propagation does not involve their culture in artificial in vitro conditions (reviewed in 4, 33). However, the in vivo xenograft environment is quite distinct from the original patient environment. First, metabolism and physiology differ between species. Second, PDXs are commonly transplanted subcutaneously, exposing the tumors to signaling cues, cellular interactions and mechanical constraints utterly different from their native microenvironment. And third, the lack of a functioning immune system in immune-compromised mice could alter tumor development and behavior82.
Indeed, PDXs undergo genomic evolution throughout their derivation and propagation. When engraftment and early passage propagation of breast cancer PDX models was followed using single-cell sequencing, extensive clonal dynamics were observed, which drastically changed the abundance of mutation clusters (and thus the allele fraction of mutations) throughout serial PDX passaging83. Similarly, the engraftment of human acute lymphoblastic leukemia (ALL) in mouse xenografts was shown to be associated with genomic evolution into a more aggressive malignancy84, and engraftment propensity varied considerably among genetically-distinct clones of acute myeloid leukemia (AML)85, 86. Our analysis of CNAs in 543 unique PDX models across 24 cancer types found that ~60% of the models acquired at least one large chromosomal aberration within a single passage, and ~90% acquired at least one such aberration within four passages81. Similar to the observation in PDCLs, genomic evolution was most rapid during PDX initiation and early passaging, and its rate considerably decreased at later passages81, consistent with strong selection pressures being associated with model initiation.
Importantly, the rate of genomic evolution, defined as the fraction of the genome altered per passage, was similar between PDXs and PDCLs81. Comparing matched PDXs, a median of ~12.5% of the genome was differentially affected by CNAs within four passages. One should note, however, that an in vitro passage normally involves many fewer cell divisions than an in vivo passage, so the rate of change per cell division may be smaller in vivo than in vitro. Nevertheless, these findings challenge the notion that PDXs better preserve the genomic landscapes of primary tumors.
It is worth noting that PDXs are also susceptible to epigenomic evolution throughout their propagation. A recent study that compared the methylation patterns between primary NSCLC tumors and their derived PDXs reported relatively low genome-wide Spearman correlations (ranging from 0.37 to 0.49)87. While many of the observed differences were related to tumor purity, others potentially reflect epigenetic instability87.
Patient-derived organoids
PDOs have emerged as physiologically relevant 3D model systems to study cancer in vitro (reviewed in 3). Long-term organoid cultures have been established from multiple epithelial cancer types, by embedding cancer cells into a 3D matrix in medium containing tissue-specific growth factors that recapitulate the tumor niche. Cancer organoids have several advantages over 2D cell lines, including more complex cellular composition. The culture conditions also permit expansion of normal epithelial cells, providing the ability to obtain matched normal organoids from the same patients3.
Several arguments suggest that tumor organoids may experience less genomic evolution than their 2D counterparts and thus better retain tumor genomic features. First, organoid derivation from primary tumors is more efficient than that of PDCLs3, suggesting that their generation might be associated with less of a population bottleneck. Second, organoid culture conditions better recapitulate those of the original tissue3, potentially alleviating some of the selection pressures entailed in the in vitro transition. Third, genomic profiles of PDOs were found to be highly similar to those of the tumors of origin23, 28, 30, 31, and PDO drug response could recapitulate patient response in the clinic31, 88.
Nonetheless, PDOs are not exempt from in vitro model evolution. Extensive genetic diversity has been recently described in colorectal PDOs89; the derivation and long-term culture of such highly heterogeneous organoids are likely to be associated with clonal dynamics that will alter the clonal composition of the model, similar to that observed in cell lines and PDXs68, 81, 83, 84. Indeed, PDOs are not perfect genomic representations of their tumors of origin, and all of the major PDO cohorts reported to date have provided examples for differences in the status of both somatic point mutations and CNAs between matched primary tumors and PDOs23, 28, 30, 31, 90. Importantly, strong clonal dynamics have been recently described in PDOs, leading to rapid expansion of pre-existing minor subclones90. As in all other models, the inherent genomic instability of cancer cells is also likely to result in de novo genetic alterations on continuous propagation of organoid models28. Future studies will be required to characterize the extent of genomic evolution in PDOs, as matched genomic data from multiple time points throughout PDO passaging become available.
Mechanisms of cancer model evolution
The genomic evolution observed in cancer models could be the result of pre-existing heterogeneity, as clonal dynamics of pre-existing tumor subclones would lead to changes in their relative abundance. A rare primary tumor subclone could expand and become the dominant subclone in the model, thereby altering the molecular landscape of the model. Alternatively, genomic evolution could be the result of ongoing genomic instability, which could lead to the accumulation and fixation of new genetic alterations following propagation of the model (Fig. 1a). Regardless of whether it reflects expansion of pre-existing subclones, the emergence of a new one, or both, genomic evolution could be either a stochastic or a deterministic process, depending on whether it is a consequence of genetic drift or of genetic selection (Fig. 1b). In either case, bottlenecks associated with the model derivation and propagation would enable and expedite genomic evolution (Fig. 1c). However, if selection is involved, an important question is whether evolutionary trajectories in cancer models reflect evolutionary trajectories in the patients from which they are derived (Fig. 1b).
Clonal dynamics
Molecular heterogeneity has emerged as a fundamental characteristic of most tumor types2. Patient-derived cancer models would thus begin as heterogeneous cell populations. The extent to which models remain heterogeneous, and the strength of clonal dynamics during model establishment and passaging, could determine the stability of the models and whether they accurately mirror their parental tumors.
Clonal dynamics play an important role in the genomic evolution of cell lines, PDXs and PDOs. Stochastic cell transitions have shown clonal dynamics within cell line populations, at the phenotypic level91. Genetic clonal dynamics has been recently described in cancer ECLs, where the relative abundance of genetic subclones varies across strains of the same cell line68. Single-cell genomic analyses of breast cancer PDXs revealed that expansion of minor subclones could drastically change the clonal composition of the model compared to the original tumor83. Such clonal dynamics were also identified in PDXs of multiple additional cancer types81, as well as in esophageal cancer PDOs90, and were not limited only to the model derivation stage, as significant differences were also observed between early- and late-passage PDXs81, 83.
Genetic drift is likely to be involved in the clonal dynamics observed in models, especially during model initiation, given that models are typically generated from a biopsy taken from a small, randomly-selected tumor region. However, both in cell lines and in PDXs, selection has been shown to be a major driver of clonal dynamics. The expansion of minor subclones in PDXs was found to be reproducible; when the same tumor population was transplanted into different mice, expansion of the same minor subclones was observed, indicating non-stochastic, directional genomic evolution81, 83. Similarly, DNA barcoding experiments in ECLs showed that minor changes in culture conditions, such as changing the medium from RPMI to DMEM, resulted in reproducible changes in barcode abundance68. Moreover, stronger selection pressures, such as drug exposure, resulted in stronger clonal dynamics. Therefore, selection-driven clonal dynamics clearly play an important role in genomic evolution of cancer models.
