

SUMMARY
Aims: To evaluate the acute effects of the mitochondrial complex I inhibitor rotenone on rat hippocampal synaptic plasticity. Methods: Electrophysiological field potential recordings were used to measure basal synaptic transmission and synaptic plasticity in rat coronal hippocampal slices. Synaptic long‐term potentiation (LTP) was induced by high‐frequency stimulation (100 Hz, 1 second × 3 at an interval of 20 seconds). In addition, mitochondrial complex I function was measured using MitoSOX imaging in mitochondrial preparations. Results: Acute exposure of hippocampal slices to 50 nM rotenone for 1 h did not alter basal CA3–CA1 synaptic transmission though 500 nM rotenone significantly reduced basal synaptic transmission. However, 50 nM rotenone significantly impaired LTP and this rotenone's effect was prevented by co‐application of rotenone plus the ketones acetoacetate and β‐hydroxybutyrate (1 mM each). Finally, we measured mitochondrial function using MitoSOX imaging in mitochondrial preparations and found that 50 nM rotenone partially reduced mitochondrial function whereas 500 nM rotenone completely eliminated mitochondrial function. Conclusions: Our findings suggest that mitochondrial activity driven by complex I is a sensitive modulator of synaptic plasticity in the hippocampus. Acute exposure of the hippocampus to rotenone eliminates complex I function and in turn impairs LTP.
Keywords: Hippocampal slice, Ketone, Long‐term potentiation, Mitochondrial dysfunction, Rotenone
Introduction
Mitochondrial dysfunction is a common finding in neurodegenerative diseases. In particular, impairment of respiratory chain complex I has been implicated in the pathogenesis of Parkinson's disease (PD) [1, 2]. Rotenone, a natural complex I inhibitor extracted from the roots of lonchocarpus species, induces oxidative stress, impairs mitochondrial energy production and causes a PD‐like syndrome in rodents [3]. Dopaminergic neurons in the substantia nigra (SN) gradually degenerate in PD, leading to a variety of motor manifestations [3, 4]. PD patients also exhibit nonmotor abnormalities, including cognitive deficits, but the underlying mechanisms are largely unknown. The hippocampus is an important structure for several forms of learning and memory and is commonly affected in neurological diseases that impair cognition [5, 6, 7].
Previous studies have reported that micromolar concentrations of rotenone induce neuronal degeneration in primary neuronal and hippocampal slice cultures [8, 9] and disrupt basal synaptic transmission in acute hippocampal slices [10, 11]. It has been reported that the dendritic mitochondria of hippocampal neurons play an important role in the morphogenesis and plasticity of spines and synapses [12]. However, the effects of acute impaired mitochondrial function by rotenone on hippocampal synaptic plasticity—a cellular correlate of learning and memory—have not been well‐investigated. This gap in knowledge might preclude understanding of the mechanisms underlying cognitive deficits in PD. In the present study, we ascertained the effects of acute exposure to rotenone on hippocampal synaptic activities, including basal synaptic transmission, paired‐pulse facilitation (PPF) and long‐term potentiation (LTP). We found that a low concentration of rotenone (50 nM), which was enough to inhibit complex I‐driven mitochondrial respiration without affecting basal synaptic transmission, significantly altered short‐ and long‐term synaptic plasticity in the CA1 region of rat hippocampal slices. Complex I involvement was confirmed by pretreatment of hippocampal slices with the ketones acetoacetate and β‐hydroxybutyrate (1 mM each), a combination previously shown to prevent calcium‐mediated disruption of complex I activity [11]. Ketones are produced by the liver when caloric intake is restricted and are used by mitochondria to satisfy cellular energy requirements in the absence of glucose. Our results revealed that ketones enhanced complex I‐driven mitochondrial respiration and prevented rotenone‐induced toxicity.
