

Summary
Aims
Alzheimer's disease (AD) and Parkinson's disease (PD) are the most prevalent neurodegenerative disorders that may share some overlapping etiologies. Mutations within leucine‐rich repeat kinase 2 (LRRK2) have been reported to be responsible for PD, and the location of LRRK2 is within a linkage peak for sporadic AD (SAD). The aim of this study was to investigate two Asian‐specific LRRK2 variants, R1628P and G2385R, with the association of Han Chinese SAD.
Methods
Genotyping of R1628P and G2385R was performed by PCR‐restriction fragment length polymorphism (RFLP) analysis in 390 patients with SAD and 545 unrelated age‐ and sex‐matched healthy controls.
Results
The frequency of the C allele within R1628P was more than three times higher in control group (1.7%) than in patients with SAD (0.5%) (OR 0.264; 95% CI, 0.088–0.792, P = 0.018). After stratification by the presence of one or two apolipoprotein E ε4 alleles, the protective effect becomes stronger (ε44: OR 0.028; 95% CI, 0.003–0.303, P = 0.003; ε4: OR 0.104; 95% CI, 0.013–0.818, P = 0.031). However, no difference was found in G2385R variant.
Conclusion
Our study suggested that R1628P variant within LRRK2 plays a protective role in Han Chinese population with SAD and such effect has an interaction with the APOE genotype.
Keywords: Alzheimer's disease, Association, Chinese, LRRK2
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disorder and presents with progressive and irreversible memory loss and cognitive decline. The great majority of AD is sporadic (SAD) although early‐onset familial AD (EOFAD) can represent up to 5% of the AD cases assessed in memory clinics 1. The role of genes in the pathogenesis/cause of a proportion (~50%) of EOFAD is now known to be the result of mutations (at least 230 to date) in three virtually fully penetrant genes—amyloid precursor protein (APP), presenilins 1 and 2 (PS1 and PS2, respectively) (http://www.molgen.ua.ac.be/ADMutations). Conversely, to date, no single gene mutation has been found in SAD, and at least in the majority of such cases, gene–environment interactions may play an important role in pathogenesis. To date, the only well‐replicated genetic locus for susceptibility to (but not causal for) SAD is the apolipoprotein E (APOE) gene, which has three alleles—ε4, ε3, and ε2 2. Research continues to identify and confirm other potential susceptibility factors for SAD.
Parkinson's disease (PD) is the second most prevalent neurodegenerative disease after AD 3. Epidemiological studies show that siblings of demented patients with PD (PDD) have an higher risk of developing AD compared with siblings of normal subjects 4, and conversely, it has been shown that first‐degree relatives of patients with AD have an increased risk of developing PD 5. In addition, there was a coexistent Alzheimer pathology in some PD patients with or without dementia 6.
In the current study, we hypothesize a common (or at least overlapping) etiology between SAD and PD with the leucine‐rich repeat kinase 2 (LRRK2). LRRK2, a large gene located on chromosome 12: 40,590,546–40,763,087, has 51 exons and encodes a multifunctional protein. Mutations within LRRK2 have been reported to be responsible for both familial and sporadic PD 7, 8. The location of LRRK2 is within a linkage peak for late‐onset SAD 9 and close to the 12q13 risk locus identified in a recent genome‐wide association study (GWAS) 10. Thus, it has been speculated that variants within LRRK2 may be associated with the risk of developing SAD. Here, we present a case–control study in the Han Chinese population to investigate two Asian‐specific LRRK2 variants, R1628P (rs33949390) and G2385R (rs34778348), with the association of SAD.
Materials and Methods
Ethics Approval
The study protocol was approved by the Ethics Committee of Huashan Hospital.
Subjects
This study included two subject groups: 390 patients with SAD (228 women and 162 men; mean age 69.99 ± 9.907; range 47–92) and 545 unrelated age‐ and sex‐matched healthy controls (336 women and 209 men; mean age 68.77 ± 9.192; range 47–93). The detailed enrollment procedure as well as inclusion and exclusion criteria for cases and controls was described previously 11. All participants were of Han Chinese descent, which accounts for approximately 90% of the entire Chinese population. A signed informed consent was obtained from each case (substitute decision maker/guardian) and control.
Genotyping
Genomic DNA was extracted from peripheral blood using a Blood Genomic DNA Extraction Kit (TIANGEN, Beijing, China). Genotyping of R1628P (forward primer: 5′‐TTCTGACTACTTTCACTGAG‐3′ and reverse primer: 5′‐GGAGGTTTACACTAGAAGC‐3′) and G2385R (forward primer: 5′‐TAGCCCTGTTGTGGAAGTG‐3′ and reverse primer: 5′‐TTCAGAGGCAGAAAGGAAG‐3′) was performed by polymerase chain reaction‐restriction fragment length polymorphism (PCR‐RFLP) analysis. PCR amplification was performed using a GeneAmp PCR system 9600 (Applied Biosystems, Foster City, CA, USA). The PCR products were digested with the restriction enzyme AccI for G2385R and BstUI for R1628P according to the manufacturer's recommendations. Digestion was followed by 2.5% agarose gel electrophoresis. The minor alleles of R1628P were further confirmed by DNA sequencing using an ABI 3730 Automated DNA Sequencer (Applied Biosystems). The APOE genotypes were determined by multiplex amplification refractory mutation system PCR as previously described 12.
