

Summary
Aims
Flax Lignan (FLL), a chemical widespread within the plant and animal kingdoms, has antioxidant, antiinfectious, and antitumor activities. However, little is known about the effects of FLL on the central nervous system (CNS).
Methods
The neuroprotective actions of FLL against N‐methyl‐d‐aspartate (NMDA) are investigated in primary cultured cortical neurons by MTT assay. The expression levels of proteins related to apoptosis and GluN2‐containing receptor were detected by Western blot analysis. Intracellular Ca2+ was measured under a confocal laser scanning microscope.
Results
After challenged with 100 μM NMDA for 30 min, loss of cell viability and excessive apoptotic cell death were observed in cultured cortical neurons. FLL protected the neurons against the NMDA‐induced cell loss in a concentration‐dependent manner. FLL also significantly inhibited the neuronal apoptosis induced by NMDA exposure through reversing intracellular concentration of Ca2+ overload and balancing of Bcl‐2 and Bax expression. Furthermore, FLL significantly reversed the upregulation of GluN2B‐containing NMDA receptors by exposure to NMDA, but did not affect the expression of GluN2A‐containing NMDA receptor.
Conclusions
These findings suggest that FLL protects cortical neurons by inhibiting the expression of GluN2B‐containing NMDA receptor and regulating the Bcl‐2 family.
Keywords: Apoptosis, Flax Lignan (FLL), GluN2B‐containing NMDA receptor, N‐methyl‐D‐aspartate (NMDA)
Introduction
Flax Lignan (FLL) is a natural chemical widespread within the plants. Flaxseed is one of the most significant sources of plant lignans 1. The structure of FLL is very similar to human estrogen and is considered as a phytoestrogen. As a phytoestrogens, FLL performs antitumor, estrogen, and antiestrogenic effects by the inhibition of aromatase enzyme activity 2, DNA and RNA synthesis, and oxidative activities 3. Increasing attentions for lignans show their potential roles in preventing lipid disorders against lead acetate–induced oxidative damage 4 and reducing serum malondialdehyde (MDA) 3. However, few data are available regarding the impact of flaxseed on neuroprotection.
The N‐methyl‐d‐aspartate receptor (NMDAR) is involved in synaptic plasticity, learning, memory, and neurological diseases 5, 6, 7, 8, 9, 10. NMDAR is the major mediator of excitotoxicity because of the high permeability of calcium. NMDAR is a heteromeric complex formed by three types of subunits: GluN1, GluN2 (A, B, C, and D), and GluN3 (A and B) 11, 12. NMDAR typically consists of GluN1 and GluN2 subunits, in which GluN1 subunits are essential for the function of NMDAR channels 13. Excitotoxicity triggered by the selective activation of NMDAR‐containing GluN2B subunit has been suggested to play an important role in the pathogenesis of neurodegenerative disorders associated with glutamate excitotoxicity 14. NMDAR meditates the flow of calcium evoking the downstream signal molecules and causing the excitotoxicity of neurons. Excessive stimulation of NMDAR is the main cause of excitotoxicity in the central nervous system (CNS) correlated with neuronal cell death. Present study investigates the possible neuroprotective properties of FLL against excitatory neurotoxicity mediated by NMDA in primary cortical neurons. We found that FLL performed significant protective effects by downregulating GluN2B expression levels and calcium overload, as well as regulating the Bcl‐2 family, including Bcl‐2 and Bax expression.
Materials and Methods
Chemicals and Reagents
Flax Lignan (FLL) (purity >98%) was purchased from Shanghai PureOne Biotechnology (Shanghai, China). 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium broidme (MTT), poly‐D‐lysine, trypsin, propidium iodide (PI), Hoechst 33258, and anti‐β‐actin antibody were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Neurobasal medium, B27 supplement, and glutamine were provided by Invitrogen (Carlsbad, CA, USA). Anti‐MAP2, anti‐GluN2A, anti‐GluN2B, anti‐Bax, and anti‐Bcl‐2 antibodies were purchased from Chemicon (Temecula, CA, USA). Dulbecco's modified Eagle's medium (DMEM) was purchased from Hyclone (Logan, UT, USA). Secondary antibodies conjugated with horseradish peroxidase (HRP) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). M‐PER protein extraction buffer and enhanced chemiluminescent solution (ECL) were obtained from Pierce (Pierce, Rockford, IL, USA). All of the other chemicals and reagents were of standard commercially available biochemical quality.
