

Summary
Aim
Till date, the mode of action of β‐PEA on neurons is not well illustrated. We tested the hypothesis that β–PEA has the ability to cause oxidative stress by inhibiting the antioxidant enzyme DT‐diaphorase and mitochondrial complexes (Complex‐I and complex‐III).
Methods
Using molecular docking as a tool, we here studied and compared the inhibitory capacity of β‐PEA on DT‐diaphorase and mitochondrial complexes. Three‐dimensional structures of mitochondrial complexes and DT‐diaphorase and their ligands were downloaded from the respective data banks, and free energy of binding (docking scores) were determined.
Results
The present finding demonstrated for the first time that β‐PEA potentiates reactive oxygen species generation by inhibiting the antioxidant enzyme DT‐diaphorase, in addition to the mitochondrial complex‐I and complex‐III.
Conclusion
As lowering of cellular antioxidant molecules is evident in many neurodegenerative disorders, β‐PEA‐induced lowering of DT‐diaphorase activity may have the capability to cause neurodegeneration, which may be potentiated by its ability to inhibit mitochondrial complexes. Thus, β‐PEA—due to its cumulative actions—may be more potent in causing neurodegeneration as compared to other endogenous neurotoxins.
Keywords: DT‐diaphorase, Mitochondrial complex‐I, Mitochondrial complex‐III, Molecular docking, Neurodegeneration, Oxidative stress, Parkinson's disease
Introduction
One of the less described amines the β‐phenethylamine (β‐PEA), which is a phenylalanine derivate in brain 1, 2 and a constituent of coca products such as chocolates and wines 3, have come to limelight because of its possible potential to cause parkinsonian symptoms in animals 3. Although the exact mechanism of action of β‐PEA in brain is not known, its role in brain has been also attributed to its ability to act like dopamine agonist 4, 5 and its administration has been found to cause several psychomotor disorders 6, 7, 8. In addition to its dopamine agonist function 9, β‐PEA‐induced parkinsonian symptom has been reported to be due to the loss of striatal dopamine and its metabolites 3. Moreover, β‐PEA is known to promote oxidative stress either by inhibiting mitochondrial complex‐I 3 or by its ability to produce.OH radicals itself 10. In dopamine‐rich neurons, the concentration of β‐PEA correlates with the concentration of dopamine 11 and is reported to be relatively high in these regions 6, 12, which suggest the vulnerability of these neurons to β‐PEA‐induced neurochemical alterations.
Similar to other parkinsonian neurotoxins like rotenone and MPTP (1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine), the molecular mechanism underlying β‐PEA‐induced neurochemical changes may be suggested to be due to generation of oxidative stress in brain 3 via inhibition of mitochondrial complex‐I 3. Likewise, inhibition of mitochondrial complex‐III has also been linked to generation of oxidative stress 13 and neurotoxicity in various neurodegenerative disorders like Parkinson's disease 14 and Huntington's disease 15, 16. However, there is no report suggesting the inhibitory action of β‐PEA on mitochondrial complex‐III.
Diminished antioxidant molecule or enzymes has also been implicated in the pathology of neurodegeneration and, expectedly, overexpression of DT‐diaphorase, an antioxidant enzyme 17 is found to protect dopaminergic neurons from neurotoxic insult 18. DT‐diaphorase provides the reducing environment in the cell, thereby preventing the oxidation of aminochromes 19 to semiquinone radicals 20, 21, 22, 23, 24. DT‐diaphorase has also been reported to prevent aminochrome‐induced disruption of actin, alpha‐, and beta‐tubulin in cellular model 20.
As β‐PEA has been reported to cause oxidative stress and produce parkinsonian symptoms as like rotenone or MPTP, we hypothesized that the principal mode of β‐PEA‐induced neurotoxicity is generation of oxidative stress via inhibition of the activity of DT‐diaphorase and mitochondrial complex‐III, in addition to complex‐I. We tested our hypothesis using molecular docking as a tool to study the inhibitory action of β‐PEA on these three enzymes and its possible molecular mechanism in inducing neurotoxicity.
