

Summary
Aim
Cerebral ischemic postconditioning has emerged recently as a kind of endogenous strategy for neuroprotection. We set out to test whether hypoxia or glucose deprivation (GD) would substitute for ischemia in postconditioning.
Methods
Adult male C57BL/6J mice were treated with postconditioning evoked by ischemia (bilateral common carotid arteries occlusion) or hypoxia (8% O 2) after 45‐min middle cerebral arterial occlusion. Corticostriatal slices from mice were subjected to 1‐min oxygen‐glucose deprivation (OGD), GD, or oxygen deprivation (OD) postconditioning at 5 min after 15‐min OGD.
Results
Hypoxic postconditioning did not decrease infarct volume or improve neurologic function at 24 h after reperfusion, while ischemic postconditioning did. Similarly, OGD and GD but not OD postconditioning attenuated the OGD/reperfusion‐induced injury in corticostriatal slices. The effective duration of low‐glucose (1 mmol/L) postconditioning was longer than that of OGD postconditioning. Moreover, OGD and GD but not OD postconditioning reversed the changes of glutamate, GABA, glutamate transporter‐1 protein expression, and glutamine synthetase activity induced by OGD/reperfusion.
Conclusions
These results suggest that the transient lack of glucose but not oxygen plays a key role in ischemic postconditioning‐induced neuroprotection, at least partly by regulating glutamate metabolism. Low‐glucose postconditioning might be a clinically safe and feasible therapeutic approach against cerebral ischemia/reperfusion injury.
Keywords: Glucose deprivation postconditioning, Hypoxic postconditioning, Ischemic postconditioning
Introduction
Over the past two decades, a variety of clinical trials of pharmacological neuroprotective strategies in stroke have been disappointing, and attention has turned to the brain's own endogenous strategies for neuroprotection, such as preconditioning 1. The unpredictable occurrence of stroke, however, greatly limits the clinical application of preconditioning. Ischemic postconditioning, another endogenous neuroprotective strategy, is attracting attention. In 2006, Zhao et al. 2 first reported that ischemic postconditioning reduces infarct volume in experimental cerebral ischemia/reperfusion injury. After that, its cerebral protective effects have been confirmed in several distinct models of ischemia 3, 4, 5, 6. Considering the unpredictability of cerebral ischemia, postconditioning would provide more therapeutic opportunities than preconditioning.
Hypoxia and glucose deprivation (GD) are two key factors in ischemia. We proposed to substitute hypoxia or GD for ischemia in postconditioning. In theory, if hypoxia or GD postconditioning alone is neuroprotective, it may be safer than ischemic postconditioning in clinical application. So far, however, little is known about whether and how hypoxia or GD postconditioning alone produces neuroprotection against ischemic injury. Serviddio et al. 7 found that brief hypoxic postconditioning before normoxic reperfusion protects the heart against ischemia‐reperfusion injury. Recently, the existence of delayed hypoxic postconditioning has also been described in brains. A late application of hypoxia (5 days) after transient middle cerebral artery occlusion (MCAO) reduces delayed thalamic atrophy, although its action is limited 8. Similarly, Zhan et al. 9 also demonstrated the neuroprotection of delayed hypoxic postconditioning against transient global cerebral ischemia. In contrast, exposure to hyperoxia during reperfusion after hypoxic‐ischemic brain injury increases secondary neural injury and ultimately impairs functional recovery 10. However, the effect of rapid hypoxic postconditioning which is applied at the beginning of reperfusion has not been demonstrated in brains.
On the other hand, Els et al. 11 reported that hyperglycemia in patients with focal middle cerebral artery (MCA) ischemia can cause a worse clinical outcome despite the recanalization of the occluded vessel by thrombolysis therapy. And in animal models, during acute focal and global ischemia, insulin therapy which lowers blood glucose levels reduces ischemic brain damage and can be neuroprotective 12, 13. These results suggest that it is important to control glucose levels after reperfusion for neuroprotection. However, whether GD postconditioning is neuroprotective or not is still unknown. We hypothesize that the transient lack of glucose in ischemic postconditioning may play a role in neuroprotection.
Materials and Methods
Ethics Statement
Full details of the study have been approved by the Zhejiang University Animal Experimentation Committee; the approval number is Zju2009‐1‐01‐005. All experiments were carried out in accordance with the ethical guidelines of the Zhejiang University Animal Experimentation Committee and were in complete compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Every effort was made to minimize any pain or discomfort, and the minimum number of animals was used.
Animals
Male adult C57BL/6 mice (n = 120) weighing 22–25 g were used in vivo and in vitro.