Ongoing genomic instability
Genomic instability is another fundamental trait of tumors, which is carried over from the primary tumors to the models derived from them. The nature and rate of the genetic alterations that arise in the model depend on the genome integrity mechanisms that are perturbed in the tumor of origin. Moreover, some types of genomic instability are exacerbated in model systems. For example, the fidelity of chromosome segregation was recently shown to depend on integrin function and thus diminish when cells were grown in 2D culture92.
Evidence for ongoing genomic instability in cancer models is extensive. As expected, GEMMs generated by perturbation of genes directly related to genome integrity maintenance (most notably, p53) are more genomically perturbed than GEMMs generated with other classes of oncogenes or tumor suppressors52, 93, 94. Similarly, PDXs from p53-null tumors experience more copy-number evolution compared to those from p53-WT tumors81. And the mutation landscape variability across ECL strains is higher in ECLs with microsatellite instability (MSI) than in ECLs without it68, in line with the hyper-mutation phenotype of MSI human tumors.
We recently demonstrated that single-cell clones derived from ECLs quickly became genetically, transcriptionally and phenotypically heterogeneous68. This means that ongoing genomic instability leads to the constant emergence of new subclones, which can eventually expand and alter the genomic composition of the tumor.
Bottlenecks of model propagation
The magnitude of genomic evolution depends on the heterogeneity of the population and on the stringency of the bottlenecks cells need to go through. Some of these bottlenecks are inherent to the nature of cancer models, whereas others depend on experimental practices that could be modified. It is therefore imperative to understand the bottlenecks associated with model derivation and propagation.
The first strong bottleneck that every model encounters is the founder effect associated with its establishment. The tumor biopsy from which a model is derived represents only a specific tumor region, and therefore a local clonal composition2. At the same time, the need to survive in a new environment and adapt to markedly different conditions makes model initiation a highly selective process5.
Routine propagation of cancer models continues to present bottlenecks for the cell population. Passaging of cell lines, PDOs or PDXs involves continuous competition that favors the fitter, more rapidly dividing cells. Propagation conditions inevitably vary, and the exact conditions (e.g., media composition, batches of reagents or cellular densities) may affect model evolution. Bottlenecks are also introduced by freezing and thawing, another routine practice in the propagation of in vitro cultured models. Finally, genetic manipulations, including those considered to be neutral (such as the introduction of a reporter gene) introduce bottlenecks in the form of viral infection and antibiotic selection.
We recently found that in ECLs, most of the variability across strains was introduced through extensive passaging or genetic manipulations, whereas multiple freeze-thaw cycles did not seem to induce extensive genomic evolution68. However, the generalizability of this finding is yet to be confirmed.
Distinct trajectories of tumor evolution
As selection plays a major role in shaping the genomic landscapes of cancer models, it is important to assess whether model evolution mirrors the tumor evolution that naturally occurs in patients. Data from both cell lines and PDXs indicate divergent trajectories of tumor evolution in cancer models and in patients. An analysis of genomic and functional heterogeneity in AML revealed that the AML founding clone was not necessarily the AML-initiating clone in the mouse model85. In solid tumors, recurrent cancer type-specific CNAs that are commonly observed in primary tumors, tend to become even more recurrent during cancer progression (i.e., metastases and recurrences); however, the same events tend to disappear in PDXs and in cell lines68, 81. Therefore, genomic landscapes of tumors are shaped by distinct selection pressures during their evolution in the natural human environment and in the artificial model environment, leading to the gradual genomic divergence of cancer models from their tumors of origin.
Implications for research and medicine
The implications of genomic evolution of cancer models for research and clinical translation depend on whether it can alter their performance in functional assays. Different model applications are expected to be differentially affected by this phenomenon. Once potential risks are identified, mitigation strategies can be devised and implemented, to properly assess, alleviate, and control for genomic evolution.
Phenotypic consequences
It is conceivable that while extensive genetic and epigenetic evolution occurs within cancer models, such evolution might not translate into biologically meaningful cellular properties. That is, this phenomenon could represent a molecular curiosity with little practical implication. Unfortunately, this does not seem to be the case. Genomic changes that arise throughout model propagation have been associated with marked phenotypic changes across all cancer model types (Fig. 2a).
Figure 2: Perils of cancer model evolution.
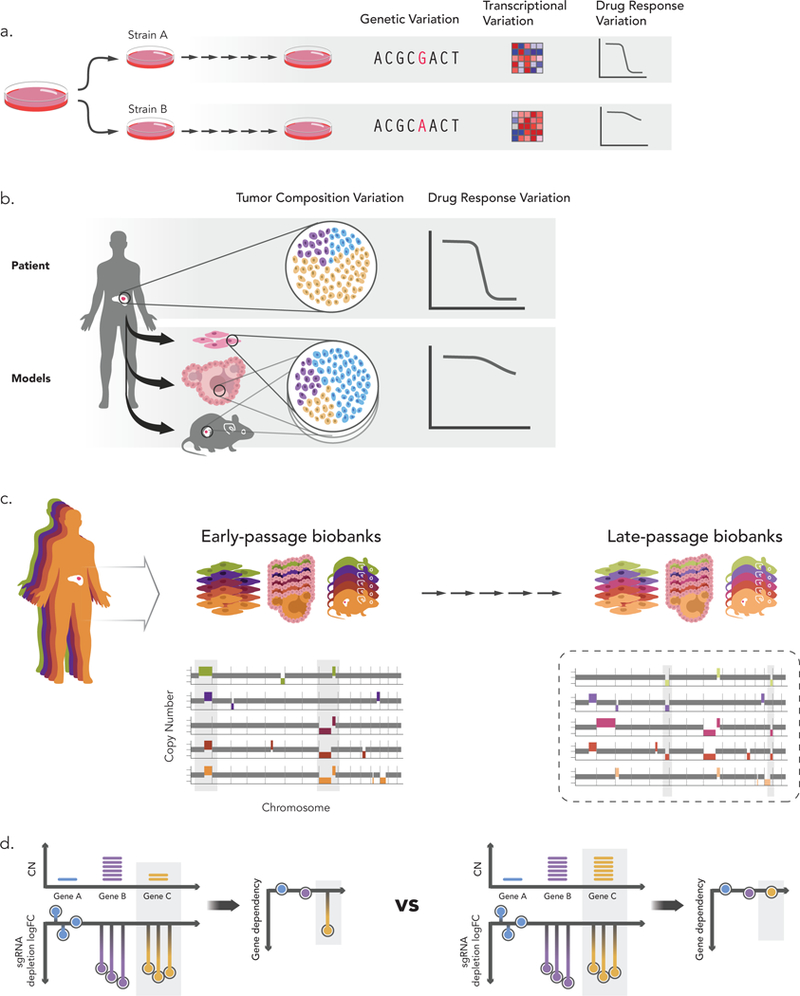
(a) Genomic evolution leads to variability across strains of the same model. Genetic alterations acquired during model propagation can translate into differential gene expression patterns and result in disparate drug response68. (b) Cancer models are used as tumor “avatars” to predict drug response in the patient from which the model was derived. However, if a rare subclone expands in the model and becomes dominant, its drug response may not reflect that of the primary tumor81. (c) Cancer models are also used as cohorts, also known as biobanks, to characterize genotype-phenotype relationships (e.g., cancer dependencies associated with specific mutations). Models are usually characterized upon their derivation, but functional experiments keep being conducted throughout model life. Continued passaging of the models may alter their genomic profiles, thus confounding analyses that make use of early-passage genomic profiles and late-passage functional experiments81. (d) CRISPR/Cas9 screen results need to be corrected for copy-number to account for the number of cuts (a phenomenon known as the CRISPR copy-number effect). Changes in aneuploidy (and other copy-number alterations) as a result of genomic evolution can jeopardize the accuracy of this computational correction. For example, if an amplification of gene C occurred between genomic characterization and CRISPR screening, and the original genomic characterization is used for correcting the copy-number effect, this gene could be mis-identified as a genetic dependency.