Cell Culture and Treatment
Primary cultures of cortical neurons were prepared from the brain of E15–E16 C57 mouse embryos. Briefly, dissociated brain tissue from embryonic 15‐ to 16‐day mouse was incubated with 0.125% trypsin in Ca2+‐ and Mg2+‐free Hank's balanced salt solution for 10 min at 37°C. Then the cortex was washed in DMEM supplemented with 10% FBS to stop trypsin activity and further dissociated by trituration. The single‐cell suspension was cultured on poly‐D‐lysine‐coated plates in Neurobasal media supplemented with 2% B27, 0.5 mM glutamine, 100 U/ml penicillin, and 100 U/ml streptomycin. It took 7 days for re‐incubation, the time required for maturation of cortical neurons, and half of the medium was changed every 2 days. The cells were characterized by immunohistochemistry staining for anti‐MAP2 antibody, revealing that this culture procedure yields more than 95% neurons 15. Neurons were seeded at a density of 5 × 104 cells/well in 96‐well plate, 3 × 105 cells/well in 24‐well plate, and 2 × 106 cells/well in 6‐well plate, respectively, for different treatment. FLL was added 24 h prior to and after the addition of NMDA and were present throughout the excitotoxic insult. The experimental protocol used in the present study was approved by the Animal Care and Use Committee of the Fourth Military Medical University.
Cell Viability Analysis
Neuronal cell viability was determined by MTT assay as described 16 with some modifications. Briefly, cortical neurons were cultured in 96‐well plates at 5 × 104 cells/well for 7 days before each treatment. For NMDA‐induced injury, cells were incubated with different concentrations of NMDA (0, 25, 50, 100, and 200 μM) for 30 min and then subjected to MTT assay. For FLL‐mediated protection assay, NPCs were pretreated with FLL (0, 0.01, 0.1, 1, and 10 μM) 24 h before subjected to NMDA (100 μM) stimulation for 30 min. At the end of each treatment, the culture medium was replaced with fresh medium containing 0.5 mg/ml MTT for 4 h at 37°C. After incubation, the medium was replaced by 150 μl/well dimethyl sulfoxide (DMSO) to resolve the formazan crystals. The optical density (OD) was read on a Universal Microplate Reader (Elx 800; Bio‐TEKinstruments Inc., Winooski, VT, USA) at 570 nm (630 nm as a reference). The data were expressed as a percent of control value and mean ± SEM of three experiments, and six wells were included in each group.
Hoechst/PI Double Staining
Apoptotic cell death was determined by PI and Hoechst 33258 double fluorescent staining as described previously 15. Neurons were cultured in 24‐well plates at a density of 600 cells/mm2. After the excitotoxic injury, the cells were stained with PI (1 μg/ml) and Hoechst 33258 (10 μg/ml) for 15 min and then fixed by 4% paraformaldehyde for 10 min. Cells were observed under a fluorescence microscope (Olympus BX61, Tokyo, Japan). The Hoechst and PI dye were excited at 340 and 620 nm, respectively. For each well, six visual fields were selected randomly.
Western Blot Analysis
To further explore the mechanisms involved in FLL‐mediated neuroprotection, we examined the effects of FLL on signaling pathways related to apoptosis by Western blot analysis. After each treatment, cells were rinsed twice with PBS and lysed by M‐PER protein extraction buffer containing 1 × protease inhibitor cocktail. Cell proteins were quantified by a BCA Kit, and equal amounts of protein (50 μg) were separated on 10% polyacrylamide gel followed by transferred onto an Immun‐Blot PDVF membrane. The membrane was blocked for 1 h with 5% non‐fat milk in Tris‐phosphate buffer containing 0.05% Tween 20 (TBS‐T). It was further incubated overnight at 4°C with primary antibodies including anti‐GluN2A (1:1000), anti‐GluN2B (1:1000), anti‐Bax (1:1000), anti‐Bcl‐2 (1:1000), and β‐actin (1:10,000) served as a loading control. After five washes with TBS‐T, membranes were further incubated with HRP‐conjugated secondary antibodies for 1–2 h and followed by four TBS‐T washes. The target protein signals were detected and digitized using ECL and Image J program.