Statistical Analysis
The genotypes and allele frequencies in patients with SAD versus controls were compared using the standard chi‐square test or the Fisher's exact test, where appropriate. Binary logistic regression analyses were used to estimate odds ratios (ORs) and the 95% confidence interval (CI). Covariates were age, gender, and APOE genotype. All statistical analyses were performed using SPSS 14.0 (SPSS Inc., Chicago, IL, USA). The criterion for a significant difference was P < 0.05.
Results
Characteristics of Participants
The general data of the participants are shown in Table 1. No statistically significant differences were observed for age and gender (P > 0.05) between cases and controls. As expected, the Mini Mental State Examination (MMSE) score 13 was significantly lower in patients with SAD than in controls (P < 0.0001). The APOE ε4 allele frequency and the APOE ε44 genotype were significantly different between patients with SAD and control subjects (P < 0.0001), being higher for the SAD group as expected.
Table 1.
Age, gender, and score of MMSE in patients with AD and control
| Control (n = 545) | AD (n = 390) | P | |
|---|---|---|---|
| Age (years ± SD) | 68.77 ± 9.192 | 69.99 ± 9.907 | 0.056 |
| Male/female | 209/336 | 162/228 | 0.343 |
| MMSE (means ± SD) | 27.81 ± 4.225 | 14.70 ± 5.835 | <0.0001 |
| APOE ε4 carrier (%) | 191 (35.05) | 180 (46.15) | 0.001 |
| APOE ε4ε4 genotype (%) | 7 (0.01) | 44 (11.28) | <0.0001 |
| APOE ε2 carrier (%) | 121 (22.2) | 31 (7.9) | <0.0001 |
| APOE ε2ε2 genotype (%) | 17 (3.1) | 3 (0.8) | 0.020 |
AD, Alzheimer's disease; APOE, apolipoprotein E; MMSE, Mini Mental State Examination.
Genotype and Allele Frequency Distribution
Polymorphisms of R1628P and G2385R were identified using PCR‐RFLP analysis, and the minor alleles of R1628P were further confirmed by DNA sequencing (Figure 1). The allele and genotype distributions of R1628P and G2385R polymorphisms are shown in Table 2, and the corresponding logistic regression analyses are shown in Tables 3 and 4, respectively. To our surprise, the frequency of the C allele within the R1628P variant was more than three times higher in control group (1.7%) than in patients with SAD (0.5%), and this difference was significant (OR 0.264; 95% CI, 0.088–0.792, P = 0.018). After stratifying by the presence of one or two APOE ε4 alleles, it was found that in APOE ε44 carriers, the C allele frequency in the control group was more than 28 times higher than in the patient group (ε44: OR 0.028; 95% CI, 0.003–0.303, P = 0.003; ε4: OR 0.104; 95% CI, 0.013–0.818, P = 0.031). In addition, the C allele was totally absent in cases and controls who were carriers of APOE ε22. However, we did not observe a difference in the frequencies of the G2385R between the SAD and control group (AA: absent; AG: P = 0.401, OR 1.306, 95% CI 0.700–2.434; allele A: P = 0.382, OR 1.315, 95% CI 0.711–2.431).
Figure 1.
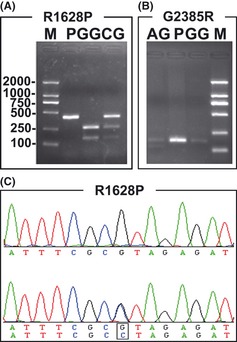
Genotypes of R1628P and G2385R. (A) Electrophoresis of BstUI‐digested R1628P PCR‐amplified products on a 2.5% agarose gel. M: marker (D2000); P: PCR product of 419 bp; GG: genotype GG, represented by two fully digested fragments of 263 bp and 156 bp; CG: genotype CG, represented by the undigested PCR product of 419 bp and two smaller fragments of 263 bp and 156 bp. (B) Electrophoresis of AccI‐digested G2385R PCR‐amplified products on a 2.5% agarose gel. M: marker (D2000); GG: genotype GG, represented by an undigested 170‐bp fragment; P: PCR product of 170 bp; AG: genotype AG, represented by an undigested PCR product of 170 bp and a shorter fragment of 123 bp, the digested smaller piece of 47 bp cannot be observed. (C) DNA sequence chromatogram of R1628P. The upper panel indicates genotype GG, whereas the genotype CG is shown in the bottom one.
Table 2.