In mice, a comparative phenotypic analysis of two substrains of the C57BL/6 strain demonstrated significant phenotypic differences between these strains in multiple physiological, biochemical and behavioral systems, ranging from blood pressure and eye morphology to bone structure and spatial memory55. Variation in morphological and behavioral traits was described among multiple C57BL/6 strains, and specific DNA-level differences were suggested to underlie these phenotypic differences56, 57. Such genomically-driven phenotypic differences can be carried over to GEMMs generated on the genetic backgrounds of these mice: for example, a Dock2 copy-number variant that spontaneously arose in C57BL/6 was inadvertently backcrossed into multiple mutant mouse lines; as a result, these mice exhibited several immune phenotypes previously described in the context of Dock2 deficiency60.
In human CLs, considerable phenotypic variation across strains of the same cell line has been described and linked to genomic variation. In our studies, 27 strains of MCF7 collected from multiple laboratories varied in their doubling time by as much as 3.5-fold under identical culture conditions. Morphological features, such as cell size and shape, largely varied as well, and morphological variation correlated well with genomic variation68. Similar differences in doubling time and morphological features were observed across 14 HeLa strains69. These HeLa strains also varied considerably in their susceptibility to Salmonella infection, which could be explained by differential expression of proteins associated with bacterial infection69.
One of the most common and important applications of cancer models is their use for testing of drug sensitivity and resistance. Variability in drug sensitivity of the same cell line to the same drug is common in the literature: for example, reported IC50 values of tamoxifen in wildtype MCF7 cells differ by >100-fold10, 95. In order to evaluate whether such differences could result from genomic evolution, we screened 27 strains of MCF7 against 321 cancer-related compounds and compared the basal gene expression of sensitive and resistant strains (forward genetics approach). A striking variability was observed: for 48 of 55 (87%) drugs for which at least one strain was highly sensitive, another completely resistant strain was also identified. In the majority (82%) of cases, a differential gene expression signature of the drug mechanism of action was observed between the sensitive and resistant strains68, highlighting that genomic variation indeed underlies the disparate drug response. Similarly, PDX response to some targeted therapies was associated with the existence of specific CNAs, and these associations were confirmed in ECLs81. Therefore, the functional effect of genomic evolution on drug response in PDXs may be as pronounced as the effect observed in CLs.
Implications for tumor “avatars”
“Avatar” experiments, also known as “co-clinical trials”, match patients’ drug responses to those of the models derived from them. The idea is that testing multiple drugs against a patient-derived cancer model could help direct the course of clinical treatment for that patient. Several such “co-clinical trials” reported high degree of concordance between the drug response of patients and their PDXs and PDOs24, 31, 88, 96, 97.
While this strategy is intriguing and useful, several cases where model drug response did not match patient’s response have been described24, 31, 88. Such discrepancies could result from changes in clonal composition of the tumor model due to genomic evolution (Fig. 2b). This could be especially important if genomic evolution affected driver events, such as ESR1 status in ER+ breast cancer (as observed throughout MCF7 evolution68) or trisomy 7 and monosomy 10 in GBM (as observed in GBM PDXs81). In addition, differences in tumor microenvironment (e.g., exposure to ligands and cytokines, matrix stiffness, ECM protein composition, etc.) can profoundly influence cell growth and therapeutic response (reviewed in 98, 99). Further analyses are warranted to evaluate the extent to which evolution of cancer models, together with differences in the tumor microenvironment, limit their use as “avatars”, and how these limitations can be overcome.
Implications for tumor model “biobanks”
Efforts to generate large panels of cancer models aim to represent the molecular diversity of patient populations, and are commonly queried to identify phenotypes associated with specific molecular features (e.g., drug sensitivity associated with a gene mutation). For this type of application, the similarity of a specific model to its tumor of origin is much less relevant. However, two important questions emerge in this setting: a) To what extent does the cohort as a whole represent the patient population? b) How stable and reliable are the molecular features characterized in such cohorts?
Overall, large cohorts of cell lines, PDXs and PDOs mirror the genomic landscapes of their respective cancer types23–27, 30, 100. A couple of important concerns remain, however. First, continuous passaging can distance the model population from the patient population and/or skew the represented populations. Indeed, some hallmark genetic alterations were found to be less prevalent in high-passage than in low-passage PDXs81. Second, biobanks of cancer models are usually characterized only once, on their derivation, and the resultant data sets of molecular features are then widely used as “lookup tables” by individual investigators. However, as models evolve, these “lookup tables” may be misleading – a given genetic alteration characterized in the model may be absent from the specific strain that happens to be available to an investigator, and vice versa68(Fig. 2c).
As discussed above, genomic evolution is most rapid during the derivation and early propagation of cancer models. Genomic evolution then continues to shape the genomic landscape of the model on continuous passaging, albeit at a slower rate. It would therefore be optimal to characterize and use the models at a point when they are already sufficiently stable, but are still good representations of their tumors of origin. In PDCLs and PDXs, such optimal time point might be between p5 to p1081, but this optimum would clearly be context- and model-specific.
Recently, a new practice emerged to first generate PDXs and then derive PDOs from them. This practice aims to have the best of both worlds, as the derivation success rate is generally higher in xenografts, whereas organoids are easier to manipulate and study at high-throughput23, 101. However, this strategy also adds strong bottlenecks to the process, as the model is being transferred twice, from a patient to a mouse and then from mouse into cell culture. It is likely that this multi-step derivation strategy would be associated with genomic evolution, especially given that the opposite transition – the transplantation of established CLs into mice – was shown to induce such evolution81.
Implications for screening purposes
Genomic evolution of cancer models can greatly affect chemical and genetic screens. In a typical primary chemical screen, compounds are screened at a single high dose against one randomly-selected strain of a cell line. The striking variability in drug response observed across strains of the same cell line suggests that very often, whether or not a compound is identified as a “hit” in a screen may depend on the particular cell line strain that was screened. This problem may be exacerbated in genetic loss-of-function screens, which typically involve genetic manipulations and antibiotic selection that can act as strong bottlenecks that increase diversification.
CRISPR screens are likely most susceptible to this problem. First, the introduction of Cas9 and gRNAs into cells often involves two selection steps, enabling more clonal selection. Second, genetic alterations that enable cells to tolerate DNA cutting may be selected in the process; for example, two recent studies showed that genome editing by CRISPR-Cas9 leads to a selection against cells with a functional p53 pathway102, 103. Third, CRISPR screen results need to be corrected based on the copy-number landscapes of the screened cells, and this is commonly done using CNA profiles of the parental cell lines13, 104, 105; the more CNA evolution that took place from the time of the original profiling of the WT strain until the actual screen was performed with the CRISPR-Cas9 strain, the less accurate the computational correction of the “copy-number effect” would be (Fig. 2d). Indeed, genome-wide CRISPR screens of two remote MCF7 strains revealed distinct gene essentiality patterns, and some of the differential dependencies could be readily explained by genomic alterations of the underlying genes68.