Transient Focal Cerebral Ischemia
Mice were anaesthetized with an intraperitoneal injection of sodium pentobarbital (45 mg/kg). Transient focal cerebral ischemia was induced by MCAO as previously described 14. Briefly, a 6‐0 nylon monofilament suture, blunted at the tip and coated with 1% poly‐l‐lysine, was advanced 10 mm into the internal carotid to occlude the origin of the MCA. Reperfusion was allowed after 45 min by monofilament removal. Achievement of ischemia was confirmed by monitoring regional cerebral blood flow by laser Doppler flowmetry (Model moorVMS‐LDF2; Moor Instruments Ltd., Axminster, UK). Animals with less than 80% reduction in cerebral blood flow in the core regions of the MCA territory were excluded from the study. Body temperature was maintained at 37°C by a heat lamp (FHC, Bowdoinham, ME, USA) during surgery and for 2 h after the start of reperfusion.
Neurological deficit scores were evaluated as previously described 15 at 24 h of reperfusion as follows: 0, no deficit; 1, flexion of the contralateral forelimb on lifting of the whole animal by the tail; 2, circling to the contralateral side; 3, falling to contralateral side; and 4, no spontaneous motor activity.
Infarct volume was determined 24 h after MCAO. The brains were quickly removed, sectioned coronally at 2‐mm intervals, and stained by immersion in the vital dye 2,3,5‐triphenyltetrazolium hydrochloride (TTC; 0.25%) at 37°C for 30 min. Extents of the normal and infracted areas were analyzed using the ImageJ software (National Institutes of Health, Bethesda, MD, USA) and determined by the indirect method, which corrects for edema (contralateral hemisphere volume minus nonischemic ipsilateral hemisphere volume). The percentage of the corrected infarct volume was calculated by dividing the infarct volume by the total contralateral hemispheric volume, and this ratio was then multiplied by 100.
Ischemic and Hypoxic Postconditioning In Vivo
For ischemic preconditioning, bilateral common carotid arteries (CCA) were transiently occluded for 5 min at 10 min after reperfusion using aneurysm clips. For hypoxic postconditioning, the mice were maintained by inhaling 8% O2 (92% N2). According to the design of ischemic postconditioning in the previous studies 3, 4, to characterize the temporal profile of hypoxic postconditioning, animals were assigned to six groups randomly (see Figure 1A), as following: (1) the control group: MCA was occluded for 45 min; (2) the first postconditioning group: after 45 min of MCAO, reperfusion was established for 30 s after which mice were subjected to 30‐s hypoxia followed by another four cycles of 30‐s/30‐s reoxygenation/hypoxia (total of five cycles); (3) the second postconditioning group: after 45 min of MCAO, mice were subjected to five cycles of 1‐min/1‐min reoxygenation/hypoxia; (4) the third postconditioning group: after 45 min of MCAO, mice were subjected to five cycles of 2‐min/2‐min reoxygenation/hypoxia; (5) the fourth postconditioning group: after 45 min of MCAO, mice were subjected to a cycle of 10‐min/10‐min reoxygenation/hypoxia; and (6) the fifth postconditioning group: after 45 min of MCAO, mice were subjected to a cycle of 30‐min/30‐min reoxygenation/hypoxia. All the animals were killed at 24 h after reperfusion.
Figure 1.
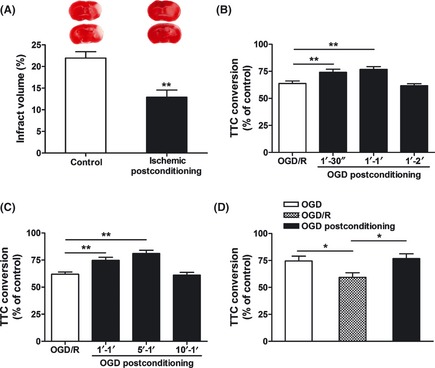
Ischemic postconditioning model in vivo and in vitro. (A) In control group, adult male mice were subjected to 45‐min middle cerebral artery occlusion (MCAO). For ischemic postconditioning group, bilateral common carotid arteries (CCA) were occluded for 5 min at 10 min after reperfusion. Infarct volume was quantified by TTC staining at 24 h after reperfusion. The upper panel shows the representative slices from each group. Data are expressed as percentage of infarct volume (n = 5). (B) Time‐course of oxygen‐glucose deprivation (OGD) postconditioning (an in vitro ischemic postconditioning model). Corticostriatal slices from adult male mice were postconditioned with OGD for 30 s, 1 min or 2 min at 1 min after 15 min of OGD. (C) Time windows of OGD postconditioning. Corticostriatal slices were postconditioned with OGD for 1 min at 1, 5, or 10 min after 15 min of OGD. (D) Comparison of corticostriatal slice injury among the acute OGD group without reperfusion, OGD/reperfusion group and OGD postconditioning group (OGD postconditioning was applied for 1 min at 5 min after 15 min of OGD). Slice injury was quantified by TTC conversion at 1 h after reperfusion. Data are expressed as percentage of TTC conversion compared with control (n = 4–5). Bars represent mean ± SEM, *P < 0.05, **P < 0.01. OGD/R, OGD/reperfusion.