Genotypes and allele frequencies of R1628P and G2385R
| R1628P | Control (%) | AD (%) | P | G2385R | Control (%) | AD (%) | P |
|---|---|---|---|---|---|---|---|
| Total | 545 (%) | 390 (%) | Total | 545 (%) | 390 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | AA | 0 (0.0) | 0 (0.0) | ||
| CG | 18 (3.3) | 4 (1.0) | AG | 22 (4.0) | 21 (5.4) | ||
| GG | 527 (96.7) | 386 (99.0) | 0.027 | GG | 523 (96.0) | 369 (94.6) | 0.346 |
| C frequency | 18 (1.7) | 4 (0.5) | A frequency | 22 (2.0) | 21 (2.7) | ||
| G frequency | 1072 (98.3) | 776 (99.5) | 0.028 | G frequency | 1068 (98.0) | 759 (97.3) | 0.351 |
| Male | 209 (%) | 162 (%) | Male | 209 (%) | 162 (%) | ||
| CC | 0.00 | 0.00 | AA | 0 (0.0) | 0 (0.0) | ||
| CG | 8 (3.8) | 1 (0.6) | AG | 12 (5.7) | 9 (5.6) | ||
| GG | 201 (96.2) | 161 (99.4) | 0.084 | GG | 197 (94.3) | 153 (94.4) | 1.000 |
| C frequency | 8 (1.9) | 1 (0.3) | A frequency | 12 (2.9) | 9 (2.8) | ||
| G frequency | 410 (98.1) | 323 (99.7) | 0.086 | G frequency | 406 (97.1) | 315 (97.2) | 1.000 |
| Female | 336 (%) | 228 (%) | Female | 336 (%) | 228 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | AA | 0 (0.0) | 0 (0.0) | ||
| CG | 10 (3.0) | 3 (1.3) | AG | 10 (3.0) | 12 (5.3) | ||
| GG | 326 (97.0) | 225 (98.7) | 0.259 | GG | 326 (97.0) | 216 (94.7) | 0.188 |
| C frequency | 10 (1.5) | 3 (0.7) | A frequency | 10 (1.5) | 12 (2.6) | ||
| G frequency | 662 (98.5) | 453 (99.3) | 0.261 | G frequency | 662 (98.5) | 444 (97.4) | 0.192 |
| EOAD | 193 (%) | 134 (%) | EOAD | 193 (%) | 134 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | AA | 0 (0.0) | 0 (0.0) | ||
| CG | 6 (3.1) | 1 (0.7) | AG | 10 (5.2) | 7 (5.2) | ||
| GG | 187 (96.9) | 133 (99.3) | 0.247 | GG | 183 (94.8) | 127 (94.8) | 1.000 |
| C frequency | 6 (1.6) | 1 (0.4) | A frequency | 10 (2.6) | 7 (2.6) | ||
| G frequency | 380 (98.4) | 267 (99.6) | 0.250 | G frequency | 376 (97.4) | 261 (97.4) | 1.000 |
| LOAD | 352 (%) | 256 (%) | LOAD | 352 (%) | 256 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | AA | 0 (0.0) | 0 (0.0) | ||
| CG | 12 (3.4) | 3 (1.2) | AG | 12 (3.4) | 14 (5.5) | ||
| GG | 340 (96.6) | 253 (98.8) | 0.111 | GG | 340 (96.6) | 242 (94.5) | 0.229 |
| C frequency | 12 (1.7) | 3 (0.6) | A frequency | 12 (1.7) | 14 (2.7) | ||
| G frequency | 692 (98.3) | 509 (99.4) | 0.113 | G frequency | 692 (98.3) | 498 (97.3) | 0.234 |
| APOE ε4 carrier | 191 (%) | 180 (%) | APOE ε4 carrier | 191 (%) | 180 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | AA | 0 (0.0) | 0 (0.0) | ||
| CG | 10 (5.2) | 1 (0.6) | AG | 7 (3.7) | 10 (5.6) | ||
| GG | 181 (94.8) | 179 (99.4) | 0.011 | GG | 184 (96.3) | 170 (94.4) | 0.460 |
| C frequency | 10 (2.6) | 1 (0.3) | A frequency | 7 (1.8) | 10 (2.8) | ||
| G frequency | 372 (97.4) | 359 (99.7) | 0.012 | G frequency | 375 (98.2) | 350 (97.2) | 0.466 |
| APOE ε4 noncarriers | 354 (%) | 210 (%) | APOE ε4 noncarriers | 354 (%) | 210 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | AA | 0 (0.0) | 0 (0.0) | ||
| CG | 8 (2.3) | 3 (1.4) | AG | 15 (4.2) | 11 (5.2) | ||