Calcium Imaging
Calcium imaging was operated as described previously 17, 18. Neurons were cultured in 3.5 mm plates, which were made especially for laser scanning microscope at a density 3 × 105 per plate. Cultured cells were washed twice using Mg2+‐free extracellular solution (ECS) containing (in mM) NaCl 140, KCl 3, CaCl2 2, HEPES 10, glucose 10, adjusted to pH 7.2–7.3 with NaOH, and osmotic pressure 310 ± 5 with sucrose. The neurons were incubated with 2.5 μM fluo‐3/AM at 37°C for 30 min. Then the cultures were washed twice and returned to the original culture medium for additional 30 min. The dye‐loaded cells were measured for fluorescence with a confocal laser scanning microscope (Olympus). Prior to NMDA application, the dye‐loaded cells were scanned for about 1 min to obtain a basal level of intracellular Ca2+, then 100 μM NMDA was applied to the cultures, and equal amount of ECS was added as a control. FLL was added 24 h before the experiments and exists in the whole experiment process. The change in Ca2+ concentration was estimated by the fluorescence ratio of the fluo‐3/AM‐loaded neurons for another 4 min. The results expressed as the change from the basal level, and cells were picked out randomly for analysis from three independent experiments, and six wells included in each group.
Statistical Analysis
Data were expressed as mean ± SEM. A t‐test was performed for statistical comparisons, and one way ANOVA was used for comparison between multiple groups followed by Tukey's multiple comparison tests as a post hoc comparison. In all cases, P < 0.05 was considered statistically significant.
Results
Protective Effects of FLL on Cell Viability
It was observed that treatment of primary cultured cortical neurons with NMDA for 30 min induced a decrease in number of cell viability and an increase in apoptotic cells. To determine whether FLL protects neurons from injury, we evaluated the effect of FLL in cultured cortical neurons firstly. Neurons were exposed to an increasing concentration of NMDA (0, 25, 50, 100, 200 μM) for 30 min. NMDA decreased cell viability in a concentration‐dependent manner as measured by MTT assay (Figure 1A). An exposure to 100 μM NMDA for 30 min was used in subsequent experiments as cell insult was significant in this paradigm (cell viability in 100 μM NMDA‐treated: 68.4 ± 2.1%, P < 0.01 vs. control alone). Pretreatment with FLL at 1 μM for 24 h showed effective neuroprotection against NMDA injury (77.4 ± 6.1%, P < 0.05 vs. NMDA alone) as well as at 10 μM (85.2 ± 4.3%, P < 0.01 vs. NMDA alone, Figure 1B). Cytotoxicity evoked by NMDA was blocked by pretreatment with the NR2B‐selective antagonist Ro 25‐6981 (0.3 μM) (Figure 1B). FLL (0.01–10 μM) itself did not affect the cell viability (Figure 1B).
Figure 1.
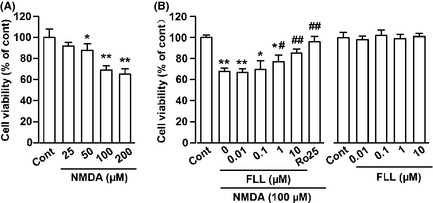
FLL promotes cell viability upon NMDA injury. (A) Dose‐dependent cytotoxic effects of NMDA on the cell viability of cortical neurons. Primary cultures of mouse neurons were treated with NMDA for 30 min, and cell viability was determined by MTT method. *P < 0.05, **P < 0.01 versus control. (B) Effects of FLL on the cell viability after exposure to NMDA. Cells were pretreated with FLL at different concentrations followed by exposure to 100 μM NMDA for 30 min. FLL alone did not change the cell viability. *P < 0.05, **P < 0.01 versus control; # P < 0.05, ## P < 0.01 versus NMDA alone.