Materials and Methods
The Receptor
The three‐dimensional structure of yeast NADH‐Quinone oxidoreductase–Mitochondrial complex‐I (PDB id: 4G73), fitted model of bovine mitochondrial complex‐III (PDB id: 2YBB) and Human DT‐diaphorase (PDB id: 1DXO) were downloaded from RCSB Protein Data Bank (www.rcsb.org/pdb) in PDB format. The selection of the structures was based on the receptor model, resolution, and source organism. The structure 4G73 has 2 chains, determined by X‐ray diffraction at a resolution of 2.52Å and has the bound ligand—quinone; 2YBB has 48 chains, determined by Cryo‐Electron microscopy at a resolution of 19Å and it has bound ligand—ubiquinone; while 1DXO is crystallographic structure determined at 2.5Å resolution having four chains and the bound ligand—duroquinone.
The Ligands
The selected ligands for docking study with the receptors are as follows: ubiquinone‐1 (CID_4462), duroquinone, and β‐PEA (CID_1001). The structure of duroquinone was available with its receptor, while other two were downloaded from NCBI PubChem Compound database (www.ncbi.nlm.nih.gov/pccompound).
Stereochemical Quality Assessment of the Receptor
For assessing the stereochemical quality of the receptors, we generated Ramachandran plots using PROCHECK 3.6.2 module 25 available at PDBSum server (www.ebi.ac.uk).
Detection of Available Cavities in the Receptors
For detecting available cavities including the cavity volumes, the amino acids present and the cavity coordinates we used Q‐SiteFinder 26. Cavity detection for mitochondrial complex‐III was performed using Molegro Virtual Docker (MVD) 27 as the structure could not be loaded into Q‐SiteFinder owing to its larger size.
Pharmacophore Generation for the Ligands
For visualization and comparison of the potential interactions groups on the three ligands used in the study, we generated their pharmacophores used LigandScout 3.0 28.
The Docking Simulation
The receptors (4G73 and 1DXO) were loaded in BioSolve IT FlexX 1.3.0 software 29. Correction of amino acids for protonations, flips, and rotations was carried out. The binding site were selected to be the active binding sites that bind duroquinone (for DT‐Diaphorase) and ubiquinone‐binding site (for mitochondrial complex‐I). The amino acids within a sphere of 20Å were included in the simulation. Docking was performed between the receptor and the said ligands with the described simulation details. The modes in which the ligands bind to the receptor were identified by iteratively evaluating a huge number of ligand conformations and estimating their interaction with the active site of the receptor.
For docking mitochondrial complex‐III from the structure—2YBB, the receptor was loaded in Molegro Virtual Docker (MVD) software 27. Correction of amino acids for protonations was carried out using MVD, followed by neighborhood minimization. Scoring function was selected to be MolDock Score with Grid Resolution set to 0.30Å. Two binding sites for ubiquinone in mitochondrial complex‐III were selected for docking which are: binding site of ubiquinone‐c (X: 11.13, Y: 71.36, Z: −26.89) and ubiquinone‐c (X: 10.75, Y: 43.65, Z: −11.04). Amino acids within a radius of 30Å were included in docking simulation. For visualizing the interactions of the ligands with the active ubiquinone‐binding sites, we injected the best ligand conformations, in terms of MolDock score, into both the ubiquinone‐binding sites of the receptor using Ligandscout 3.0 28 followed by generation of pharmacophores.
Results
Stereochemical Quality of the Receptors
Ramachandran plot (not shown) generated for complex‐I, complex‐III, and DT‐diaphorase, respectively, shows that 90.0%, 89.9%, and 85.4% of the residues fall in the most favoured regions, whereas 9.5%, 9.3%, and 13.2% fall in the additionally allowed regions of the plot.
Available Cavities in the Receptors
Ten sites each in the complex‐I and DT‐diaphorase and two sites in complex‐III were detected (Figure 1). These cavities are the active sites in the enzymes.
Figure 1.
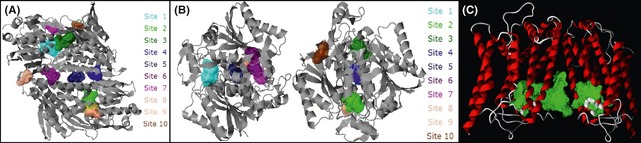
Available cavities in the different receptors. (A): Mitochondrial complex‐I; (B): DT‐diaphorase; (C): Mitochondrial complex‐III. The different colored regions show the cavities. The cavities on complex‐I and DT‐diaphorase have been determined using QSiteFinder, while cavities on complex‐III have been determined using Molegro Virtual Docker.