Implications for reproducibility
When results obtained in one laboratory cannot be replicated in another laboratory, one should consider the potential contribution of genomic evolution to the observed discrepancies. For example, mispairing C57BL/6 substrains of GEMMs and WT controls led to opposite results related to the role of Jnk2 in livery injury59. Of note, many publications that make use of C57BL/6 mice do not report which specific substrain was used.
The recent genomic and phenotypic comparisons of multiple MCF7 and HeLa strains provide another compelling demonstration of the potential of genomic evolution to jeopardize cancer research reproducibility. For example, studying sensitivity to proteasome inhibition using MCF7 can clearly lead to different conclusions, based on the strain used68. Importantly, however, while disagreements between large data sets of chemical and genetic dependencies do exist38–40, it is not clear yet to what extent this reflects genomic differences between strains.
Mitigation strategies
Many of the risks associated with cancer model evolution can be mitigated by adjusting model propagation strategies and experimental designs. Mitigation strategies can be divided into three classes: 1) Tracking and reporting model propagation; 2) Routine assessment of model genetic diversification; and 3) Alleviating propagation bottlenecks. In addition, robustness of findings should be confirmed across collections of model systems that represent the relevant aspects of cancer being modelled in different genomic and environmental contexts.
Track and report propagation
A requirement for a clear nomenclature of models – taking into account the model history and strain – could make investigators more aware of the phenomenon and prevent mispairing of controls. Keeping track of passage and generation information and reporting it in publications is a desirable practice; the “absolute” passage number of a model does not necessarily carry useful information, but matching passage numbers can guarantee a uniform and appropriate within-study use of the model.
Assess diversification
Routine monitoring of genomic evolution could alleviate its detrimental outcomes. Assessment of diversification is needed after prolonged propagation, or when models go through strong bottlenecks such as genetic manipulations or single-cell cloning. As the genetic distance between cell line strains correlates very well with the expression distance and the drug response distance68, relatively inexpensive commonly-used genomic technologies, such as low-pass whole-genome sequencing, can be applied to accurately assess cell line diversification. Characterizing the genomic features of the model at the same time that it is subjected to functional experiments is therefore both desirable and feasible.
We developed Cell STRAINER (Cell STRAin Instability profilER; see Related links) to facilitate the routine assessment of cell line diversification. Users can upload cell line genomic data of their cell line strain (currently in the format of CNA profiles), and these data are compared to reference genomic data of the same cell line, as characterized by the Cancer Cell Line Encyclopedia8. Large genetic distance between the two strains is indicative of considerable cell line diversification, suggesting that other genomic features of the CCLE strain may also not apply to the tested strain.
When cell line misidentification and mycoplasma contamination emerged as major problems in cell line research, journals began to require authors to confirm cell line identity (using DNA fingerprinting) and mycoplasma contamination status. Similarly, we can envision that cell line genetic diversification assessment may become a pre-requisite for publication in the future. Of note, the same genomic analysis can also confirm cell line identity, and therefore replace DNA fingerprinting in most cases. As biobanks of PDXs and PDOs become more widely used, tools similar to Cell STRAINER should be developed for these models as well.
Minimize genomic evolution
Experimental practices can be adjusted to minimize genomic evolution and its phenotypic effects. First, it is advisable to avoid unnecessary passaging by using techniques that minimize cell culture after the initial characterization106. Second, bottlenecks should be reduced as much as possible. Practically, this means keeping culture conditions as constant as possible, as even minor changes can lead to clonal selection68. This also means that single-cell cloning should be avoided when population-based assays can be used instead. Better mimicking of the human tumor environment in the model – a desirable goal regardless – could also alleviate selection pressures and minimize genomic evolution.
Given that prolonged passaging leads to continuous model evolution, multiple frozen stocks should be prepared for large studies, so that models be used at comparable passage numbers throughout the entire course of the study. This could be especially important for large-scale screens, where downstream follow-up experiments are often performed many months after the original screen.
Finally, some models are inherently unstable and are thus prone to much more rapid genomic evolution81. Sometimes the use of such models is unavoidable or even desirable (e.g., when one wishes to study genomic instability); however, for some applications these models can simply be replaced by alternative, more stable ones.
Emerging opportunities
While the perils of genomic evolution of cancer models warrant caution, and applying mitigation strategies is recommended, this natural phenomenon could also be harnessed in creative ways. One might take advantage of the dynamic nature of cancer models to pursue novel avenues of research, or to address longstanding open questions in new ways.
Panels of near-isogenic cancer models
In mouse models, the realization that different substrains differ in their genomic and phenotypic features was successfully used to illuminate genotype-phenotype associations. For example, a genomic comparison of two C57BL6 substrains that differ significantly in their alcohol consumption, identified candidate genes that explain this trait56. Application of a similar strategy uncovered the genetic alterations underlying differences in immune phenotypes60.
The same approach can be applied to patient-derived models (Fig. 3a). Genomic evolution leads to the generation of genetically-matched, near-isogenic models. These models are not fully isogenic, as more than a single event would normally separate each pair of strains. Nonetheless, panels of such near-isogenic models can be used to study specific molecular features that are variable across strains. As a proof of concept, comparison of gene expression and drug response between cell line strains with and without a given genetic alteration correctly identified cellular consequences of, and pathways perturbed by, that alteration68. This reverse-genetics approach would be especially useful for studying the consequence of genetic events that are difficult to introduce experimentally, such as large chromosomal changes (Box 2).
Figure 3: New research opportunities presented by cancer model evolution.