Flax Lignan Protection of Neuronal Cells Against NMDA‐Induced Apoptosis
Hoechst 33258 and PI double staining were performed to further determine the neuroprotective effects of FLL. There was 7.9 ± 2.1% cells in control group that underwent apoptosis or cell death, whereas NMDA markedly induced apoptotic/necrotic cells in 35.1 ± 4.1% (P < 0.01 vs. control, Figure 2A,B). FLL (1 and 10 μM) significantly attenuated excitotoxicity of NMDA on cortical neurons. The percentage of cells undergoing apoptosis decreased to 23.4 ± 4.2% and 9.8 ± 6.1% respectively (P < 0.01 vs. NMDA alone; Figure 2A,B).
Figure 2.
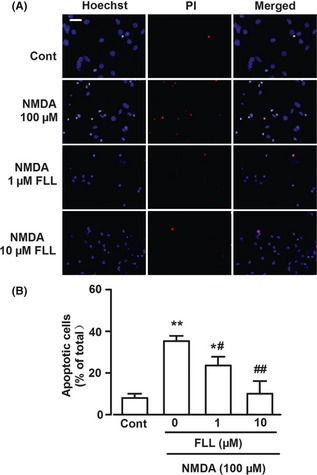
Hoechst 33258 and PI double staining in cultured cortical neurons. (A) Representative fluorescence images obtained after Hoechst 33258 and PI double staining in control, NMDA‐treated, and NMDA + FLL‐treated groups. Scale bar: 20 μm. (B) The percentage of apoptotic neurons in total neurons for control, NMDA‐treated, and NMDA + FLL‐treated groups. The cell numbers were counted from the Hoechst 33258 and PI staining in three independent observations. *P < 0.05, **P < 0.01 versus control group; # P < 0.05, ## P < 0.01 versus NMDA alone.
Effect of FLL on Apoptotic Proteins Expression
B‐cell lymphoma/leukemia‐2 (Bcl‐2) and Bcl‐2 associated X protein (Bax) expressions were determined to investigate the possible relation between NMDA‐induced cell death and potential intracellular mediators. Western blot analysis showed that B‐cell lymphoma / leukemia‐2 (Bcl‐2) and Bcl‐2 associated X protein (Bax) both were expressed in non‐injured cortical neurons, and FLL (10 μM) treatment alone did not alter the expression of these proteins (data not shown). NMDA stimulation significantly changed the expression of Bax (143.0 ± 8.6% of control, P < 0.05; Figure 3A,B) and Bcl‐2 (41.1 ± 6.6% of control, P < 0.01; Figure 3A,C), increasing the ratio of Bax/Bcl‐2 (3.48 ± 0.11 of control, P < 0.05; Figure 3D). These changes were significantly reversed by treatment of FLL (ratio of Bax/Bcl‐2, 1 μM: 2.40 ± 0.19, 10 μM 1.19 ± 0.10 of control, P < 0.05; Figure 3D). The effect of FLL on the Bax/Bcl‐2 ratio might constitute an important element responsible for neuroprotection.
Figure 3.
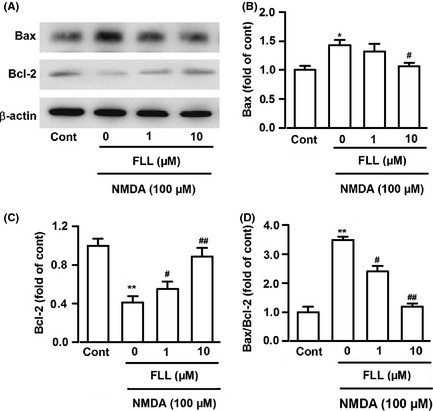
Effects of FLL on Bax and Bcl‐2 protein expression. (A) Representative Western blots showing expression of Bax and Bcl‐2 protein. (B‐D) The intensity of the bands was quantified by scanning densitometry, normalized with respect to endogenous β‐actin, and expressed as fold change. *P < 0.05, **P < 0.01 versus control group; # P < 0.05, ## P < 0.01 versus NMDA alone. Data were expressed as mean ± SEM of three independent experiments.