| GG | 346 (97.7) | 207 (98.6) | 0.754 | GG | 339 (95.8) | 199 (94.8) | 0.679 |
| C frequency | 8 (1.1) | 3 (0.7) | A frequency | 15 (2.1) | 11 (2.6) | ||
| G frequency | 700 (98.9) | 417 (99.3) | 0.755 | G frequency | 693 (97.9) | 409 (97.4) | 0.682 |
| APOE ε44 carrier | 7 (%) | 44 (%) | APOE ε44 carrier | 7 (%) | 44 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | AA | 0 (0.0) | 0 (0.0) | ||
| CG | 4 (57.1) | 1 (2.3) | AG | 0 (0.0) | 1 (2.3) | ||
| GG | 3 (42.9) | 43 (97.7) | 0.001 | GG | 7 (100.0) | 43 (97.7) | 1.000 |
| C frequency | 4 (28.6) | 1 (1.1) | A frequency | 0 (0.0) | 1 (1.1) | ||
| G frequency | 10 (71.4) | 87 (98.9) | 0.001 | G frequency | 14 (100.0) | 87 (98.9) | 1.000 |
| APOE ε44 noncarriers | 538 (%) | 346 (%) | APOE ε44 noncarriers | 538 (%) | 346 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | AA | 0 (0.0) | 0 (0.0) | ||
| CG | 14 (2.6) | 3 (0.9) | AG | 22 (4.1) | 20 (5.8) | ||
| GG | 524 (97.4) | 343 (99.1) | 0.080 | GG | 516 (95.9) | 326 (94.2) | 0.260 |
| C frequency | 14 (1.3) | 3 (0.4) | A frequency | 22 (2.0) | 20 (2.9) | ||
| G frequency | 1062 (98.7) | 689 (99.6) | 0.082 | G frequency | 1054 (98.0) | 672 (97.1) | 0.266 |
| APOE ε2 carrier | 121 (%) | 31 (%) | APOE ε2 carrier | 121 (%) | 31 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | AA | 0 (0.0) | 0 (0.0) | ||
| CG | 2 (1.7) | 0 (0.0) | AG | 3 (2.5) | 3 (9.7) | ||
| GG | 119 (98.3) | 31 (100.0) | 1.000 | GG | 118 (97.5) | 28 (90.3) | 0.100 |
| C frequency | 2 (0.8) | 0 (0.0) | A frequency | 3 (1.2) | 3 (4.8) | ||
| G frequency | 240 (99.2) | 62 (100.0) | 1.000 | G frequency | 239 (98.8) | 59 (95.2) | 0.102 |
| APOE ε2 noncarriers | 424 (%) | 359 (%) | APOE ε2 noncarriers | 424 (%) | 359 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | AA | 0 (0.0) | 0 (0.0) | ||
| CG | 16 (3.8) | 4 (1.1) | AG | 19 (4.5) | 18 (5.0) | ||
| GG | 408 (96.2) | 355 (98.9) | 0.022 | GG | 405 (95.5) | 341 (95.0) | 0.738 |
| C frequency | 16 (1.9) | 4 (0.6) | 0.023 | A frequency | 19 (2.2) | 18 (2.5) | |
| G frequency | 832 (98.1) | 714 (99.4) | G frequency | 829 (97.8) | 700 (97.5) | 0.741 | |
| APOE ε22 carrier | 17 (%) | 3 (%) | APOE ε22 carrier | 17 (%) | 3 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | AA | 0 (0.0) | 0 (0.0) | ||
| CG | 0 (0.0) | 0 (0.0) | AG | 0 (0.0) | 1 (33.3) | 0.150 | |
| GG | 17 (100.0) | 3 (100.0) | – | GG | 17 (100.0) | 2 (66.7) | |
| C frequency | 0 (0.0) | 0 (0.0) | A frequency | 0 (0.0) | 1 (16.7) | 0.150 | |
| G frequency | 34 (100.0) | 6 (100.0) | – | G frequency | 34 (100.0) | 5 (83.3) | |
| APOE ε22 noncarriers | 528 (%) | 387 (%) | APOE ε22 noncarriers | 528 (%) | 387 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | AA | 0 (0.0) | 0 (0.0) | ||
| CG | 18 (3.4) | 4 (1.0) | AG | 22 (4.2) | 20 (5.2) | 0.524 | |
| GG | 510 (96.6) | 383 (99.0) | 0.027 | GG | 506 (95.8) | 367 (94.8) | |
| C frequency | 18 (1.7) | 4 (0.5) | A frequency | 22 (2.1) | 20 (2.6) | ||
| G frequency | 1038 (98.3) | 770 (99.5) | 0.028 | G frequency | 1034 (97.9) | 754 (97.4) | 0.529 |
AD, Alzheimer's disease; APOE, apolipoprotein E; EOAD, early‐onset AD.
Table 3.