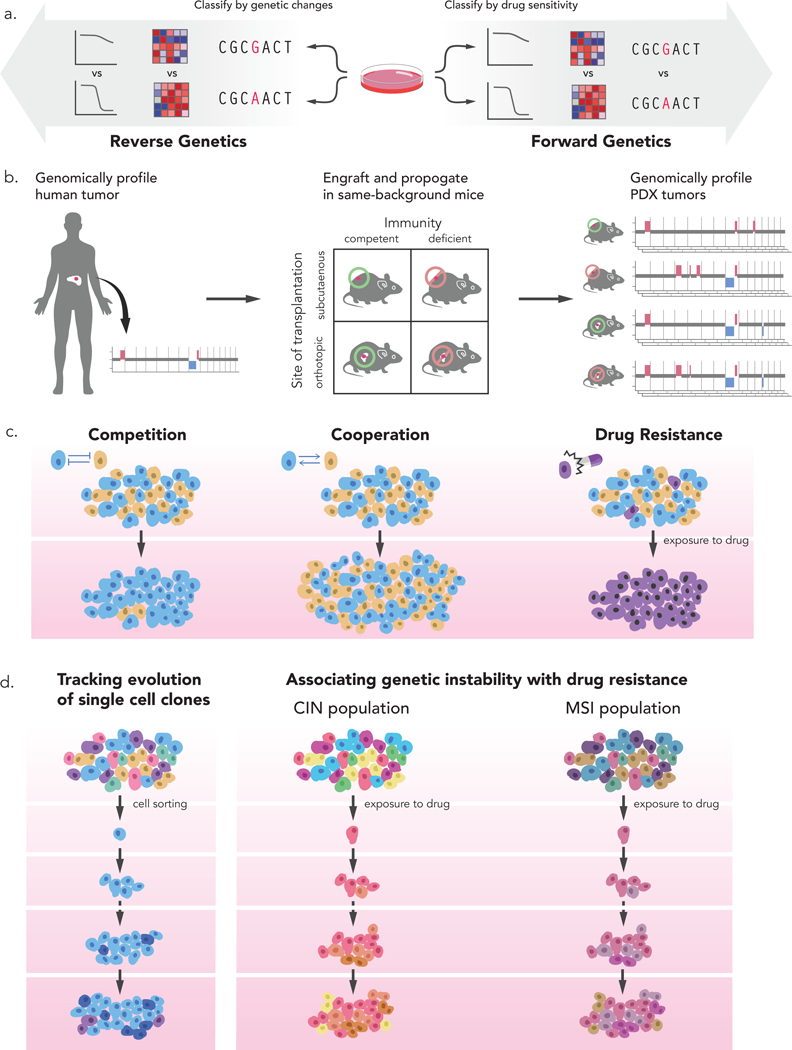
(a) The natural genomic evolution of cancer models generates semi-isogenic model strains that can be used both for reverse genetics and for forward genetics. In reverse genetics experiments, the gene expression profiles and drug response patterns of model strains with a genetic alteration of interest can be compared to those of strains without that alteration. In forward genetics experiments, the genetic landscapes and gene expression profiles of model strains that are sensitive to a drug of interest can be compared to those of strains that are resistant to that drug. (b) Genomic evolution can be used to study the selection pressures that shape the genetic landscapes of tumors. For example, PDXs can be generated in mice hosts that share their genetic background, but differ in their immune status and transplantation sites. The genomic profiles of these PDX models following propagation can be characterized and compared to that of the primary human tumor. The rate, extent and identity of genomic alterations in the various models can help identify the components that determine the evolutionary trajectory of tumorigenesis. (c) Cancer model evolution can be used to study cancer heterogeneity and cellular interactions. Competitive and cooperative interactions between tumor subclones can be dissected using single-cell genomics, genome editing and cell barcoding technologies. Drug sensitivity and resistance can also be studied by following tumor evolution and clonal dynamics following drug exposure. (d) Mechanisms of genomic instability itself can be studied by following how single-cell clones become heterogeneous (left), or by following how drug resistance mechanisms differ between models that harbor distinct deficiencies in genome maintenance pathways (right). CIN, chromosomal instability; MSI, Microsatellite instability.
Box 2: Model evolution and cancer aneuploidy.
Aneuploidy demonstrates both the perils and the opportunities presented by cancer model evolution. Aneuploidy provides a unique lens through which one could follow tumor evolution, as this is a discrete event at the cellular level, is unique to cancer cells, and can be detected by multiple methods127, 128. In addition, due to the high fitness cost associated with specific aneuploidies under most cellular circumstances129, these events may be particularly sensitive to changes in selection pressures.
The study of aneuploidy in cancer models is exposed to all the risks associated with model evolution. Variability in arm-level and whole-chromosome copy-number status exists within ECLs, and aneuploidy landscapes rapidly evolve in PDXs and PDCLs81. These genetic changes are associated with very consistent changes of gene expression along the affected chromosome(s), in the expected direction68, 81. Both in ECLs and in PDXs, the existence of specific aneuploidies was associated with the response to specific anticancer drugs81. The gradual disappearance of recurrent cancer aneuploidies throughout the derivation and propagation of PDXs81 emphasizes the potential risk that model evolution poses to accurately using these models as tumor “avatars”.
At the same time, model evolution can be used to advance the study of cancer aneuploidy. The artificial introduction of extra chromosomes into cells is tumor-suppressive130, whereas naturally-occurring aneuploidy can be tumor-promoting131, as also suggested by the cancer type-specific patterns of aneuploidy recurrence132. Therefore, naturally-occurring aneuploid variants that arise during model evolution may be uniquely suitable for studying aneuploidy in a more cancer-relevant context.
Indeed, following aneuploidy landscapes in developing tumors in GEMMs enabled us to identify aneuploidies that are associated with specific drivers, narrow down regions of interest within altered chromosomes, and identify candidate genes that cooperate with the initial transgene to drive tumorigenesis52 (see box figure, part a). Another promising direction is to use naturally-occurring variation in aneuploidy landscapes within ECLs to study the cellular consequences of recurrent aneuploidies, and to identify synthetic lethalities associated with these events (see box figure, part b).
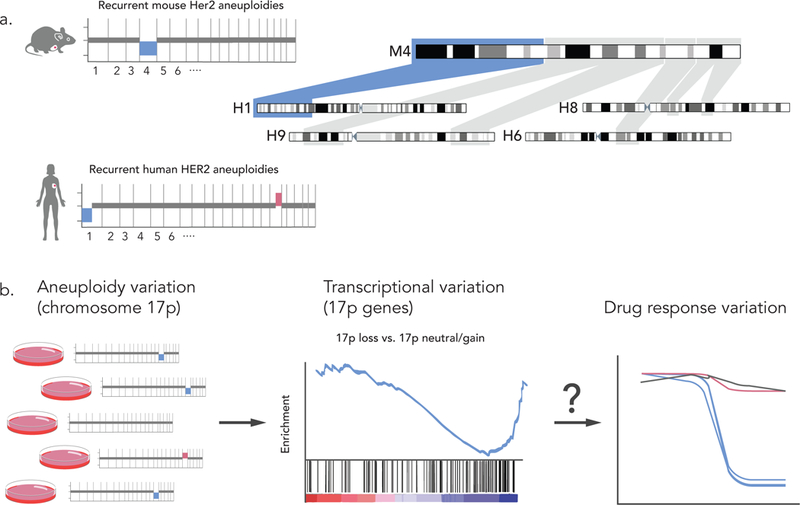
Similarly, taking a forward-genetics approach as described above when comparing global gene expression levels between drug sensitive and drug resistant MCF7 strains68, such comparisons could uncover the genomics underlying variable phenotypes. Importantly, such comparisons provide more statistical power than similar comparisons of non-genetically-matched cancer models. However, this approach is limited to studying differential phenotypes, and so is not useful for studying drugs that do not affect that particular cell line. Another limitation is that ongoing clonal dynamics may affect the stability of the panel, rendering the genomic profiling of the strains a moving target.
Understanding selection pressures
Since both positive and negative selection can alter the genomic landscapes of patient-derived tumors, cancer models could potentially be used to study selection itself. For example, the immune-deficient subcutaneous mouse environment of PDXs is different from the immune-competent organ-specific patient environment. Which of these components is most important for shaping the trajectory of tumor evolution? Following the rate, extent and identity of genomic alterations in subcutaneous vs. orthotopic models, and in immune-deficient vs. “humanized” mice, should help clarify the relative importance of each of these factors (Fig. 3b). Similarly, following clonal dynamics in PDOs or cell lines cultured under various defined culture conditions may illuminate the role of specific media components (e.g., growth factors) in shaping the genomic landscape of the tumor.