Effects of FLL on Expression of GluN2A‐and GluN 2B‐Containing NMDARs
GluN2A‐ and GluN2B‐containing NMDARs are linked to different intracellular cascades and participate in different functions in neuronal cell survival or death 19. Blockade of GluN2B‐containing NMDA receptor promotes neuronal survival, exerting a protective action against NMDA receptor–mediated neuronal damage 17, 19, 20. In contrast, activation of GluN2A‐containing NMDAR promotes neuronal survival and exerts neuroprotection against either NMDAR‐mediated or non‐NMDAR‐mediated neuronal damage 19. Western blot analysis was performed to detect effects of FLL on the expression of NMDAR subtypes in the primary cultures. FLL (10 μM) alone did not change the basal expression levels of GluN2A and GluN2B (data not shown). GluN2B subtype expression was notably increased in cultured cortical neuron after exposure to NMDA (301.3 ± 14.0% of control, P < 0.05; Figure 4A,B), while the GluN2A subtype expression was not changed notably (76.2% ± 21.1% of control; Figure 4A,B). Upregulation of GluN2B subtype by NMDA stimuli was significantly reduced in the presence of FLL (247.0 ± 14.1%, 182.0 ± 13.3%; P < 0.05 vs. NMDA alone; Figure 4A,B). However, FLL had no effects on the GluN2A subtype expression (101.0 ± 4.1%, 105.0 ± 11.1% of control; P > 0.05 vs. NMDA alone; Figure 4A,C). Thus, downregulated GluN2B subtype expression by FLL is, at least partly, responsible for the neuroprotective effects of FLL against NMDA‐evoked excitotoxic injury.
Figure 4.
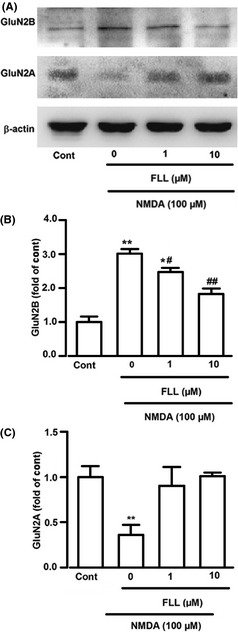
Effects of FLL on GluN2A and GluN2B expression. (A) Representative Western blots showing the expression level of GluN2A and GluN2B subtypes with different treatment. (B) The intensity of GluN2B subtypes level was normalized with endogenous β‐actin and expressed as fold change compared with control. **P < 0.01 versus control group; # P < 0.05, ## P < 0.01 versus NMDA alone. (C) The intensity of GluN2A subtypes level was normalized with endogenous β‐actin and expressed as fold change compared with control. Data were expressed as mean ± SEM of three independent experiments.
Pretreatment of FLL inhibited NMDA‐induced Ca2+ overload in cortical neurons
NMDAR activation increases cytoplasmic Ca2+ concentration in cultured neurons 21 and Ca2+ overload triggers multiple intracellular catabolic processes, followed by an irreversible death of neuronal cells in the brain 22. We, next, focused on the effect of FLL on the Ca2+ overload. The fluorescence intensity can be regarded as an indicator of cytoplasmic Ca2+ concentration 23. Ca2+ fluorescence in cultured neuron was stable during detection time (Figure 5A,B), and perfusion of FLL (10 μM) alone did not induce the change in Ca2+ fluorescence (Figure 5B). NMDA (100 μM) evoked a fast elevation of Ca2+ concentration in cultured neurons (Figure 5A,C). The amplitude and speed of Ca2+ concentration induced by NMDA perfusion were significantly decreased in the neurons with pretreatment of FLL (1 or 10 μM) for 24 h as compared to the control neurons (Figure 5A,C). To test whether FLL interacts with NMDA receptors and directly inhibits the calcium influx from the NMDA receptors, we perfused NMDA first to induce the calcium fluorescence and added the FLL 1 min later. We found that subsequent FLL (10 μM) did not change the calcium fluorescence intensity by NMDA any more (Figure 5D). This suggests that FLL does not interact directly with NMDA receptors and inhibit the calcium influx from NMDA receptors.
Figure 5.