Logistic regression analysis of R1628P
| R1628P | Control | AD | P | OR (95% CI) |
|---|---|---|---|---|
| Total | 545 (%) | 390 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | ||
| CG | 18 (3.3) | 4 (1.0) | 0.017 | 0.261 (0.086–0.788) |
| GG | 527 (96.7) | 386 (99.0) | Reference | |
| C | 18 (1.7) | 4 (0.5) | 0.018 | 0.264 (0.088–0.792) |
| G | 1072 (98.3) | 776 (99.5) | Reference | |
| Male | 209 (%) | 162 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | ||
| CG | 8 (3.8) | 1 (0.6) | 0.058 | 0.131 (0.016–1.072) |
| GG | 201 (96.2) | 161 (99.4) | Reference | |
| C | 8 (1.9) | 1 (0.3) | 0.061 | 0.135 (0.017–1.098) |
| G | 410 (98.1) | 323 (99.7) | Reference | |
| Female | 336 (%) | 228 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | ||
| CG | 10 (3.0) | 3 (1.3) | 0.172 | 0.396 (0.105–1.496) |
| GG | 326 (97.0) | 225 (98.7) | Reference | |
| C | 10 (1.5) | 3 (0.7) | 0.169 | 0.396 (0.106–1.481) |
| G | 662 (98.5) | 453 (99.3) | Reference | |
| EOAD | 193 (%) | 134 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | ||
| CG | 6 (3.1) | 1 (0.7) | 0.125 | 0.184 (0.021–1.600) |
| GG | 187 (96.9) | 133 (99.3) | Reference | |
| C | 6 (1.6) | 1 (0.4) | 0.128 | 0.188 (0.022–1.616) |
| G | 380 (98.4) | 267 (99.6) | Reference | |
| LOAD | 352 (%) | 256 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | ||
| CG | 12 (3.4) | 3 (1.2) | 0.072 | 0.306 (0.084–1.112) |
| GG | 340 (96.6) | 253 (98.8) | Reference | |
| C | 12 (1.7) | 3 (0.6) | 0.072 | 0.307 (0.085–1.109) |
| G | 692 (98.3) | 509 (99.4) | Reference | |
| APOE ε4 carriers | 191 (%) | 180 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | ||
| CG | 10 (5.2) | 1 (0.6) | 0.031 | 0.102 (0.013–0.810) |
| GG | 181 (94.8) | 179 (99.4) | Reference | |
| C | 10 (2.6) | 1 (0.3) | 0.031 | 0.104 (0.013–0.818) |
| G | 372 (97.4) | 359 (99.7) | Reference | |
| APOE ε4 noncarriers | 354 (%) | 210 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | ||
| CG | 8 (2.3) | 3 (1.4) | 0.475 | 0.613 (0.160–2.349) |
| GG | 346 (97.7) | 207 (98.6) | Reference | |
| A | 8 (1.1) | 3 (0.7) | 0.473 | 0.613 (0.161–2.333) |
| G | 700 (98.9) | 417 (99.3) | Reference | |
| APOE ε44 carriers | 7 (%) | 44 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | ||
| CG | 4 (57.1) | 1 (2.3) | 0.003 | 0.015 (0.001–0.229) |
| GG | 3 (42.9) | 43 (97.7) | Reference | |
| C | 4 (28.6) | 1 (1.1) | 0.003 | 0.028 (0.003–0.303) |
| G | 10 (71.4) | 87 (98.9) | Reference | |
| APOE ε44 noncarriers | 538 (%) | 346 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | ||
| CG | 14 (2.6) | 3 (0.9) | 0.079 | 0.324 (0.092–1.138) |
| GG | 524 (97.4) | 343 (99.1) | Reference | |
| C | 14 (1.3) | 3 (0.4) | 0.078 | 0.324 (0.093–1.135) |
| G | 1062 (98.7) | 689 (99.6) | Reference | |
| APOE ε2 carriers | 121 (%) | 31 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | ||
| CG | 2 (1.7) | 0 (0.0) | 0.999 | 0.000 (0.000) |
| GG | 119 (98.3) | 31 (100.0) | Reference | |
| C | 2 (0.8) | 0 (0.0) | 0.999 | 0.000 (0.000) |
| G | 240 (99.2) | 62 (100.0) | Reference | |
| APOE ε2 noncarriers | 424 (%) | 359 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | ||
| CG | 16 (3.8) | 4 (1.1) | 0.027 | 0.288 (0.095–0.870) |
| GG | 408 (96.2) | 355 (98.9) | Reference | |
| C | 16 (1.9) | 4 (0.6) | 0.028 | 0.291 (0.097–0.876) |
| G | 832 (98.1) | 714 (99.4) | Reference | |
| APOE ε22 carriers | 17 (%) | 3 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | ||
| CG | 0 (0.0) | 0 (0.0) | – | – |
| GG | 17 (100.0) | 3(100.0) | Reference | |
| C | 0 (0.0) | 0 (0.0) | – | – |
| G | 34 (100.0) | 6 (100.0) | Reference | |
| APOE ε22 noncarriers | 528 (%) | 387 (%) | ||
| CC | 0 (0.0) | 0 (0.0) | ||
| CG | 18 (3.4) | 4 (1.0) | 0.028 | 0.293 (0.098–0.875) |
| GG | 510 (96.6) | 383 (99.0) | Reference | |
| C | 18 (1.7) | 4 (0.5) | 0.028 | 0.296 (0.099–0.878) |
| G | 1038 (98.3) | 770 (99.5) | Reference |
AD, Alzheimer's disease; APOE, apolipoprotein E; EOAD, early‐onset AD.
Table 4.