Studying heterogeneity
Tumor heterogeneity is a fundamental aspect of tumor biology, with profound implications for drug response and clinical outcome2, 107. Single-cell technologies have revolutionized the way cancer heterogeneity is studied and understood, and deeper sequencing allows more accurate reconstruction of tumor clonal composition108, 109. The heterogeneous, dynamic nature of cancer models raises interesting opportunities for research (Fig. 3c).
Cooperative and competitive interactions
There is growing evidence that cooperative and competitive interactions between tumor subclones can influence disease progression and clinical outcome (reviewed in 110). Using engineered subclones of a human breast cancer ECL, intra-tumor clonal heterogeneity was shown to drive tumor growth and dissemination111. Similarly, cooperation between subclones was found to promote invasion in a zebrafish melanoma xenograft model112, and differentiated cell populations could increase invasiveness and growth of cancer stem cell populations through factor secretion, in pancreatic and glioblastoma cell lines, respectively113, 114. Most recently, functional cooperativity was described between genetically-distinct subclones derived from human pediatric brain tumors115.
The realization that cancer models are naturally heterogeneous suggests that they can be used to dissect the mechanistic basis of clonal interactions. Single-cell profiling of cell lines might help determine the abundance of existing subclones, and clonal dynamics throughout various types of perturbations and challenges can then be followed. Experiments like the one described above can potentially be performed without cell engineering, taking advantage of the natural variation in the cell population to study non-cell-autonomous interactions.
Determinants of drug sensitivity
The pre-existence of drug-resistant subclones was recently shown to be a major mechanism of drug resistance in cancer cell line populations116, 117. Drug treatment of barcoded cell populations reproducibly induced the enrichment of the same specific barcodes, indicating selection of pre-existing resistant subclones68, 116. Molecular, biochemical and functional analyses of sensitive and resistant subclones within a heterogeneous population could therefore be used to study drug sensitivity in cancer models.
Studying genomic instability
As ongoing genomic instability keeps shaping the genomic landscape of cancer models, these models can be used to study the cellular mechanisms underlying genomic instability. Indeed, cancer models have been used to study such mechanisms for many years (reviewed in 118–120). However, the recent advances in the field suggest new ways to study genomic instability in cancer models (Fig. 3d).
Single-cell clones of genomically unstable cell lines become genetically and transcriptionally heterogeneous. Studying the genomic evolution of single-cell clones from their initiation can therefore be a promising way to study how diversity is generated. Another idea would be to perform DNA barcoding experiments with cell lines that are deficient in distinct mechanisms of genome integrity maintenance, in order to identify associations between mechanisms of genomic instability and drug resistance.
Concluding remarks
Genomic evolution is inevitable in living model systems. Like other sources of research irreproducibility, model diversification should be assessed routinely, reported appropriately and controlled for experimentally. It should not be viewed as a disaster, nor should it be ignored. The continuous improvement of cancer models over the past few years has yielded significantly better modeling of the human disease. Moving from a static to a more dynamic way of thinking about the genomics of cancer models could be another important step in this direction. It would help minimize the detrimental effects of genomic evolution on cancer research and cancer precision medicine, and at the same time open novel avenues of research that take advantage of these dynamics. Genomic evolution of cancer models can therefore contribute to the evolution of cancer research.
Acknowledgements
The authors thank Iris Fung for her assistance with designing and preparing the figures. This work was supported by Human Frontiers Science Program (U.B.-D.), Howard Hughes Medical Institute (T.R.G.), US National Institutes of Health (R01 CA188228; R.B.), Gray Matters Brain Cancer Foundation (R.B.), Bridge Project (R.B.), Broad Institute SPARC award (R.B.), and Broad Institute BroadNext10 grant (U.B.-D.).
Glossary
- Patient-derived xenografts (PDXs)
Models generated by the direct engraftment of resected human tumors into immune-deficient mice, followed by their serial transplantation between mice.
- Patient-derived organoids (PDOs)
Models generated by the embedment of tumor (or normal) cells into a 3D matrix, using culture conditions that mimic the in vivo tumor niche.
- Patient-derived cell lines (PDCLs)
Models generated by the transferring of tumor cells into a 2D plastic dish, using culture conditions that enable cells to proliferate.
- Genetically-engineered mouse models (GEMMs)
Models generated by genetically manipulating mice, using genetic alterations that characterize human tumors.
- Established cancer cell lines (ECLs)
Models generated as PDCLs, followed by prolonged culture propagation. These models are not assumed to represent the specific tumors from which they were derived.
- Clonal dynamics
Changes in the relative abundance of tumor subclones throughout model propagation.
- Pre-existing heterogeneity
Genetic diversity within the original tumor, contributing to the genomic evolution of the model.
- Ongoing genomic instability
Generation of de novo genetic alterations throughout model propagation, contributing to the genomic evolution of the model.
- Genetic drift
Stochastic changes in the clonal composition of the cancer cell population, due to chance disappearance and/or expansion of particular subclones.
- Genetic selection
Directional changes in the clonal composition of the cancer cell population, due to growth advantage and/or disadvantage of particular subclones.
- Founder effect
Genetic diversity that results when a cell population is descended from a small number of original cells.
- Copy-number effect
In CRISPR screens, copy number changes result in gene-independent anti-proliferative effect of Cas9-mediated DNA cleavage, confounding the measurement of gene essentiality. This effect can be corrected computationally using genome-wide copy-number measurements.
Footnotes
Competing interests
The authors declare no competing financial interests.