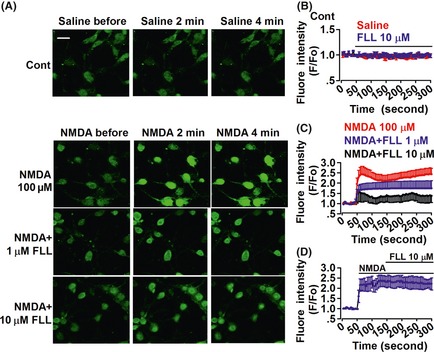
Effects of FLL on intracellular calcium overload. (A) Cultured neurons were loaded with fluo‐3, followed by successive perfusion of NMDA for 4 min, and fluorescence images were obtained; saline served as control. Scale bar: 20 μm. (B) Fluorescence intensity was normalized according to the fluorescence detected, and values represent the mean ± SEM in four separate experiments. Saline (n = 27 neurons) or FLL alone (n = 21 neurons) did not change the fluorescence intensity. (C) Fluorescence intensity induced by NMDA perfusion in the control (n = 32 neurons) and FLL‐pretreated neurons (n = 22 neurons in FLL 1 μM group; n = 25 neurons in FLL 10 μM group). (D) Subsequent FLL did not change the calcium fluorescence intensity by NMDA (n = 20 neurons).
Discussion
In this study, we demonstrated that Flax Lignan prevented the neuronal injury induced by NMDA stimuli and increased the cell viability, as showed by the results from the MTT assay, apoptotic staining, and Western blot analysis. The results suggest that the neuronal protection of Flax Lignan is correlated with the suppression of apoptosis induced by activation of GluN2B‐containing NMDAR and regulation of the Bcl‐2 family.
Glutamate is responsible for basal excitatory synaptic transmission and synaptic plasticity including long‐term potentiation and long‐term depression associated with cognitive processes 24. Excessive glutamate accumulation, however, induces neuronal death both in vitro and in vivo, and whether cells undergo apoptosis or necrosis depend on the dosage and duration of glutamate stimulation 25, 26. Most investigators agree that pathologic activation of subtype NMDAR contributes to neuronal death after acute excitotoxic trauma, such as brain ischemia and acts as the major mediator 27, 28. NMDAR consists of GluN2A subunit that promotes neuron protection, whereas GluN2B‐containing NMDAR mediates excitotoxicity 29. In the present study, the data showed that FLL reverses the upregulation of GluN2B induced by NMDA, implicating the neuroprotection of FLL is likely to antagonize a particular NMDAR subunit.
The key step in NMDA‐induced neuronal cell apoptosis is the overload of intracellular Ca2+, followed by overstimulation of NMDAR 30. Ca2+ overload triggers several downstream lethal reactions, including nitrosative stress, oxidative stress, and mitochondrial dysfunction 31. In this study, the elevation of Ca2+ stimulated by NMDA is inhibited by FLL in a dose‐dependent manner to support neuroprotection.
Glutamate evoked different intracellular cytotoxic signals; among these, Bcl‐2 family proteins play critical roles in apoptotic cell death 32. Bcl‐2 family proteins consist of anti‐ and proapoptotic families. Antiapoptotic protein, Bcl‐2, has been reported to inhibit caspases activation in cell apoptosis, whereas the proapoptotic protein, Bax, promotes cell apoptosis via translocation to the mitochondrial membrane as one of the major causes of some neurological disorders 33. Accordingly, the balance between Bcl‐2 and Bax determines the fate of survival or death of cells in response to cell insults 34. In this study, we found that FLL treatment increased the ratio of Bcl‐2/Bax in NMDA‐injured neurons, suggesting FLL rescued cortical neurons from cell apoptosis, possibly through regulation of apoptosis‐related proteins.
In summary, our results suggest that the neuroprotective effects of FLL are partially associated with the downregulation of GluN2B‐containing NMDAR. However, we could not exclude the possibility that FLL may take neuroprotective activities through other pathways. The results provide further insights into the neuroprotective functions of Flax Lignan. This may be helpful for the Traditional Chinese Medicine in the treatment of neurodegenerative disorders.
Conflict of Interest
The authors declare no conflict of interests.
Acknowledgments
This work was supported by National Natural Science Foundation of China No. 31070923.
The first two authors contributed equally to this work.