Brain Slice Preparation
Mice were anesthetized with ether and then decapitated. The brains were rapidly removed and placed in ice‐cold artificial cerebrospinal fluid (ACSF; in mmol/L, 124 NaCl, 5 KCl, 1.25 KH2PO4, 2 MgSO4, 26 NaHCO3, 2 CaCl2, 10 glucose, pH 7.4) bubbled with 5% CO2 and 95% O2. The corticostriatal slices (400 μm thick) were prepared using a vibratome (World Precision Instruments, Sarasota, FL, USA) in ice‐cold ACSF bubbled with 5% CO2 and 95% O2. Slices were preincubated on a nylon net in the wells of an interface style holding chamber thoroughly wetted with ACSF at 25°C, and gassed with warmed humidified 5% CO2 and 95% O2. After incubation for 1 h, the slices were incubated at 37°C for 20 min before they were used for experiments.
Oxygen‐glucose Deprivation In Vitro
Ischemia was simulated in vitro by oxygen‐glucose deprivation (OGD). As described previously 16, slices were transferred into an incubator containing glucose‐free ACSF at 37°C bubbled with 5% CO2 and 95% N2. Bubbling with these gases for 30 min before the placement of slices reduced the oxygen content in the solution. OGD was maintained for 15 min. The control slices were incubated in normal ACSF bubbled with 5% CO2 and 95% O2. After the OGD, the slices were allowed to recover in oxygenated ACSF at 37°C for 1 h. Slice injury was assessed by TTC (0.25%) conversion at 37°C for 30 min. Formazan in slices was extracted by 500 μL mixture of ethanol/dimethylsulfoxide (1:1), and absorbance was detected at 490 nm. Viability was determined by absorbance normalized by the dry weight of the slice and expressed as the percentage of control slices included in each experiment.
Postconditioning and Study Groups of Brain Slices
For time‐course experiments, 30‐s, 1‐min, or 2‐min OGD postconditioning was applied at 1 min after 15 min of OGD. For time‐window experiments, slices were postconditioned with OGD for 1 min at 1, 5, or 10 min after 15 min of OGD. In the GD postconditioning group, the slices were postconditioned with GD (5% CO2 and 95% O2, incubating in glucose‐free ACSF) for 1 min at 5 min after 15 min of OGD. In the oxygen deprivation (OD) postconditioning group, the slices were postconditioned with hypoxia (5% CO2 and 95% N2, incubating in ACSF) for 1 min at 5 min after 15 min of OGD. For the low‐glucose concentration‐response study, 0, 1, 2, or 5 mmol/L glucose postconditioning was applied for 1 min at 5 min after 15 min of OGD. For the low‐glucose time‐course experiments, 1, 5, or 30 min of 1 mmol/L glucose postconditioning was applied at 5 min after 15 min of OGD, and 5 min of OGD postconditioning was applied at the same time point simultaneously.
HPLC Determination of Glutamate and GABA Concentrations
At 1 h after reperfusion, the corticostriatal slices were homogenized with 0.4 mol/L perchloric solution, centrifuged at 15,000 g for 20 min at 4°C, and the supernatant was collected. Analysis of glutamate and GABA in each sample was performed by high‐performance liquid chromatography (HPLC) as we described previously 17, 18. The HPLC was controlled, and the data acquired and analyzed using CoulArray® software (ESA, Chelmsford, MA, USA). All of the equipment was from ESA (CoulArray software).
Immunoblotting and Glutamine Synthetase Activity Analysis
At 1 h after reperfusion, the corticostriatal slices were homogenized, and the total proteins were purified using cell and tissue protein extraction reagents according to the manufacturer's instructions (KC‐415; KangChen, Shanghai, China). Forty‐five micrograms protein equivalent of each sample was electrophoresed on polyacrylamide gel, transferred onto nitrocellulose, and probed with monoclonal antiglutamate transporter‐1 (GLT‐1) antibodies (1:500; Cell Signaling, Beverly, MA, USA) or monoclonal antiglutamine synthetase (GS) antibodies (1:800; Millipore, Billerica, CA, USA) followed by IRDye 800‐coupled antirabbit IgG (1:10,000; LI‐COR Biosciences, Lincoln, NE, USA) or IRDye 700‐coupled antimouse IgG (1:10,000; LI‐COR Biosciences) secondary antibodies. Antitubulin antibody (1:300; Boster, Wuhan, China) was used as the control. Blots were visualized using an Odyssey infrared imaging system (LI‐COR Biosciences) and analyzed using Odyssey software. Relative optical densities were obtained by comparing measured values with the mean values from the control group. GS activity was assayed by a GS activity kit (Jiancheng, Nanjing, China) according to the manufacturer's instructions.