Possible Interactions and Pharmacophores of the Ligands
Duroquinone can form a total of 5 hydrophobic interactions: four with its four methyl groups and one with its aromatic ring. The aromatic ring can also form aromatic interactions. Each –OH group can donate as well as accept hydrogen bonds (Figure 2A). The side chain of ubiquinone can form two hydrophobic interactions, while all the four oxygen atoms can accept one hydrogen bond each (Figure 2B). Similarly, the aromatic ring of β‐PEA can form aromatic as well as hydrophobic interactions. The amine group is positively ionizable and can donate hydrogen bond (Figure 2C).
Figure 2.
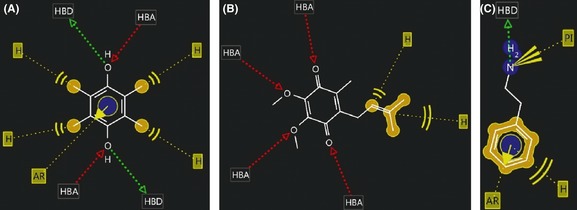
The structures and possible interactions of the ligands used in docking with the receptors are shown: (A) Duroquinone; (B) ubiquinone, and (C) β‐PEA. Different interacting groups on the ligands are denoted as: HBA, Hydrogen bond acceptor; HBD, Hydrogen bond donor; H, hydrophobic interactions; AR, aromatic interactions; PI, Positively ionizable group.
Interactions of Mitochondrial Complex‐I with the Ligands
From the docking study, we found that ubiquinone binds to mitochondrial complex‐I by forming hydrogen bonds with lysine 506 (with which two hydrogen bonds are formed) and lysine 117, while forming weak interactions with lysine 506, phenylalanine 505, phenylalanine 504, asparagine 113, and lysine 117 (Figure 3A; Table 1). β‐PEA interacts at the same site forming hydrogen bonds with lysine 506, arginine 507, and asparagine 508, while weak interactions with proline 110, phenylalanine 510, asparagine 508, phenylalanine 258, lysine 506, and asparagine 113 (Figure 3B; Table 1). The binding site for both the ligands falls in the active site of the receptor; site 4 (Figure 1)—as determined by Q‐SiteFinder.
Figure 3.

Interactions of mitochondrial complex‐I and DT‐diaphorase with their respective ligands. (A) ubiquinone and (B) β‐PEA, docked at the ubiquinone‐binding site of complex‐I; (C) Duroquinone and (D) β‐PEA, docked at the active duroquinone‐binding site on DT‐diaphorase. The dotted lines represent hydrogen bonding, while the green lines represent weak interactions.
Table 1.
The binding affinities (free energies of binding) and the types of interactions of different ligands with their respective receptors: mitochondrial complex‐I, complex‐III, and DT‐diaphorase. The free energy of binding indicates the amount of energy liberated when the receptors and the ligands bind or dock, and thus is an estimation of the affinity between them. Free energies of binding determined by different softwares are not same, because they use different algorithms, and thus are not comparable
| Receptor | Ligand | Docking score | Residues involved in hydrogen bonding | Residues involved in weak interactions |
|---|---|---|---|---|
| Complex‐I | Ubiquinone | −12.7297a | Lys 506 (2), Lys 117 | Lys 506, Phe 505, Phe 504, Asn 113, Lys 117 |
| β‐PEA | −15.8417a | Lys 506, Arg 507, Asp 508 | Pro 110, Phe 510, Asp 508, Phe 258, Lys 506, Asn 113 | |
| DT‐diaphorase | Duroquinone | −19.9065a | Tyr l26, Tyr l28, FAD‐301 | Tyr 126, Tyr 128, FAD‐301 |
| β‐PEA | −16.6305a | Tyr 126 | Tyr 126, Tyr 128, Phe 178, FAD‐301 | |
| Complex‐III (site: UQ‐C) | Ubiquinone‐C | −8O.2565b | HOH 2044 (2) | Phe 220C, Phe 18C, Leu 197C, Hem‐502C |
| β‐PEA | −74.127b | None | Phe220C, Leu 21C, Leu 197C, Hem‐502C | |
| Complex‐III (site: UQ‐c) | Ubiquinone‐C | −79.8071b | HOH 3023c (3) | Phe 220c, Leu 197c, Phe 18c, Hem‐502c |
| β‐PEA | −52.7069b | His 201c | Ile 27c, Phe 220c, Hem‐502c |
Docking scores obtained by FlexX docking.