References
- 1. Tingley WG, Roche KW, Thompson AK, Huganir RL. Regulation of NMDA receptor phosphorylation by alternative splicing of the C‐terminal domain. Nature 1993;364:70–73. [DOI] [PubMed] [Google Scholar]
- 2. Wang C, Makela T, Hase T, Adlercreutz H, Kurzer MS. Lignans and flavonoids inhibit aromatase enzyme in human preadipocytes. J Steroid Biochem Mol Biol 1994;50:205–212. [DOI] [PubMed] [Google Scholar]
- 3. Kitts DD, Yuan YV, Wijewickreme AN, Thompson LU. Antioxidant activity of the flaxseed lignan secoisolariciresinol diglycoside and its mammalian lignan metabolites enterodiol and enterolactone. Mol Cell Biochem 1999;202:91–100. [DOI] [PubMed] [Google Scholar]
- 4. Newairy AS, Abdou HM. Protective role of flax lignans against lead acetate induced oxidative damage and hyperlipidemia in rats. Food Chem Toxicol 2009;47:813–818. [DOI] [PubMed] [Google Scholar]
- 5. Badanich KA, Doremus‐Fitzwater TL, Mulholland PJ, Randall PK, Delpire E, Becker HC. NR2B‐deficient mice are more sensitive to the locomotor stimulant and depressant effects of ethanol. Genes Brain Behav 2011;10:805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cao X, Cui Z, Feng R, et al. Maintenance of superior learning and memory function in NR2B transgenic mice during ageing. Eur J Neurosci 2007;25:1815–1822. [DOI] [PubMed] [Google Scholar]
- 7. Chang LR, Liu JP, Zhang N, Wang YJ, Gao XL, Wu Y. Different expression of NR2B and PSD‐95 in rat hippocampal subregions during postnatal development. Microsc Res Tech 2009;72:517–524. [DOI] [PubMed] [Google Scholar]
- 8. Cui Y, Jin J, Zhang X, et al. Forebrain NR2B overexpression facilitating the prefrontal cortex long‐term potentiation and enhancing working memory function in mice. PLoS ONE 2011;6:e20312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Day DE, Cooper MA, Markham CM, Huhman KL. NR2B subunit of the NMDA receptor in the basolateral amygdala is necessary for the acquisition of conditioned defeat in Syrian hamsters. Behav Brain Res 2011;217:55–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. D'Mello R, Marchand F, Pezet S, McMahon SB, Dickenson AH. Perturbing PSD‐95 interactions with NR2B‐subtype receptors attenuates spinal nociceptive plasticity and neuropathic pain. Mol Ther 2011;19:1780–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Farrant M, Feldmeyer D, Takahashi T, Cull‐Candy SG. NMDA‐receptor channel diversity in the developing cerebellum. Nature 1994;368:335–339. [DOI] [PubMed] [Google Scholar]
- 12. Gielen M, Siegler Retchless B, Mony L, Johnson JW, Paoletti P. Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature 2009;459:703–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Papadia S, Hardingham GE. The dichotomy of NMDA receptor signaling. Neuroscientist 2007;13:572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kew JN, Kemp JA. An allosteric interaction between the NMDA receptor polyamine and ifenprodil sites in rat cultured cortical neurones. J Physiol 1998;512(Pt 1):17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shi TY, Feng SF, Xing JH, et al. Neuroprotective effects of Salidroside and its analogue tyrosol galactoside against focal cerebral ischemia in vivo and H2O2‐induced neurotoxicity in vitro . Neurotox Res 2012;21:358–367. [DOI] [PubMed] [Google Scholar]
- 16. Yang Q, Yang ZF, Liu SB, et al. Neuroprotective effects of hydroxysafflor yellow A against excitotoxic neuronal death partially through down‐regulation of NR2B‐containing NMDA receptors. Neurochem Res 2010;35:1353–1360. [DOI] [PubMed] [Google Scholar]