Statistical Analysis
All data were collected and analyzed in a blind fashion. Data are presented as mean ± SEM. One‐way ANOVA (analysis of variance) with least significant difference (LSD) or Dunnett's T3 post‐hoc test (in which equal variances were not assumed) was applied for multiple comparisons, whereas Student's t‐test was used for comparisons between two groups. Neurologic deficit scores were analyzed with the nonparametric Mann–Whitney U‐test. P < 0.05 was considered statistically significant.
Results
Neuroprotective Effects of Ischemic Postconditioning In Vivo and In Vitro
To determine whether ischemic postconditioning protects the brain against MCAO/reperfusion‐induced injury in mice, we subjected animals to postconditioning with 5 min of bilateral CCA occlusion at 10 min after reperfusion. Forty‐five minutes of MCAO in control group resulted in a significant infarct at 24 h after reperfusion, as determined by TTC staining (Figure 1A). Ischemic postconditioning significantly reduced infarct volume by 41.22% (P < 0.01 vs. control group). Similarly, postconditioned animals exhibited lower neurologic deficit scores at 24 h after reperfusion relative to control animals (P < 0.05, data not shown).
Furthermore, an in vitro ischemic postconditioning model was established in corticostriatal slices from adult male mice. Exposure of the slices to 15‐min OGD produced a significant injury, as determined by TTC conversion, at 1 h after reperfusion. For OGD postconditioning, we subjected the slices to 30 s, 1 min, or 2 min of OGD at 1 min after reperfusion (Figure 1B). Both the 30‐s and 1‐min treatments significantly increased the viability of brain slices (P < 0.01). But postconditioning evoked with the longer duration (2 min) of OGD failed to induce neuroprotection. Then, postconditioning evoked with 1 min of OGD conducted at 1 or 5 min after reperfusion reduced the slice injury caused by 15‐min OGD in a time‐dependent manner (P < 0.01; Figure 1C). However, with 1‐min OGD initiated at 10 min after reperfusion, postconditioning did not block the injury of slices. The effective time window and duration of postconditioning in brain slices were shorter than that in vivo, and the differences may be partly attributed to the different tolerance to ischemia/reperfusion injury between in vivo and in vitro models. In addition, postconditioning with 1‐min OGD initiated at 5 min after reperfusion retained slice viability to the level of 15‐min OGD without reperfusion, which implies that this postconditioning procedure almost completely prevented the reperfusion process‐induced injury (Figure 1D).
Hypoxic Postconditioning did not Decrease Infarct Volume or Neurological Deficit Scores after Transient MCAO
To determine the effect of hypoxic postconditioning, mice were subjected to hypoxia (8% O2) with various permutations after transient MCAO (Figure 2A). Five cycles of 30‐s/30‐s and 1‐min/1‐min reoxygenation/hypoxia had no effect on infarct volume compared with the control group (Figure 2B), whereas five cycles of 2‐min/2‐min reoxygenation/hypoxia and one cycle of 10‐min/10‐min or 30‐min/30‐min reoxygenation/hypoxia increased infarct volume (P < 0.05 vs. control group). Similarly, the neurological deficit scores also increased after one cycle of 10‐min/10‐min or 30‐min/30‐min reoxygenation/hypoxia (P < 0.05 vs. control group, Figure 2C). And the neurological deficit scores of the other three hypoxic postconditioning groups did not significantly change compared with control group.
Figure 2.

Effects of hypoxic postconditioning on infarct volume and neurologic deficit scores at 24 h after transient middle cerebral artery occlusion (MCAO). (A) Experimental groups and protocols for hypoxic postconditioning studies. The middle cerebral artery (MCA) was occluded for 45 min, and reperfusion lasted for 24 h; Post 30″/30″ indicates five cycles of 30‐s/30‐s reoxygenation/hypoxia; Post 1′/1′, five cycles of 1‐min/1‐min reoxygenation/hypoxia; Post 2′/2′, five cycles of 2‐min/2‐min reoxygenation/hypoxia; Post 10′/10′, a cycle of 10‐min/10‐min reoxygenation/hypoxia; Post 30′/30′, a cycle of 30‐min/30‐min reoxygenation/hypoxia. (B) Quantification of infarct volume by TTC staining after various permutations of ischemia and hypoxic postconditioning. Data are expressed as percentage of infarct volume. (C) Quantification of neurologic deficit scores. Bars represent mean ± SEM (n = 6–8). *P < 0.05 vs. control group.