Docking scores obtained by Molegro Virtual Docker.
Interactions of DT‐Diaphorase with the Ligands
The binding site of the ligands, duroquinone and β‐PEA with DT‐diaphorase, is found to be the site 3 of the receptor. Duroquinone has been found to form hydrogen bonds with tyrosine 126, tyrosine 128, and FAD‐301 (Figure 3C). Interestingly, weak interactions are also formed with the same residues and the cofactor FAD‐301. It is important to note that the cofactor FAD is directly involved in holding the ligand at the active site. Similarly, β‐PEA forms hydrogen bond with tyrosine 126, while weak interactions with tyrosine 128, tyrosine 126, FAD‐301, and one additional weak interaction with phenylalanine 178 (Figure 3D; Table 1).
Interactions of Mitochondrial Complex‐III with the Ligands
Ubiquinone‐c has been found to form two hydrogen bonds with water molecule 2044C; and weak interactions with phenylalanine 220C, phenylalanine 18C, leucine 197C, and the cofactor Heme‐502C (Figure 4A; Table 1). At this site, β‐PEA forms no hydrogen bonds but weak interactions with phenylalanine 220C, leucine 21C, leucine 197C, and the cofactor Heme‐502C (Figure 4B; Table 1). Similarly, ubiquinone‐c forms three hydrogen bonds with the water molecule HOH 3023c, and weak interactions with phenylalanine 220c, leucine 197c, phenylalanine 18c and cofactor‐502c (Figure 4C; Table 1). At this site, β‐PEA forms hydrogen bonds with histidine 201c, and weak interactions with isoleucine 27c, phenylalanine 220c, and cofactor Heme‐502c (Figure 4D; Table 1).
Figure 4.

Interactions of β‐PEA and ubiquinone at the two active sites on mitochondrial complex‐III determined using LigandScout 3.0. The interactions of ubiquinone (A) and β‐PEA (B) at the active site on chain C; and ubiquinone (C) and β‐PEA (D) at the active site on chain c are shown. The red dotted lines represent hydrogen bond donation in the direction of the arrow, green dotted lines represent hydrogen bond acceptance in the shown direction, while yellow lines represent weak interactions.
Free Energy of Binding of the Ligands with the Respective Receptors
From our study, we find that the free energy of binding for ubiquinone with mitochondrial complex‐I is −12.7297; while with β‐PEA, it is −15.8417 (Table 1). With DT‐diaphorase, duroquinone binds with a binding score of −19.9065, and β‐PEA has a binding score of −16.6305 (Table 1). From the docking study, we find that ubiquinone‐c binds with the mitochondrial complex‐III with a score of −67.3081, while β‐PEA has a score of −67.4708 at this site. Ubiquinone‐c binds with a score of −64.1951, while β‐PEA has a score of −44.8554 (Table 1).
Discussion
Inhibition of mitochondrial complex‐I and complex‐III has been reported to cause leakage of electrons 30 and thereby results in the production of reactive oxygen species like ˙OH radicals 31, 32 and hydrogen peroxide 13 generating oxidative stress in the brain. Also, inhibition of DT‐diaphorase has been linked to oxidative stress in neurodegenerative diseases, and the enzyme has been reported to impart neuroprotection in aminochrome‐induced nigral cell death 18 through its quinone reductase activity 33, 34. Meanwhile, oxidative stress causes mitochondrial complex‐I inhibition 35 that results in the drop of cellular ATP thereby leading to ubiquitin‐proteasome system (UPS) dysfunction 36, 37. Inhibition of the UPS leads to accumulation of misfolded proteins like α‐synuclein aggregates or Lewy bodies, which are regarded as the hallmarks of neurodegenerative diseases like PD 38. Lewy bodies, in turn, are reported to inhibit mitochondrial complexes and exaggerate oxidative stress 39. It is also known that oxidative stress causes activation of Caspases and thereby lead to neurodegeneration via apoptotic mode of cell death 40. As oxidative stress is one of the prime causes of neurodegeneration, the present findings that β‐PEA inhibit mitochondrial complexes and DT‐diaphorase, elucidated a novel mode of neurotoxicity of the trace amine. Our finding that β‐PEA actively interacts with the quinone‐binding site of complex‐I, complex‐III, and DT‐diaphorase suggested that β‐PEA may act as a competitive inhibitor.