Logistic regression analysis of G2385R
| G2385R | Control | AD | P | OR (95% CI) |
|---|---|---|---|---|
| Total | 545 (%) | 390 (%) | ||
| AA | 0 (0.0) | 0 (0.0) | ||
| AG | 22 (4.0) | 21 (5.4) | 0.401 | 1.306 (0.700–2.434) |
| GG | 523 (96.0) | 369 (94.6) | Reference | |
| A | 22 (2.0) | 21 (2.7) | 0.382 | 1.315 (0.711–2.431) |
| G | 1068 (98.0) | 759 (97.3) | Reference | |
| Male | 209 (%) | 162 (%) | ||
| AA | 0 (0.0) | 0 (0.0) | ||
| AG | 12 (5.7) | 9 (5.6) | 0.811 | 0.896 (0.365–2.202) |
| GG | 197 (94.3) | 153 (94.4) | Reference | |
| A | 12 (2.9) | 9 (2.8) | 0.852 | 0.919 (0.380–2.225) |
| G | 406 (97.1) | 315 (97.2) | Reference | |
| Female | 336 (%) | 228 (%) | ||
| AA | 0 (0.0) | 0 (0.0) | ||
| AG | 10 (3.0) | 12 (5.3) | 0.127 | 1.983 (0.824–4.773) |
| GG | 326 (97.0) | 216 (94.7) | Reference | |
| A | 10 (1.5) | 12 (2.6) | 0.126 | 1.970 (0.827–4.692) |
| G | 662 (98.5) | 444 (97.4) | Reference | |
| EOAD | 193 (%) | 134 (%) | ||
| AA | 0 (0.0) | 0 (0.0) | ||
| AG | 10 (5.2) | 7 (5.2) | 0.884 | 1.078 (0.392–2.971) |
| GG | 183 (94.8) | 127 (94.8) | Reference | |
| A | 10 (2.6) | 7 (2.6) | 0.866 | 1.090 (0.402–2.955) |
| G | 376 (97.4) | 261 (97.4) | Reference | |
| LOAD | 352 (%) | 256 (%) | ||
| AA | 0 (0.0) | 0 (0.0) | ||
| AG | 12 (3.4) | 14 (5.5) | 0.266 | 1.575 (0.707–3.506) |
| GG | 340 (96.6) | 242 (94.5) | Reference | |
| A | 12 (1.7) | 14 (2.7) | 0.258 | 1.577 (0.716–3.473) |
| G | 692 (98.3) | 498 (97.3) | Reference | |
| APOE ε4 carriers | 191 (%) | 180 (%) | ||
| AA | 0 (0.0) | 0 (0.0) | ||
| AG | 7 (3.7) | 10 (5.6) | 0.470 | 1.443 (0.534–3.902) |
| GG | 184 (96.3) | 170 (94.4) | Reference | |
| A | 7 (1.8) | 10 (2.8) | 0.444 | 1.467 (0.550–3.911) |
| G | 375 (98.2) | 350 (97.2) | Reference | |
| APOE ε4 noncarriers | 354 (%) | 210 (%) | ||
| AA | 0 (0.0) | 0 (0.0) | ||
| AG | 15 (4.2) | 11 (5.2) | 0.622 | 1.223 (0.549–2.725) |
| GG | 339 (95.8) | 199 (94.8) | Reference | |
| A | 15 (2.1) | 11 (2.6) | 0.618 | 1.223 (0.554–2.697) |
| G | 693 (97.9) | 409 (97.4) | Reference | |
| APOE ε44 carriers | 7 (%) | 44 (%) | ||
| AA | 0 (0.0) | 0 (0.0) | ||
| AG | 0 (0.0) | 1 (2.3) | 1.000 | 4.181E7 (0.000) |
| GG | 7 (100.0) | 43 (97.7) | Reference | |
| A | 0 (0.0) | 1 (1.1) | 1.000 | 6.241E7 (0.000) |
| G | 14 (100.0) | 87 (98.9) | Reference | |
| APOE ε44 noncarriers | 538 (%) | 346 (%) | ||
| AA | 0 (0.0) | 0 (0.0) | ||
| AG | 22 (4.1) | 20 (5.8) | 0.310 | 1.381 (0.740–2.578) |
| GG | 516 (95.9) | 326 (94.2) | Reference | |
| A | 22 (2.0) | 20 (2.9) | 0.302 | 1.383 (0.747–2.558) |
| G | 1054 (98.0) | 672 (97.1) | Reference | |
| APOE ε2 carriers | 121 (%) | 31 (%) | ||
| AA | 0 (0.0) | 0 (0.0) | ||
| AG | 3 (2.5) | 3 (9.7) | 0.323 | 2.431 (0.418–14.131) |
| GG | 118 (97.5) | 28 (90.3) | Reference | |
| A | 3 (1.2) | 3 (4.8) | 0.325 | 2.350 (0.428–12.898) |
| G | 239 (98.8) | 59 (95.2) | Reference | |
| APOE ε2 noncarriers | 424 (%) | 359 (%) | ||
| AA | 0 (0.0) | 0 (0.0) | ||
| AG | 19 (4.5) | 18 (5.0) | 0.789 | 1.096 (0.565–2.122) |
| GG | 405 (95.5) | 341 (95.0) | Reference | |
| A | 19 (2.2) | 18 (2.5) | 0.759 | 1.108 (0.576–2.129) |
| G | 829 (97.8) | 700 (97.5) | Reference | |
| APOE ε22 carriers | 17 (%) | 3 (%) | ||
| AA | 0 (0.0) | 0 (0.0) | ||
| AG | 0 (0.0) | 1 (33.3) | 0.999 | 2.531E18 (0.000) |
| GG | 17 (100.0) | 2 (66.7) | Reference | |
| A | 0 (0.0) | 1 (16.7) | 1.000 | 7.685E9 (0.000) |
| G | 34 (100.0) | 5 (83.3) | Reference | |
| APOE ε22 noncarriers | 528 (%) | 387 (%) | ||
| AA | 0 (0.0) | 0 (0.0) | ||
| AG | 22 (4.2) | 20 (5.2) | 0.554 | 1.207 (0.648–2.249) |
| GG | 506 (95.8) | 367 (94.8) | Reference | |
| A | 22 (2.1) | 20 (2.6) | 0.531 | 1.217 (0.658–2.249) |
| G | 1034 (97.9) | 754 (97.4) | Reference |
AD, Alzheimer's disease; APOE, apolipoprotein E; EOAD, early‐onset AD.