- 17. Shen H, Yuan Y, Ding F, Liu J, Gu X. The protective effects of Achyranthes bidentata polypeptides against NMDA‐induced cell apoptosis in cultured hippocampal neurons through differential modulation of NR2A‐ and NR2B‐containing NMDA receptors. Brain Res Bull 2008;77:274–281. [DOI] [PubMed] [Google Scholar]
- 18. Nishimura Y, Yamaguchi JY, Kanada A, et al. Increase in intracellular Cd(2+) concentration of rat cerebellar granule neurons incubated with cadmium chloride: cadmium cytotoxicity under external Ca(2+)‐free condition. Toxicol In Vitro 2006;20:211–216. [DOI] [PubMed] [Google Scholar]
- 19. Liu Y, Wong TP, Aarts M, et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo . J Neurosci 2007;27:2846–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters CA, Andrews SB. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc Natl Acad Sci USA 2009;106:9854–9859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA‐receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature 1986;321:519–522. [DOI] [PubMed] [Google Scholar]
- 22. Liu S, Lau L, Wei J, et al. Expression of Ca(2+)‐permeable AMPA receptor channels primes cell death in transient forebrain ischemia. Neuron 2004;43:43–55. [DOI] [PubMed] [Google Scholar]
- 23. Matsumoto Y, Yamamoto S, Suzuki Y, Tsuboi T, Terakawa S, Ohashi N, Umemura K. Na+/H+ exchanger inhibitor, SM‐20220, is protective against excitotoxicity in cultured cortical neurons. Stroke 2004;35:185–190. [DOI] [PubMed] [Google Scholar]
- 24. Mark LP, Prost RW, Ulmer JL, et al. Pictorial review of glutamate excitotoxicity: fundamental concepts for neuroimaging. AJNR Am J Neuroradiol 2001;22:1813–1824. [PMC free article] [PubMed] [Google Scholar]
- 25. Chen X, Liu J, Gu X, Ding F. Salidroside attenuates glutamate‐induced apoptotic cell death in primary cultured hippocampal neurons of rats. Brain Res 2008;1238:189–198. [DOI] [PubMed] [Google Scholar]
- 26. Li N, Liu B, Dluzen DE, Jin Y. Protective effects of ginsenoside Rg2 against glutamate‐induced neurotoxicity in PC12 cells. J Ethnopharmacol 2007;111:458–463. [DOI] [PubMed] [Google Scholar]
- 27. Arundine M, Tymianski M. Molecular mechanisms of glutamate‐dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci 2004;61:657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guo H, Barrett TM, Zhong Z, Fernandez JA, Griffin JH, Freeman RS, Zlokovic BV. Protein S blocks the extrinsic apoptotic cascade in tissue plasminogen activator/N‐methyl D‐aspartate‐treated neurons via Tyro3‐Akt‐FKHRL1 signaling pathway. Mol Neurodegener 2011;6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cho SI, Park UJ, Chung JM, Gwag BJ. Neu2000, an NR2B‐selective, moderate NMDA receptor antagonist and potent spin trapping molecule for stroke. Drug News Perspect 2010;23:549–556. [DOI] [PubMed] [Google Scholar]
- 30. Sattler R, Tymianski M. Molecular mechanisms of calcium‐dependent excitotoxicity. J Mol Med (Berl) 2000;78:3–13. [DOI] [PubMed] [Google Scholar]
- 31. Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr 2000;130:1007S–1015S. [DOI] [PubMed] [Google Scholar]
- 32. Zeng KW, Wang XM, Ko H, Kwon HC, Cha JW, Yang HO. Hyperoside protects primary rat cortical neurons from neurotoxicity induced by amyloid beta‐protein via the PI3K/Akt/Bad/Bcl(XL)‐regulated mitochondrial apoptotic pathway. Eur J Pharmacol 2011;672:45–55. [DOI] [PubMed] [Google Scholar]
- 33. Vila M, Jackson‐Lewis V, Vukosavic S, et al. Bax ablation prevents dopaminergic neurodegeneration in the 1‐methyl‐ 4‐phenyl‐1,2,3,6‐tetrahydropyridine mouse model of Parkinson's disease. Proc Natl Acad Sci USA 2001;98:2837–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zha H, Reed JC. Heterodimerization‐independent functions of cell death regulatory proteins Bax and Bcl‐2 in yeast and mammalian cells. J Biol Chem 1997;272:31482–31488. [DOI] [PubMed] [Google Scholar]