We hereby report for the first time that β‐PEA has more free energy of binding with the active ubiquinone‐binding site of mitochondrial complex‐I compared with that of the natural substrate ubiquinone (Figure 3; Table 1). The residues lysine 506 and asparagine 113 are the residues that are involved in binding of both the ligands with the complex‐I through weak interactions, while the residue lysine 506 is involved in hydrogen bonding (Figure 3; Table 1). The higher free energy of binding is attributed to the more number of weak interactions being formed by β‐PEA compared with ubiquinone (Table 1), which might result in the competitive inhibition of the receptor. While our study confirms the findings of Sengupta and Mohanakumar (3) that β‐PEA can inhibit mitochondrial complex‐I 3, the present study demonstrates for the first time that β‐PEA may potentially interact and interfere with substrate binding at the ubiquinone‐binding site and thereby inhibit the enzyme.
Similar to the mitochondrial complex‐I inhibition, β‐PEA has the potential to inhibit mitochondrial complex‐III. We studied the mode of binding and potential inhibitory action of β‐PEA at the two ubiquinone‐binding sites present in the complex‐III of the supercomplex 41 and found that, at both the sites, β‐PEA interacts with the same residues with which ubiquinone binds with similar interactions (Figure 4). Importantly, the cofactor Heme is also involved in binding both the substrates to the active site. Ubiquinone forms more hydrogen bonds, as a result of which the free energy of binding of ubiquinone is found to be little more as compared to β‐PEA (Table 1). The residues phenylalanine 220C, leucine 197C, and the cofactor Heme 502C are involved in binding both ubiquinone and β‐PEA to the active site through weak interactions. Similarly, phenylalanine 220c and Heme 502c are involved in weak interactions with both the substrates at the other active site (Table 1). Thus, β‐PEA has ability to competitively inhibit mitochondrial complex‐III via interfering with the substrate binding at the active site of the enzyme. As mitochondrial complex‐III inhibition is known to generate oxidative stress 13, 42 and has been implicated in the pathogenesis of PD 14 as well as in HD 15, 16, the present finding is of immense importance.
The finding that β‐PEA potentially interacts with the active substrate binding site of DT‐diaphorase, reveals a novel mode of neurotoxicity. The free energy of binding of β‐PEA and duroquinone to DT‐diaphorase is comparable, and the binding involves same residues signifying that β‐PEA can compete for the active site on the enzyme (Figure 3C,D). The residues tyrosine 126 has been found to be involved in hydrogen bonding with both duroquinone and β‐PEA; and the residues tyrosine 126, tyrosine 128 and the cofactor FAD 43 are found to be involved in weak interactions with the both the ligands (Table 1). Since DT‐diaphorase provides the reducing environment in the cell 20, 21, 22, 23, 24 and is also known to protect nigral neurons from toxic insult 17, 18, inhibition of the enzyme by β‐PEA, endanger neurons to neurodegenerative changes.
Conclusion
The present finding demonstrated for the first time that β‐PEA potentiates reactive oxygen species generation by inhibiting the antioxidant enzyme DT‐diaphorase, in addition to the mitochondrial complex‐I and complex‐III (Figure 5). As lowering of cellular antioxidant molecules is evident in many neurodegenerative disorders, β‐PEA‐induced lowering of DT‐diaphorase activity may have the capability to cause neurodegeneration, which may be potentiated by its ability to inhibit mitochondrial complex‐I and complex‐III. Thus, β‐PEA—due to its cumulative actions—may be more potent in causing neurodegeneration as compared to other endogenous neurotoxins. However, as β‐PEA has rapid turn‐over rate in the brain, it might not be as much toxic as like MPTP, rotenone, and others. As β‐PEA containing food items have become an integral part of modern life, our study is of immense importance which suggests that consumption of such food items is a serious health concern, and further research may be initiated to unveil the aspects in vivo.
Figure 5.