Discussion
LRRK2 is a large gene located on chromosome 12 that has 51 exons and encodes a multifunctional protein. Recent studies found LRRK2 immunopositivity in a subset of neurofibrillary tangles in AD and the parkinsonism–dementia complex of Guam (PDCG) 14. Although the physical function of LRRK2 remains unclear, it has been suggested that it may be a cytoplasmic kinase capable of autophosphorylation as well as a GTPase. An interaction with microtubules has also been reported 15, 16, 17, suggesting that LRRK2‐induced neurodegeneration might be partly mediated by the inhibition of microtubule dynamics. Moreover, it is found that LRRK2 may have an interaction with mitochondria and is involved in pathways that elicit oxidative stress or free radical damage 18.
Despite a plausible role of LRRK2 dysfunction in neurodegenerative diseases such as PD and AD, most research to date has failed to find an association between LRRK2 mutations/variants (e.g., G2019S and I2020T, the most common mutations in PD and one Asian‐specific variant G2385R) and AD in different ethnic groups including Chinese, Brazilian, Ashkenazi Jewish, Italian, and Norwegian 19, 20, 21, 22, 23, 24, 25. To date, the only exception has been a case–control study in 217 patients with AD and 668 controls in Singapore population 26. This study identified the association between the variant R1628P within LRRK2 and AD (C allele: AD 3.5% vs. control 1.6%, OR 2.3, 95 CI 1.2–4.4, P = 0.018). However, the results we report here are diametrically opposite (C allele: AD 0.5% vs. control 1.7%, OR 0.264, 95 CI 0.088–0.792, P = 0.018). We found the C allele frequency in controls to be more than three times higher than in cases, suggesting that the minor allele C in the R1628P SNP plays a protective role in SAD, especially after stratification for the presence of one or two APOE ε4 alleles. However, these preliminary data need to be further investigated in a larger cohort.
There are several possible explanations for the different findings between the Singapore and Shanghai studies. Firstly, methodological concerns such as ascertainment bias and sample size limitations may influence the results. Here, we used a much larger sample. Our patient group is almost twofold of the Singapore study (390 vs. 217); thus, the result is more convincing. Besides, in our study, we applied very stringent enrollment criteria (patients with any cardinal sign of parkinsonism were excluded from this study) to make sure that our patient group is sufficiently representative. This may explain why the R1628P variant frequency in the controls is comparable (1.7% vs. 1.6%) in both the Shanghai and Singapore studies, while the frequency in patients is very different (0.5% vs. 3.5%). In addition, although epidemiological studies indicated that there may be an overlapping family history between AD and PD, significant association has been reported between APOE ε2 allele and sporadic PD 27, in contrast to AD where the ε2 allele functions as a protective factor. Consistent with this interesting finding, our study revealed a protective effect of the LRRK2 R1628P variant in AD although this is thought to be a risk factor in PD. It remains unclear what the underlying pathologic mechanism might be. We postulate that there must be some complex interactions between the LRRK2 and APOE genes that play an important role in the development of neurodegenerative diseases such as AD and PD. Further research is required to elucidate why the same allele could have a protective role in one neurodegenerative process, but act as a risk factor for another.
In summary, our study indicated a protective effect of the C allele in the LRRK2 R1628P variant with SAD. This protective effect was more significant among the APOE ε4 allele carriers. Thus, we propose that there may be an interaction between APOE and LRRK2 in the pathogenesis of neurodegenerative disease. This observation will no doubt provide a new research focus for studying the biological function of LRRK2.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
The authors sincerely thank the participants for their help and willingness to participate in this study and the anonymous reviewers for improving this manuscript. This work was supported by a grant from the National Natural Science Foundation of China to Zhi‐Ying Wu (81125009) and the grant from Huashan Hospital for special professorship of Fudan University to Zhi‐Ying Wu.
The first two authors contributed equally to this work.