GD but not OD Postconditioning Protected Corticostriatal Slices Against OGD/Reperfusion‐induced Injury
To compare the effects of OGD, GD, and OD postconditioning, we subjected corticostriatal slices to 1 min of OGD, GD, or OD postconditioning at 5 min after 15‐min OGD. Both OGD and GD postconditioning produced significant neuroprotection (P < 0.01, Figure 3A), while no beneficial effect was observed when OD postconditioning was applied. In addition, we tested the effects of different concentrations of low‐glucose postconditioning (Figure 3B). Postconditioning with 1 or 2 mmol/L glucose for 1 min at 5 min after reperfusion also reversed the slice injury (P < 0.01), and the beneficial effect was comparable with that of GD postconditioning. However, 5 mmol/L glucose postconditioning with the same procedure had no effect. In addition, the effective duration of 1 mmol/L glucose postconditioning applied at 5 min after reperfusion was as long as 5 min (P < 0.01), but 5 min of OGD postconditioning had no effect on slices injury (Figure 3C).
Figure 3.
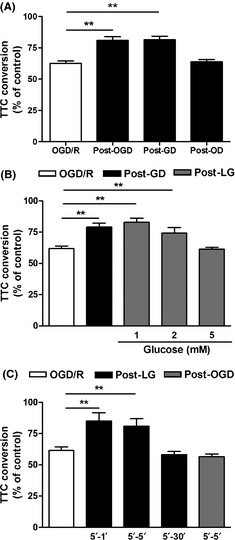
Effects of postconditioning with oxygen‐glucose deprivation (OGD), glucose deprivation (GD), oxygen deprivation (OD) and low glucose on OGD/reperfusion‐induced injury. (A) Corticostriatal slices were postconditioned with OGD, GD or OD for 1 min at 5 min after 15 min of OGD, and 1 h later slice injury was assessed by TTC conversion. (B) The concentration response of low‐glucose postconditioning. Zero, 1, 2 or 5 mmol/L glucose postconditioning was applied for 1 min at 5 min after 15 min of OGD. (C) The time‐course of low‐glucose postconditioning. One, 5, or 30‐min of 1 mmol/L glucose postconditioning was applied at 5 min after 15 min of OGD, and 5 min of OGD postconditioning was applied at the same time point simultaneously. Data are expressed as percentage of TTC conversion compared with control. Bars represent the mean ± SEM (n = 4–5), **P < 0.01. OGD/R, OGD/reperfusion; Post‐OGD, OGD postconditioning; Post‐GD, GD postconditioning; Post‐LG, low‐glucose postconditioning; Post‐OD, OD postconditioning.
Effects of Postconditioning Evoked by OGD, GD and OD on Glutamate and GABA Levels, GLT‐1 and GS Expression, and GS Activity After OGD/Reperfusion in Corticostriatal Slices
In the early stage of reperfusion, the excitatory neurotransmitter glutamate and the inhibitory neurotransmitter GABA are involved in regulating ischemia/reperfusion‐induced injury 19, 20. Here, we determined the effects of postconditioning evoked by OGD, GD and OD on glutamate and GABA levels after OGD/reperfusion. Fifteen‐min OGD followed by 1‐h reperfusion resulted in an increase of glutamate (Figure 4A) and a decrease of GABA (Figure 4B) in corticostriatal slices. Postconditioning evoked by OGD and GD abolished the increase of glutamate (P < 0.01) and the decrease of GABA (P < 0.05) induced by OGD/reperfusion. However, OD postconditioning had no effects on glutamate and GABA levels.
Figure 4.

Effects of postconditioning with oxygen‐glucose deprivation (OGD), glucose deprivation (GD) and oxygen deprivation (OD) on glutamate metabolism and GABA level. Corticostriatal slices were postconditioned with OGD, GD or OD for 1 min at 5 min after 15 min of OGD, and glutamate (A) and GABA (B) levels, GLT‐1 (C) and glutamine synthetase (GS) (D) protein expression and GS activity (E) were assayed at 1 h after reperfusion. Bars represent mean ± SEM (n = 3–4), *P < 0.05, **P < 0.01. OGD/R, OGD/reperfusion; Post‐OGD, OGD postconditioning; Post‐GD, GD postconditioning; Post‐OD, OD postconditioning.