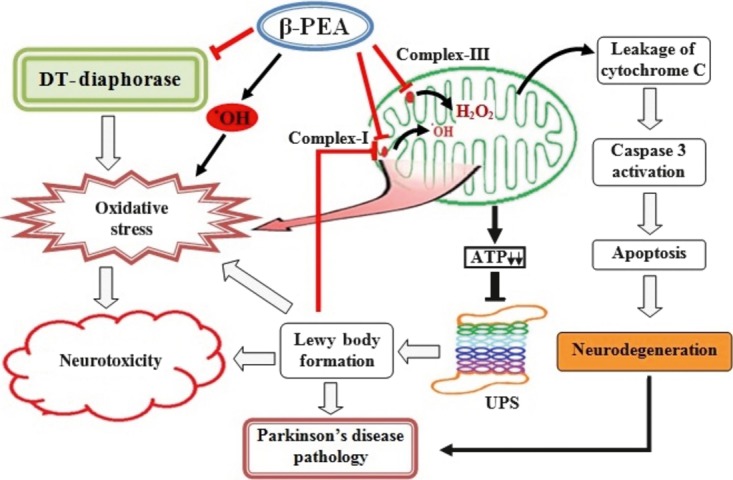
Possible mechanisms of β‐PEA‐induced neurotoxicity: β‐PEA causes oxidative stress directly by generating. OH radicals and also by inhibiting of mitochondrial complex‐I, complex‐III, and DT‐diaphorase. Inhibition of mitochondrial complexes will lead to fall in cellular ATP resulting in ubiquitin‐proteasome system (UPS) dysfunction, production of misfolded proteins like Lewy bodies that together may culminates in neuronal cell death by apoptotic mode. The neuronal cell loss together with Lewy bodies like pathology may contribute to the development of neurodegenerative disorders like Parkinson's disease.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgment
We are thankful to the developers of the softwares: Ligandscout 3.03b (available from Inte:Ligand GmbH, 2344 Maria Enzersdorf, Austria; www.inteligand.com) and MVD (available from http://www.molegro.com) for providing us with evaluation copies. We thank the Bioinformatics Centre, Assam University, Silchar for journal access and software support.
The first two authors contributed equally to this work.
References
- 1. Durden DA, Phillips SR. Kinetic measurements of the turnover rates of phenylethylamine and tryptamine in vivo in the rat brain. J Neurochem 1980;34:1725–1732. [DOI] [PubMed] [Google Scholar]
- 2. Durden DA, Philips SR, Boulton AA. Identification and distribution of betaphenylethylamine in the rat. Can J Biochem 1973;51:995–1002. [DOI] [PubMed] [Google Scholar]
- 3. Sengupta T, Mohanakumar KP. 2‐Phenylethylamine, a constituent of chocolate and wine, causes mitochondrial complex‐I inhibition, generation of hydroxyl radicals and depletion of striatal biogenic amines leading to psycho‐ motor dysfunctions in Balb/c mice. Neurochem Int 2010;57:637–646. [DOI] [PubMed] [Google Scholar]
- 4. Barroso N, Rodriguez M. beta‐Phenylethylamine regulation of dopaminergic nigrostriatal cell activity. Brain Res 1995;12:201–204. [DOI] [PubMed] [Google Scholar]
- 5. Barroso N, Rodriguez M. Action of β‐phenylethylamine and related amines on nigrostriatal dopamine neurotransmission. Eur J Pharmacol 1996;297:195–203. [DOI] [PubMed] [Google Scholar]
- 6. Paterson IA, Juorio AV, Boulton AA. 2‐Phenylethylamine: A modulator of catecholamine transmission in the mammalian central nervous system? J Neurochem 1990;55:1827–1837. [DOI] [PubMed] [Google Scholar]
- 7. Sabelli HC, Fink P, Fawcett J, Tom C. Sustained antidepressant effect of PEA replacement. J Neuropsychiatry Clin Neurosci 1996;8:168–171. [DOI] [PubMed] [Google Scholar]
- 8. Burchett SA, Hicks TP. The mysterious trace amines: Protean neuromodulators of synaptic transmission in mammalian brain. Prog Neurobiol 2006;79:223–246. [DOI] [PubMed] [Google Scholar]
- 9. Janssen PA, Leysen JE, Megens AA, Awouters FH. Does phenylethylamine act as an endogenous amphetamine in some patients. Int J Neuropsychopharmacol 1999;2:229–240. [DOI] [PubMed] [Google Scholar]
- 10. Kawano T, Pinontoan R, Uozumi N, et al. Phenylethylamine‐induced generation of reactive oxygen species and ascorbate free radicals in tobacco suspension culture: Mechanism for oxidative burst mediating Ca2+ influx. Plant Cell Physiol 2000;41:1259–1266. [DOI] [PubMed] [Google Scholar]
- 11. Greenshaw AJ, Juorio AV, Nguyen TV. Depletion of striatal beta‐phenyl ethylamine following dopamine but not 5‐HT denervation. Brain Res Bull 1986;17:477–484. [DOI] [PubMed] [Google Scholar]