References
- 1.Greaves M Evolutionary determinants of cancer. Cancer Discov 5, 806–20 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGranahan N & Swanton C Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 168, 613–628 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Drost J & Clevers H Organoids in cancer research. Nat Rev Cancer 18, 407–418 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Byrne AT et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat Rev Cancer 17, 254–268 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Gillet JP, Varma S & Gottesman MM The clinical relevance of cancer cell lines. J Natl Cancer Inst 105, 452–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeggo PA, Pearl LH & Carr AM DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer 16, 35–42 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Kersten K, de Visser KE, van Miltenburg MH & Jonkers J Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol Med 9, 137–153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barretina J et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garnett MJ et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basu A et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang W et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 41, D955–61 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsherniak A et al. Defining a Cancer Dependency Map. Cell 170, 564–576 e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meyers RM et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 49, 1779–1784 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klijn C et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol 33, 306–12 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Bertotti A et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov 1, 508–23 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Laurent C et al. Patient-derived xenografts recapitulate molecular features of human uveal melanomas. Mol Oncol 7, 625–36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin D et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res 74, 1272–83 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Townsend EC et al. The Public Repository of Xenografts Enables Discovery and Randomized Phase II-like Trials in Mice. Cancer Cell 29, 574–586 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garman B et al. Genetic and Genomic Characterization of 462 Melanoma Patient-Derived Xenografts, Tumor Biopsies, and Cell Lines. Cell Rep 21, 1936–1952 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krepler C et al. A Comprehensive Patient-Derived Xenograft Collection Representing the Heterogeneity of Melanoma. Cell Rep 21, 1953–1967 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pauli C et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov 7, 462–477 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart E et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature 549, 96–100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beshiri ML et al. A PDX/organoid biobank of advanced prostate cancers captures genomic and phenotypic heterogeneity for disease modeling and therapeutic screening. Clin Cancer Res (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drapkin BJ et al. Genomic and Functional Fidelity of Small Cell Lung Cancer Patient-Derived Xenografts. Cancer Discov 8, 600–615 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao H et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med 21, 1318–25 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Bruna A et al. A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds. Cell 167, 260–274 e22 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao D et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 159, 176–187 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van de Wetering M et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee SH et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 173, 515–528 e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sachs N et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 172, 373–386 e10 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Vlachogiannis G et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Izumchenko E et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann Oncol 28, 2595–2605 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hidalgo M et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov 4, 998–1013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marangoni E et al. A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin Cancer Res 13, 3989–98 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Zhang X et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res 73, 4885–97 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prinz F, Schlange T & Asadullah K Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov 10, 712 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Begley CG & Ellis LM Drug development: Raise standards for preclinical cancer research. Nature 483, 531–3 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Haibe-Kains B et al. Inconsistency in large pharmacogenomic studies. Nature 504, 389–93 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cancer Cell Line Encyclopedia, C. & Genomics of Drug Sensitivity in Cancer, C. Pharmacogenomic agreement between two cancer cell line data sets. Nature 528, 84–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haverty PM et al. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature 533, 333–7 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Franca TF & Monserrat JM Reproducibility crisis in science or unrealistic expectations? EMBO Rep 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hunter P The reproducibility “crisis”: Reaction to replication crisis should not stifle innovation. EMBO Rep 18, 1493–1496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baker M Reproducibility crisis: Blame it on the antibodies. Nature 521, 274–6 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Loken E & Gelman A Measurement error and the replication crisis. Science 355, 584–585 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Horbach S & Halffman W The ghosts of HeLa: How cell line misidentification contaminates the scientific literature. PLoS One 12, e0186281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walrath JC, Hawes JJ, Van Dyke T & Reilly KM Genetically engineered mouse models in cancer research. Adv Cancer Res 106, 113–64 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tschaharganeh DF, Lowe SW, Garippa RJ & Livshits G Using CRISPR/Cas to study gene function and model disease in vivo. FEBS J 283, 3194–203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andrechek ER et al. Genetic heterogeneity of Myc-induced mammary tumors reflecting diverse phenotypes including metastatic potential. Proc Natl Acad Sci U S A 106, 16387–92 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Keymeulen A et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 525, 119–23 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Westcott PM et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature 517, 489–92 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nassar D, Latil M, Boeckx B, Lambrechts D & Blanpain C Genomic landscape of carcinogen-induced and genetically induced mouse skin squamous cell carcinoma. Nat Med 21, 946–54 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Ben-David U et al. The landscape of chromosomal aberrations in breast cancer mouse models reveals driver-specific routes to tumorigenesis. Nat Commun 7, 12160 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herschkowitz JI et al. Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc Natl Acad Sci U S A 109, 2778–83 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uchimura A et al. Germline mutation rates and the long-term phenotypic effects of mutation accumulation in wild-type laboratory mice and mutator mice. Genome Res 25, 1125–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simon MM et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol 14, R82 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mulligan MK et al. Alcohol trait and transcriptional genomic analysis of C57BL/6 substrains. Genes Brain Behav 7, 677–89 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Liron T, Raphael B, Hiram-Bab S, Bab IA & Gabet Y Bone loss in C57BL/6J-OlaHsd mice, a substrain of C57BL/6J carrying mutated alpha-synuclein and multimerin-1 genes. J Cell Physiol 233, 371–377 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Kalish S et al. C57BL/6N Mice Are More Resistant to Ehrlich Ascites Tumors Than C57BL/6J Mice: The Role of Macrophage Nitric Oxide. Med Sci Monit Basic Res 21, 235–40 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bourdi M, Davies JS & Pohl LR Mispairing C57BL/6 substrains of genetically engineered mice and wild-type controls can lead to confounding results as it did in studies of JNK2 in acetaminophen and concanavalin A liver injury. Chem Res Toxicol 24, 794–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mahajan VS et al. Striking Immune Phenotypes in Gene-Targeted Mice Are Driven by a Copy-Number Variant Originating from a Commercially Available C57BL/6 Strain. Cell Rep 15, 1901–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mattapallil MJ et al. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci 53, 2921–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jones C et al. Comparative genomic hybridization reveals extensive variation among different MCF-7 cell stocks. Cancer Genet Cytogenet 117, 153–8 (2000). [DOI] [PubMed] [Google Scholar]
- 63.Coser KR et al. Antiestrogen-resistant subclones of MCF-7 human breast cancer cells are derived from a common monoclonal drug-resistant progenitor. Proc Natl Acad Sci U S A 106, 14536–41 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nugoli M et al. Genetic variability in MCF-7 sublines: evidence of rapid genomic and RNA expression profile modifications. BMC Cancer 3, 13 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kleensang A et al. Genetic variability in a frozen batch of MCF-7 cells invisible in routine authentication affecting cell function. Sci Rep 6, 28994 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frattini A et al. High variability of genomic instability and gene expression profiling in different HeLa clones. Sci Rep 5, 15377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sato S, Rancourt A, Sato Y & Satoh MS Single-cell lineage tracking analysis reveals that an established cell line comprises putative cancer stem cells and their heterogeneous progeny. Sci Rep 6, 23328 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ben-David U et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu Y et al. Genomic, Proteomic and Phenotypic Heterogeneity in HeLa Cells across Laboratories: Implications for Reproducibility of Research Results. bioRxiv (2018). [Google Scholar]