Furthermore, GLT‐1 and GS specifically expressed in astrocytes are two major mediators for maintaining glutamate and GABA levels and are involved in regulating the ischemia‐induced excitotoxicity 21. GLT‐1 expression (Figure 4C) and GS activity (Figure 4E) of the slices were significantly down‐regulated at 1 h after reperfusion, but GS expression did not change (Figure 4D). Postconditioning evoked by OGD and GD, but not OD significantly reversed OGD/reperfusion‐induced down‐regulation of GLT‐1 expression and GS activity. There was no difference among the five groups in GS expression (Figure 4D).
Discussion
In the present study, we provided the first evidence that GD postconditioning inhibited the injury induced by OGD/reperfusion in cultured corticostriatal slices. This effect was comparable to that of OGD postconditioning with the same procedure. However, the beneficial effect was not observed when OD postconditioning was applied. This phenomenon was also verified in cerebral ischemia in vivo. Application of hypoxic postconditioning in the early period of reperfusion did not produce neuroprotection and even exacerbated the injury after ischemia. These results suggest that the transient lack of glucose but not oxygen in ischemic postconditioning is a key factor inducing neuroprotection during the early phase after reperfusion.
Despite the fact that reperfusion halts the process of ischemic cell death, it still leads to further cell death in the early stage. Hence, targeting cell death due to reperfusion has the potential to maximize cell salvage 22. Ischemic postconditioning, as a novel neuroprotective strategy, does limit the extent of reperfusion injury and maximizes reperfusion salvage, which has been demonstrated in several distinct models of brain ischemia 3, 4, 5, 6. In this study, we further confirmed the neuroprotection of ischemic postconditioning against MCAO/reperfusion‐induced injury in mice (Figure 1A). In addition, we established an in vitro ischemic postconditioning model in cultured corticostriatal slices from adult male mice. Postconditioning with 30 s or 1 min of OGD conducted at 1 min after reperfusion reduced slice death caused by OGD/reperfusion. However, with a longer duration of OGD postconditioning (2 min), the slice survival rate was not improved. One‐minute OGD postconditioning initiated at 1 or 5 min, but not >10 min after reperfusion‐induced robust neuroprotection. So, these results indicate that the onset time and duration of postconditioning are critical for generating neuroprotection. Compared with acute OGD injury and OGD/reperfusion injury, OGD postconditioning completely reversed the reperfusion process‐induced injury, which implies that ischemic postconditioning can induce a robust neuroprotection which inhibits progressive injury after reperfusion.
It has been demonstrated that both rapid and delayed hypoxic preconditioning offer neuroprotection 14, 23, 24. We hypothesized that rapid hypoxic postconditioning may also induce neuroprotection, just like ischemic postconditioning. Based on the OGD postconditioning model, we compared the effects of postconditioning with OGD and OD alone (Figure 3A). Surprisingly, although OGD postconditioning was significantly neuroprotective, the beneficial effect was not observed when OD postconditioning was applied. It is likely that the mechanisms of rapid hypoxic preconditioning and postconditioning are different. This phenomenon was also verified in cerebral ischemia in vivo (Figure 2). Application of hypoxic postconditioning in the early period of reperfusion did not improve neurological function or decrease infarct volume, and even exacerbated the injury after ischemia if the duration of hypoxic postconditioning was longer than 10 min. Therefore, these results suggest that, different from hypoxic preconditioning, transient hypoxia in ischemic postconditioning may not be a key factor inducing neuroprotection during the early phase after reperfusion. However, Leconte et al. 8 reported that a late application of hypoxia starting 5 days but not 1 day after transient MCAO reduces injury. This conflict may be attributed to the different onset times of hypoxic postconditioning, because in our study hypoxic postconditioning was applied rapidly within minutes after reperfusion. So, hypoxic postconditioning applied at different phases (early or delayed) may play different roles in cerebral ischemia/reperfusion injury. In addition, the neuroprotection induced by delayed hypoxic postconditioning is limited. Only delayed thalamic atrophy but not cortical or striatal damage is inhibited 8. Therefore, whether rapid or delayed, hypoxic postconditioning may be difficult to apply for treating focal cerebral ischemic/reperfusion‐induced injury.