- 12. Berry MD. Mammalian central nervous system trace amines. Pharmacologic amphetamines, physiologic neuromodulators. J Neurochem 2004;90:257–271. [DOI] [PubMed] [Google Scholar]
- 13. Rana M, de Coo I, Diaz F, Smeets H, Moraes CT. An out‐of‐frame cytochrome b gene deletion from a patient with parkinsonism is associated with impaired complex III assembly and an increase in free radical production. Ann Neurol 2000;48:774–781. [PubMed] [Google Scholar]
- 14. Haas RH, Nasirian F, Nakano K, et al. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson's disease. Ann Neurol 1995;37:714–722. [DOI] [PubMed] [Google Scholar]
- 15. Tabrizi SJ, Cleeter MW, Xuereb J, Taanman JW, Cooper JM, Schapira AH. Biochemical abnormalities and excitotoxicity in Huntington's disease brain. Ann Neurol 1999;45:25–32. [DOI] [PubMed] [Google Scholar]
- 16. Mann VM, Cooper JM, Javoy‐Agid F, Agid Y, Jenner P, Schapira AH. Mitochondrial function and parental sex effect in Huntington's disease. Lancet 1990;336:749. [DOI] [PubMed] [Google Scholar]
- 17. Cadenas E. Antioxidant and prooxidant functions of DT‐diaphorase in quinone metabolism. Biochem Pharmacol 1995;49:127–140. [DOI] [PubMed] [Google Scholar]
- 18. Muñoz P, Paris I, Sanders LH, Greenamyre JT, Segura‐Aguilar J. Overexpression of VMAT‐2 and DT‐diaphorase protects substantia nigra‐derived cells against aminochrome neurotoxicity. Biochim Biophys Acta 2012;1822:1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Segura‐Aguilar J, Lind C. On the mechanism of the Mn3+‐induced neurotoxicity of dopamine: Prevention of quinone‐derived oxygen toxicity by DT‐diaphorase and superoxide dismutase. Chem Biol Interact 1989;72:309–324. [DOI] [PubMed] [Google Scholar]
- 20. Paris I, Perez‐Pastene C, Cardenas S, et al. Aminochrome induces disruption of actin, alpha‐, and beta‐tubulin cytoskeleton networks in substantia‐nigra‐derived cell line. Neurotox Res 2010;18:82–92. [DOI] [PubMed] [Google Scholar]
- 21. Arriagada C, Paris I, Sanchez de las Matas MJ, et al. On the neurotoxicity mechanism of leukoaminochrome o‐semiquinone radical derived from dopamine oxidation: Mitochondria damage, necrosis and hydroxyl radical formation. Neurobiol Dis 2004;16:468–477. [DOI] [PubMed] [Google Scholar]
- 22. Lozano J, Muñoz P, Nore BF, Ledoux S, Segura‐Aguilar J. Stable expression of short interfering RNA for DT‐diaphorase induces neurotoxicity. Chem Res Toxicol 2010;23:1492–1496. [DOI] [PubMed] [Google Scholar]
- 23. Paris I, Muñoz P, Huenchuguala S, et al. Autophagy protects against aminochrome‐induced cell death in substantia nigra‐derived cell line. Toxicol Sci 2011;121:376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fuentes P, Paris I, Nassif M, Caviedes P, Segura‐Aguilar J. Inhibition of VMAT‐2 and DT‐diaphorase induce cell death in a substantia Nigra‐derived cell line—an experimental cell model for dopamine toxicity studies. Chem Res Toxicol 2007;20:776–783. [DOI] [PubMed] [Google Scholar]
- 25. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst 1993;26:283–291. [Google Scholar]
- 26. Laurie AT, Jackson RM. Q‐SiteFinder: An energy‐based method for the prediction of protein‐ligand binding sites. Bioinformatics 2005;21:1908–1916. [DOI] [PubMed] [Google Scholar]