Materials and Methods
Hippocampal Slice Preparation
Hippocampal slices were prepared from 4–6‐week‐old Wistar rats as previously described [13, 14, 15, 16]. Brains were rapidly removed under deep isoflurane anesthesia and bathed in cold (4°C) artificial cerebrospinal fluid (ACSF) containing (in mM): NaCl 119; KCl 2.5; NaHCO3 26; MgSO3 1.3; NaH2PO4 1.0; CaCl2 2.5 and glucose 11; pH 7.4. The ACSF was continuously bubbled with 95% O2–5% CO2 (carbogen). Coronal sections (400 μm) containing the dorsal hippocampus were cut with a vibratome (Vibroslice 725 M, WPI, Sarasota, FL, USA) and transferred to a holding chamber and incubated at room temperature (21 ± 1°C) for at least 60 min before recording. Slices were then transferred to a liquid‐air interface chamber (Fine Science Tools, Inc., Foster City, CA, USA) and suspended on a nylon net at the liquid‐air interface in a bath of continuously dripping oxygenated ACSF (2–2.5 mL/min). Humidified carbogen was passed along the upper surface of the slice, and bath temperature was set at 30 ± 1°C.
Electrophysiological Recordings
Standard extracellular field potential recordings were performed in the stratum radiatum of the hippocampal CA1 region using borosilicate glass micropipettes pulled to a tip diameter of about 1 μm and filled with 2M NaCl [13, 15]. Evoked field excitatory postsynaptic potentials (fEPSPs) were recorded using an Axoclamp‐2B amplifier (Axon Instruments Inc., Union City, CA, USA). Schaffer collaterals were stimulated with a custom bipolar platinum wire electrode (diameter 0.003”) connected to an isolated pulse stimulator (Model 2100, AM Systems, Carlsborg, WA, USA). Stimulus intensity was determined using input–output response curves and was set at 50% of the maximal response. LTP was induced by high‐frequency tetanic stimulations (100 Hz, 1 second × 3 at intervals of 20 seconds) of Schaffer collaterals following a stable 20‐min baseline. fEPSPs were subsequently recorded for an additional 60 min at 0.03 Hz.
Mitochondrial Isolation and Measurement of Mitochondrial Respiration
All experimental protocols were approved by the Institutional Animal Care and Use Committee at the Barrow Neurological Institute (A3519–01). Brains from 4–6‐week‐old Wistar rats were rapidly removed under deep isoflurane anesthesia and bathed in cold (4°C) isolation buffer (in mM: sucrose 320; EDTA 1; Tris‐HCl 10; pH adjusted to 7.4 with KOH). The cortical and hippocampal regions were isolated and manually homogenized with a dounce tissue grinder (BellCo, Vineland, NJ, USA). The homogenized tissues were initially centrifuged at 1000 rpm for 10 min (4°C) to remove large cellular debris. The supernatant was then recovered and centrifuged at 14,000 rpm (4°C) to isolate the mitochondrial fraction. The supernatant was discarded and the pellet was dissolved in respiration buffer (in mM unless otherwise stated: EDTA 0.5; MgCl2 3; KCl 40; KH2PO4 10; HEPES 20; sucrose 110; BSA 1 g/L; pH adjusted to 7.4 with KOH). Mitochondrial respiration was measured with the Oxygen Biosensory System (BD Biosciences, San Jose, CA, USA) at room temperature. The Oxygen Biosensory System consists of 96‐well plates coated with an oxygen sensitive fluorescent indicator. Higher fluorescence reflects increased oxygen consumption and, therefore, enhanced mitochondrial respiration. We added 0.5 mg of protein to each well and triggered complex I‐dependent state III respiration with 5 mM glutamate, 5 mM malate and 1 mM ADP (all in respiration buffer at pH 7.4).
Data Analysis and Drugs
Data were acquired through an Axon Digidata 1322 (Axon Instruments) interface board set to a sampling frequency of 10 kHz and controlled with pClamp 9.2 (Axon Instruments), filtered in Clampfit 9.0 (Axon Instruments) using an 8‐pole Bessel filter and a 1‐kHz low‐pass filter, and stored on hard media for subsequent off‐line analysis. Statistical comparisons using Student's t‐test (independent or paired) or analysis of variance were performed with Origin 5.0 (Microcal Software Inc., Northampton, MA, USA). P values less than 0.05 were considered to be statistically significant. Drugs used in the present study included rotenone, lithium acetoacetate and DL‐β‐hydroxybutyric acid sodium salt that were purchased from Sigma–Aldrich (St. Louis, MO, USA). Rotenone was dissolved to 100 μM stock solution using DMSO and was diluted to 50 nM concentration using incubation solution before experiment. All drugs were applied to slices via a bath‐application system.