Furthermore, we were interested to find that GD postconditioning‐induced neuroprotection was comparable with the effect of OGD postconditioning, which suggests that the transient GD in ischemic postconditioning may play a key role in inducing neuroprotection. Not only GD but also low‐glucose (1 or 2 mmol/L) postconditioning‐induced neuroprotection. These results offer the possibility for the clinical application of low‐glucose postconditioning, because complete deprivation of blood glucose is impossible. In addition, the effective duration of low‐glucose postconditioning was as long as 5 min, while 5 min of OGD postconditioning had no effect on slice injury. Therefore, the effective range of low‐glucose postconditioning duration is much wider than that of OGD postconditioning. It is likely that transiently maintaining blood glucose at a low level after thrombolysis or reperfusion is a potential and alternative therapeutic approach instead of ischemic postconditioning. Considering that it is hard to maintain low blood glucose level within minutes at the beginning of reperfusion in mice and insulin itself has direct neuroprotection besides down‐regulating blood glucose 25, in the present study, a low‐glucose postconditioning model in vivo was absent.
In addition, we found that postconditioning evoked by GD or OGD but not OD abolished the increase of glutamate and the decrease of GABA induced by OGD/reperfusion. GLT‐1 and GS are specifically expressed in astrocytes and are important for glutamate metabolism and GABA synthesis 26, 27. Both are down‐regulated by cerebral ischemia, which further aggravates the ischemic injury 28, 29, 30. Interestingly, we further found that both GD and OGD postconditioning reversed the OGD/reperfusion‐induced down‐regulation of GLT‐1 expression and GS activity. These results imply that the improved glutamate metabolism might be involved in postconditioning‐induced neuroprotection through maintaining GLT‐1 expression and GS activity in astrocytes. However, the beneficial effects were not observed when OD postconditioning was applied in the present study. Therefore, GD but not OD in ischemic postconditioning is involved in maintaining GLT‐1 expression and GS activity in astrocytes, which might further regulate glutamate and GABA levels and injury induced by ischemia/reperfusion.
Overall, the results of this study suggest that transient lack of glucose but not oxygen in ischemic postconditioning is a key factor for inducing neuroprotection. Regulating glutamate metabolism might be involved in this beneficial effect, although the underlying mechanisms deserve to be further examined. Low‐glucose postconditioning is effective for a longer duration than ischemic postconditioning, which means it might be a clinically safe and feasible therapeutic approach against cerebral ischemia/reperfusion injury.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was funded by the National Basic Research of China 973 Program (2011CB504403), the National Natural Science Foundation of China (81030061, 81273506, 81202520, 81102429), and the Fundamental Research Funds for the Central Universities (2012FZA7002). We are grateful to Dr. Iain C. Bruce for reading the manuscript.
The first three authors contributed equally to this work.
References
- 1. Pignataro G, Scorziello A, Di Renzo G, Annunziato L. Post‐ischemic brain damage: Effect of ischemic preconditioning and postconditioning and identification of potential candidates for stroke therapy. FEBS J 2009;276:46–57. [DOI] [PubMed] [Google Scholar]
- 2. Zhao H, Sapolsky RM, Steinberg GK. Interrupting reperfusion as a stroke therapy: Ischemic postconditioning reduces infarct size after focal ischemia in rats. J Cereb Blood Flow Metab 2006;26:1114–1121. [DOI] [PubMed] [Google Scholar]
- 3. Xing B, Chen H, Zhang M, et al. Ischemic postconditioning inhibits apoptosis after focal cerebral ischemia/reperfusion injury in the rat. Stroke 2008;39:2362–2369. [DOI] [PubMed] [Google Scholar]
- 4. Pignataro G, Meller R, Inoue K, et al. In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: Ischemic postconditioning. J Cereb Blood Flow Metab 2008;28:232–241. [DOI] [PubMed] [Google Scholar]
- 5. Gao XW, Ren CC, Zhao H. Protective effects of ischemic postconditioning compared with gradual reperfusion or preconditioning. J Neurosci Res 2008;86:2505–2511. [DOI] [PubMed] [Google Scholar]
- 6. Wang JY, Shen J, Gao Q, et al. Ischemic postconditioning protects against global cerebral ischemia/reperfusion‐induced injury in rats. Stroke 2008;39:983–990. [DOI] [PubMed] [Google Scholar]
- 7. Serviddio G, Di Venosa N, Federici A, et al. Brief hypoxia before normoxic reperfusion (postconditioning) protects the heart against ischemia‐reperfusion injury by preventing mitochondria peroxyde production and glutathione depletion. Faseb Journal 2005;19:354–361. [DOI] [PubMed] [Google Scholar]