- 27. Thomsen R, Christensen MH. MolDock: a new technique for high‐accuracy molecular docking. J Med Chem 2006;49:3315–3321. [DOI] [PubMed] [Google Scholar]
- 28. Wolber G, Langer T. Ligandscout: 3‐D pharmacophores derived from protein‐bound ligands and their use as virtual screening filters. J Chem Inf Model 2005;45:160–169. [DOI] [PubMed] [Google Scholar]
- 29. Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol 1996;261:470–489. [DOI] [PubMed] [Google Scholar]
- 30. Thomas B, Mohanakumar KP. Melatonin protects against oxidative stress caused by 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine in the mouse nigrostiratum. J Pineal Res 2004;36:25–32. [DOI] [PubMed] [Google Scholar]
- 31. Saravanan KS, Sindhu KM, Senthilkumar KS, Mohanakumar KP. L‐deprenyl protects against rotenone‐induced, oxidative stress‐mediated dopaminergic neurodegeneration in rats. Neurochem Int 2006;49:28–40. [DOI] [PubMed] [Google Scholar]
- 32. Thomas B, Saravanan KS, Mohanakumar KP. In vitro and in vivo evidences that antioxidant action contributes to the neuroprotective effects of the neuronal nitric oxide synthase and monoamine oxidase‐B inhibitor, 7‐nitroindazole. Neurochem Int 2008;52:990–1001. [DOI] [PubMed] [Google Scholar]
- 33. Schultzberg M, Segura‐Aguilar J, Lind L. Distribution of DT‐diaphorase in the rat brain: Biochemical and immunohistochemical studies. Neuroscience 1998;27:763–766. [DOI] [PubMed] [Google Scholar]
- 34. Segura‐Aguilar JE, Lind C, Nordström Ö, Bartfai T. Regional and subcellular distribution of DT‐diaphorase in the rat brain. Chem Scr 1987;27A:55–57. [Google Scholar]
- 35. Schapira AH, Gu M, Taanman JW, et al. Mitochondria in the etiology and pathogenesis of Parkinson's disease. Ann Neurol 1998;44:S89–S98. [DOI] [PubMed] [Google Scholar]
- 36. Hoffman L, Pratt G, Rechsteiner M. Multiple forms of 20S multi‐catalytic and the 26S ubiquitin/ATP dependent proteases from rabbit reticulocyte lysate. J Biol Chem 1992;267:22362–22368. [PubMed] [Google Scholar]
- 37. Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: A cell biologist thinks about neurodegenerative diseases. Neuron 2001;29:15–32. [DOI] [PubMed] [Google Scholar]
- 38. Glickman MH, Ciechanover A. The Ubiquitin‐Proteasome proteolytic pathway: Destruction for the sake of construction. Physiol Rev 2002;82:373–428. [DOI] [PubMed] [Google Scholar]
- 39. Sullivan PG, Dragicevic NB, Deng JH, et al. Proteasome inhibition alters neural mitochondrial homeostasis and mitochondria turnover. J Biol Chem 2004;279:20699–20707. [DOI] [PubMed] [Google Scholar]
- 40. Tatton NA. Increased caspase 3 and Bax immunoreactivity accompany nuclear GAPDH translocation and neuronal apoptosis in Parkinson's disease. Exp Neurol 2000;166:29–43. [DOI] [PubMed] [Google Scholar]
- 41. Althoff T, Mills DJ, Popot JL, Kühlbrandt W. Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J 2011;30:4652–4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Poyton RO, Ball KA, Castello PR. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinal Metab 2009;20:332–340. [DOI] [PubMed] [Google Scholar]
- 43. Faig M, Bianchet MA, Talalay P, et al. Structures of recombinant human and mouse NAD(P)H:Quinone oxidoreductases: Species comparison and structural changes with substrate binding and release. Proc Natl Acad Sci USA 2000;97:3177–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]