- 70.Fasterius E & Al-Khalili Szigyarto C Analysis of public RNA-sequencing data reveals biological consequences of genetic heterogeneity in cell line populations. Sci Rep 8, 11226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weissbein U, Plotnik O, Vershkov D & Benvenisty N Culture-induced recurrent epigenetic aberrations in human pluripotent stem cells. PLoS Genet 13, e1006979 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mekhoubad S et al. Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell 10, 595–609 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Locke WJ & Clark SJ Epigenome remodelling in breast cancer: insights from an early in vitro model of carcinogenesis. Breast Cancer Res 14, 215 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boehm JS & Golub TR An ecosystem of cancer cell line factories to support a cancer dependency map. Nat Rev Genet 16, 373–4 (2015). [DOI] [PubMed] [Google Scholar]
- 75.Liu X et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol 180, 599–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hong AL et al. Integrated genetic and pharmacologic interrogation of rare cancers. Nat Commun 7, 11987 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Peng S et al. Tumor grafts derived from patients with head and neck squamous carcinoma authentically maintain the molecular and histologic characteristics of human cancers. J Transl Med 11, 198 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gunther HS et al. Glioblastoma-derived stem cell-enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene 27, 2897–909 (2008). [DOI] [PubMed] [Google Scholar]
- 79.Schulte A et al. A distinct subset of glioma cell lines with stem cell-like properties reflects the transcriptional phenotype of glioblastomas and overexpresses CXCR4 as therapeutic target. Glia 59, 590–602 (2011). [DOI] [PubMed] [Google Scholar]
- 80.Cifola I et al. Renal cell carcinoma primary cultures maintain genomic and phenotypic profile of parental tumor tissues. BMC Cancer 11, 244 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ben-David U et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet 49, 1567–1575 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Villacorta-Martin C, Craig AJ & Villanueva A Divergent evolutionary trajectories in transplanted tumor models. Nat Genet 49, 1565–1566 (2017). [DOI] [PubMed] [Google Scholar]
- 83.Eirew P et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 518, 422–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Clappier E et al. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J Exp Med 208, 653–61 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Klco JM et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell 25, 379–92 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.de Boer B et al. Prospective Isolation and Characterization of Genetically and Functionally Distinct AML Subclones. Cancer Cell 34, 674–689 e8 (2018). [DOI] [PubMed] [Google Scholar]
- 87.Grasse S et al. Epigenomic profiling of non-small cell lung cancer xenografts uncover LRP12 DNA methylation as predictive biomarker for carboplatin resistance. Genome Med 10, 55 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tiriac H et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Roerink SF et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 556, 457–462 (2018). [DOI] [PubMed] [Google Scholar]
- 90.Li X et al. Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat Commun 9, 2983 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gupta PB et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 146, 633–44 (2011). [DOI] [PubMed] [Google Scholar]
- 92.Knouse KA, Lopez KE, Bachofner M & Amon A Chromosome Segregation Fidelity in Epithelia Requires Tissue Architecture. Cell (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rowald K et al. Negative Selection and Chromosome Instability Induced by Mad2 Overexpression Delay Breast Cancer but Facilitate Oncogene-Independent Outgrowth. Cell Rep 15, 2679–91 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hingorani SR et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–83 (2005). [DOI] [PubMed] [Google Scholar]
- 95.Sato M, Glasebrook AL & Bryant HU Raloxifene: A selective estrogen receptor modulator. Journal of Bone and Mineral Metabolism 12, S9–S20 (1994). [Google Scholar]
- 96.Kim HR et al. Co-clinical trials demonstrate predictive biomarkers for dovitinib, an FGFR inhibitor, in lung squamous cell carcinoma. Ann Oncol 28, 1250–1259 (2017). [DOI] [PubMed] [Google Scholar]
- 97.Owonikoko TK et al. Patient-derived xenografts faithfully replicated clinical outcome in a phase II co-clinical trial of arsenic trioxide in relapsed small cell lung cancer. J Transl Med 14, 111 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Klemm F & Joyce JA Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol 25, 198–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Morgan MM et al. Personalized in vitro cancer models to predict therapeutic response: Challenges and a framework for improvement. Pharmacol Ther 165, 79–92 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Neve RM et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–27 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gendoo DMA et al. Whole Genomes Define Concordance of Matched Primary, Xenograft, and Organoid Models of Pancreas Cancer. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Haapaniemi E, Botla S, Persson J, Schmierer B & Taipale J CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med (2018). [DOI] [PubMed] [Google Scholar]
- 103.Ihry RJ et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med (2018). [DOI] [PubMed] [Google Scholar]
- 104.Aguirre AJ et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov 6, 914–29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Munoz DM et al. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discov 6, 900–13 (2016). [DOI] [PubMed] [Google Scholar]
- 106.Mitra A, Mishra L & Li S Technologies for deriving primary tumor cells for use in personalized cancer therapy. Trends Biotechnol 31, 347–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Andor N et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med 22, 105–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Janiszewska M et al. In situ single-cell analysis identifies heterogeneity for PIK3CA mutation and HER2 amplification in HER2-positive breast cancer. Nat Genet 47, 1212–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Venteicher AS et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tabassum DP & Polyak K Tumorigenesis: it takes a village. Nat Rev Cancer 15, 473–83 (2015). [DOI] [PubMed] [Google Scholar]
- 111.Marusyk A et al. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 514, 54–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chapman A et al. Heterogeneous tumor subpopulations cooperate to drive invasion. Cell Rep 8, 688–95 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mateo F et al. SPARC mediates metastatic cooperation between CSC and non-CSC prostate cancer cell subpopulations. Mol Cancer 13, 237 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ouchi R, Okabe S, Migita T, Nakano I & Seimiya H Senescence from glioma stem cell differentiation promotes tumor growth. Biochem Biophys Res Commun 470, 275–281 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vinci M et al. Functional diversity and cooperativity between subclonal populations of pediatric glioblastoma and diffuse intrinsic pontine glioma cells. Nat Med (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bhang HE et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med 21, 440–8 (2015). [DOI] [PubMed] [Google Scholar]
- 117.Hata AN et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med 22, 262–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Negrini S, Gorgoulis VG & Halazonetis TD Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol 11, 220–8 (2010). [DOI] [PubMed] [Google Scholar]
- 119.Gaillard H, Garcia-Muse T & Aguilera A Replication stress and cancer. Nat Rev Cancer 15, 276–89 (2015). [DOI] [PubMed] [Google Scholar]
- 120.Sansregret L, Vanhaesebroeck B & Swanton C Determinants and clinical implications of chromosomal instability in cancer. Nat Rev Clin Oncol 15, 139–150 (2018). [DOI] [PubMed] [Google Scholar]
- 121.KA W DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). www.genome.gov/sequencingcostsdata Accessesed August 3, 2018.
- 122.Roth A et al. PyClone: statistical inference of clonal population structure in cancer. Nat Methods 11, 396–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Silva GO et al. SynthEx: a synthetic-normal-based DNA sequencing tool for copy number alteration detection and tumor heterogeneity profiling. Genome Biol 18, 66 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Carter SL, Eklund AC, Kohane IS, Harris LN & Szallasi Z A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet 38, 1043–8 (2006). [DOI] [PubMed] [Google Scholar]
- 125.Zhang S, Yuan Y & Hao D A genomic instability score in discriminating nonequivalent outcomes of BRCA½ mutations and in predicting outcomes of ovarian cancer treated with platinum-based chemotherapy. PLoS One 9, e113169 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Burrell RA et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ben-David U & Benvenisty N in StemBook (Cambridge (MA), 2008). [Google Scholar]
- 128.Ben-David U, Mayshar Y & Benvenisty N Virtual karyotyping of pluripotent stem cells on the basis of their global gene expression profiles. Nat Protoc 8, 989–97 (2013). [DOI] [PubMed] [Google Scholar]
- 129.Santaguida S & Amon A Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat Rev Mol Cell Biol 16, 473–85 (2015). [DOI] [PubMed] [Google Scholar]
- 130.Sheltzer JM et al. Single-chromosome Gains Commonly Function as Tumor Suppressors. Cancer Cell 31, 240–255 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ben-David U et al. Aneuploidy induces profound changes in gene expression, proliferation and tumorigenicity of human pluripotent stem cells. Nat Commun 5, 4825 (2014). [DOI] [PubMed] [Google Scholar]
- 132.Zack TI et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet 45, 1134–40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]