- 8. Leconte C, Tixier E, Freret T, et al. Delayed hypoxic postconditioning protects against cerebral ischemia in the mouse. Stroke 2009;40:3349–3355. [DOI] [PubMed] [Google Scholar]
- 9. Zhan L, Li D, Liang D, et al. Activation of Akt/FoxO and inactivation of MEK/ERK pathways contribute to induction of neuroprotection against transient global cerebral ischemia by delayed hypoxic postconditioning in adult rats. Neuropharmacology 2012;63:873–882. [DOI] [PubMed] [Google Scholar]
- 10. Koch JD, Miles DK, Gilley JA, Yang CP, Kernie SG. Brief exposure to hyperoxia depletes the glial progenitor pool and impairs functional recovery after hypoxic‐ischemic brain injury. J Cereb Blood Flow Metab 2008;28:1294–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Els T, Klisch J, Orszagh M, et al. Hyperglycemia in patients with focal cerebral ischemia after intravenous thrombolysis: Influence on clinical outcome and infarct size. Cerebrovasc Dis 2002;13:89–94. [DOI] [PubMed] [Google Scholar]
- 12. Hamilton MG, Tranmer BI, Auer RN. Insulin Reduction of Cerebral Infarction Due to Transient Focal Ischemia. J Neurosurg 1995;82:262–268. [DOI] [PubMed] [Google Scholar]
- 13. LeMay DR, Gehua L, Zelenock GB, D'Alecy LG. Insulin administration protects neurologic function in cerebral ischemia in rats. Stroke 1988;19:1411–1419. [DOI] [PubMed] [Google Scholar]
- 14. Fan YY, Hu WW, Dai HB, et al. Activation of the central histaminergic system is involved in hypoxia‐induced stroke tolerance in adult mice. J Cereb Blood Flow Metab 2011;31:305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible Middle Cerebral‐Artery Occlusion without Craniectomy in Rats. Stroke 1989;20:84–91. [DOI] [PubMed] [Google Scholar]
- 16. Lee JJ, Li LL, Jung HH, Zuo ZY. Postconditioning with isoflurane reduced ischemia‐induced brain injury in rats. Anesthesiology 2008;108:1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jin CL, Yang LX, Wu XH, et al. Effects of carnosine on amygdaloid‐kindled seizures in Sprague‐Dawley rats. Neuroscience 2005;135:939–947. [DOI] [PubMed] [Google Scholar]
- 18. Hu WW, Fang Q, Xu ZH, et al. Chronic h1‐antihistamine treatment increases seizure susceptibility after withdrawal by impairing glutamine synthetase. CNS Neurosci Ther 2012;18:683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matsumoto K, Lo EH, Pierce AR, Halpern EF, Newcomb R. Secondary elevation of extracellular neurotransmitter amino acids in the reperfusion phase following focal cerebral ischemia. J Cereb Blood Flow Metab 1996;16:114–124. [DOI] [PubMed] [Google Scholar]
- 20. Saransaari P, Oja SS. GABA release under normal and ischemic conditions. Neurochem Res 2008;33:962–969. [DOI] [PubMed] [Google Scholar]
- 21. Rossi DJ, Brady JD, Mohr C. Astrocyte metabolism and signaling during brain ischemia. Nat Neurosci 2007;10:1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ovize M, Baxter GF, Di Lisa F, et al. Postconditioning and protection from reperfusion injury: Where do we stand? Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology Cardiovasc Res 2010;87:406–423. [DOI] [PubMed] [Google Scholar]
- 23. Perez‐Pinzon MA, Born JG. Rapid preconditioning neuroprotection following anoxia in hippocampal slices: Role of the K‐ATP(+) channel and protein kinase C. Neuroscience 1999;89:453–459. [DOI] [PubMed] [Google Scholar]
- 24. Miller BA, Perez RS, Shah AR, Gonzales ER, Park TS, Gidday JM. Cerebral protection by hypoxic preconditioning in a murine model of focal ischemia‐reperfusion. NeuroReport 2001;12:1663–1669. [DOI] [PubMed] [Google Scholar]
- 25. Sanderson TH, Kumar R, Sullivan JM, Krause GS. Insulin blocks cytochrome c release in the reperfused brain through PI3‐K signaling and by promoting Bax/Bcl‐XL binding. J Neurochem 2008;106:1248–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rossi DJ, Brady JD, Mohr C. Astrocyte metabolism and signaling during brain ischemia. Nat Neurosci 2007;10:1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA‐glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem 2006;98:641–653. [DOI] [PubMed] [Google Scholar]
- 28. Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J Neurosci 2007;27:4253–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oliver CN, Starke‐Reed PE, Stadtman ER, Liu GJ, Carney JM, Floyd RA. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion‐induced injury to gerbil brain. Proc Natl Acad Sci U S A 1990;87:5144–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shen Y, He P, Fan YY, et al. Carnosine protects against permanent cerebral ischemia in histidine decarboxylase knockout mice by reducing glutamate excitotoxicity. Free Radic Biol Med 2010;48:727–735. [DOI] [PubMed] [Google Scholar]