References
- 1. Clarimon J, Djaldetti R, Lleo A, et al. Whole genome analysis in a consanguineous family with early onset Alzheimer's disease. Neurobiol Aging 2009;30:1986–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Saunders AM, Strittmatter WJ, Schmechel D, et al. Association of apolipoprotein E allele ε4 with late‐onset familial and sporadic Alzheimer's disease. Neurology 1993;43:1467–1472. [DOI] [PubMed] [Google Scholar]
- 3. Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal‐dominant parkinsonism with pleomorphic pathology. Neuron 2004;44:601–607. [DOI] [PubMed] [Google Scholar]
- 4. Marder K, Tang MX, Alfaro B, et al. Risk of Alzheimer's disease in relatives of Parkinson's disease patients with and without dementia. Neurology 1999;52:719–724. [DOI] [PubMed] [Google Scholar]
- 5. Hofman A, Schulte W, Tanja TA, et al. History of dementia and Parkinson's disease in 1st‐degree relatives of patients with Alzheimer's disease. Neurology 1989;39:1589–1592. [DOI] [PubMed] [Google Scholar]
- 6. Papapetropoulos S, Lieberman A, Gonzalez J, Mash DC. Can Alzheimer's type pathology influence the clinical phenotype of Parkinson's disease? Acta Neurol Scand 2005;111: 353–359. [DOI] [PubMed] [Google Scholar]
- 7. Paisan‐Ruiz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8‐linked Parkinson's disease. Neuron 2004;44:595–600. [DOI] [PubMed] [Google Scholar]
- 8. Zabetian CP, Samii A, Mosley AD, et al. A clinic‐based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology 2005;65:741–744. [DOI] [PubMed] [Google Scholar]
- 9. Pericak‐Vance MA, Bass MP, Yamaoka LH, et al. Complete genomic screen in late‐onset familial Alzheimer disease. Evidence for a new locus on chromosome 12. JAMA 1997;278:1237–1241. [PubMed] [Google Scholar]
- 10. Beecham GW, Martin ER, Li YJ, et al. Genome‐wide association study implicates a chromosome 12 risk locus for late‐onset Alzheimer disease. Am J Hum Genet 2009;84:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li HL, Shi SS, Guo QH, et al. PICALM and CR1 variants are not associated with sporadic Alzheimer's disease in Chinese patients. J Alzheimers Dis; 25: 111–117. [DOI] [PubMed] [Google Scholar]
- 12. Donohoe GG, Salomaki A, Lehtimaki T, Pulkki K, Kairisto V. Rapid identification of apolipoprotein E genotypes by multiplex amplification refractory mutation system PCR and capillary gel electrophoresis. Clin Chem 1999;45:143–146. [PubMed] [Google Scholar]
- 13. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 14. Miklossy J, Arai T, Guo JP, et al. LRRK2 expression in normal and pathologic human brain and in human cell lines. J Neuropathol Exp Neurol 2006;65:953–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gandhi PN, Wang X, Zhu X, Chen SG, Wilson‐Delfosse AL. The Roc domain of leucine‐rich repeat kinase 2 is sufficient for interaction with microtubules. J Neurosci Res 2008;86:1711–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gillardon F. Interaction of elongation factor 1‐alpha with leucine‐rich repeat kinase 2 impairs kinase activity and microtubule bundling in vitro. Neuroscience 2009;163:533–539. [DOI] [PubMed] [Google Scholar]
- 17. Gillardon F. Leucine‐rich repeat kinase 2 phosphorylates brain tubulin‐beta isoforms and modulates microtubule stability–a point of convergence in parkinsonian neurodegeneration? J Neurochem 2009;110:1514–1522. [DOI] [PubMed] [Google Scholar]
- 18. Lin TK, Liou CW, Chen SD, et al. Mitochondrial dysfunction and biogenesis in the pathogenesis of Parkinson's disease. Chang Gung Med J 2009;32:589–599. [PubMed] [Google Scholar]
- 19. Santos‐Reboucas CB, Abdalla CB, Martins PA, et al. LRRK2 p.G2019S mutation is not common among Alzheimer's disease patients in Brazil. Dis Markers 2009;27: 13–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tedde A, Bagnoli S, Cellini E, Nacmias B, Piacentini S, Sorbi S. No association between the LRRK2 G2019S mutation and Alzheimer's disease in Italy. Cell Mol Neurobiol 2007;27:877–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chang TY, Kuo HC, Lu CS, Wu‐Chou YH, Huang CC. Analysis of the LRRK2 Gly2385Arg variant in Alzheimer's disease in Taiwan. Parkinsonism Relat Disord;16: 28–30. [DOI] [PubMed] [Google Scholar]
- 22. Tan EK, Lee J, Chen CP, Wong MC, Zhao Y. Case control analysis of LRRK2 Gly2385Arg in Alzheimer's disease. Neurobiol Aging 2009;30:501–502. [DOI] [PubMed] [Google Scholar]
- 23. Saunders‐Pullman R, Lipton RB, Senthil G, et al. Increased frequency of the LRRK2 G2019S mutation in an elderly Ashkenazi Jewish population is not associated with dementia. Neurosci Lett 2006;402:92–96. [DOI] [PubMed] [Google Scholar]
- 24. Lee E, Hui S, Ho G, Tan EK, Chen CP. LRRK2 G2019S and I2020T mutations are not common in Alzheimer's disease and vascular dementia. Am J Med Genet B Neuropsychiatr Genet 2006;141B:549–550. [DOI] [PubMed] [Google Scholar]
- 25. Toft M, Sando SB, Melquist S, et al. LRRK2 mutations are not common in Alzheimer's disease. Mech Ageing Dev 2005;126:1201–1205. [DOI] [PubMed] [Google Scholar]
- 26. Zhao Y, Ho P, Yih Y, Chen C, Lee WL, Tan EK. LRRK2 variant associated with Alzheimer's disease. Neurobiol Aging 2011;32:1990–1993. [DOI] [PubMed] [Google Scholar]
- 27. Harhangi BS, de Rijk MC, van Duijn CM, Van Broeckhoven C, Hofman A, Breteler MM. APOE and the risk of PD with or without dementia in a population‐based study. Neurology 2000;54:1272–1276. [DOI] [PubMed] [Google Scholar]