Results
Effects of Rotenone on Basal Synaptic Transmission in Hippocampal Slices
Repetitive stimulation (5–10 V, 100 μs, 0.03 Hz) of Schaffer collaterals induced typical evoked fEPSPs in the hippocampal CA1 region, which were stable for 1 h (Figure 1A). Bath‐perfusion of rotenone at 50 nM (Figure 1B) did not significantly affect basal synaptic transmission, but 500 nM rotenone significantly reduced the slope of fEPSPs (Figure 1C, n = 6). Statistical analyses showed that the normalized fEPSP slope was 1.02 ± 0.02 after rotenone exposure for 1–10 min and 0.64 ± 0.02 after exposure between 70–80 min (Figure 1D, P < 0.01). These results suggest that acute exposure to rotenone impairs hippocampal synaptic transmission dependent upon concentration and exposure period.
Figure 1.
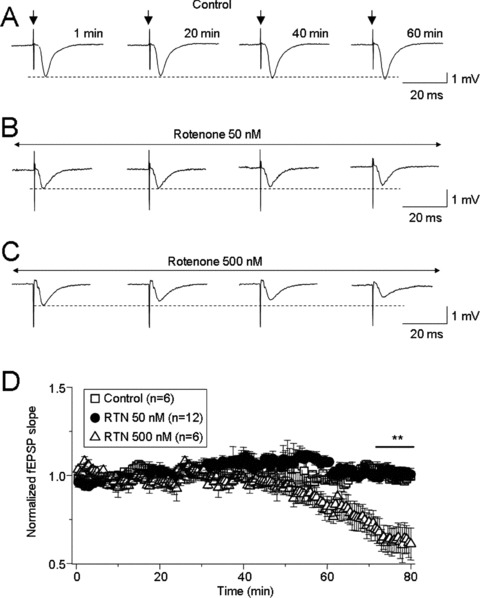
Rotenone suppressed basal synaptic transmission in concentration‐ and time‐dependent manners. Basic field excitatory postsynaptic potentials (fEPSPs) were recorded from the hippocampal CA1 region following repetitive stimulation (100 μs, 0.03 Hz for 60 min) of Schaffer collaterals. Stimulation intensity was set to that which induced 50% of the maximal response. Representative typical traces evoked by single stimulation in the absence or presence of different concentrations of rotenone are shown (A–C). Summary of the concentration‐dependent inhibition of basal fEPSPs by rotenone is shown (D). Each symbol represents the average from 6–12 slices (from 4–8 rats), and the vertical bars represent ±SE. **P < 0.01 when comparing evoked responses under control conditions versus 500 nM rotenone between 70–80 min (horizontal bar).
Effects of Rotenone on LTP in Hippocampal Slices
To assess the effects of rotenone on LTP, we measured high‐frequency stimuli (100 Hz, 1 second × 3 at 20‐second intervals)‐induced LTP in the hippocampal CA1 region. In the control group (n = 15 from 10 rats), baseline fEPSPs (50% of maximal stimuli with 0.03 Hz) were recorded for 20 min. Tetanic stimuli then were subsequently delivered only in recordings showing less than ±10% variation in baseline responses. Thereafter, baseline stimulus‐induced fEPSPs were recorded for another 60 min (Figure 2A). In the rotenone‐treated group (n = 16 from 10 rats), 50 nM rotenone was delivered throughout the whole recording session (total 80 min, Figure 2B). Results showed that rotenone impaired HFS‐induced LTP (Figure 2C). Statistical analyses revealed that the averaged slopes of fEPSPs were significantly reduced at 10–60 min following LTP induction in the rotenone‐treated group compared to the control group (n = 15 from 10 rats, Figure 2D). These results suggest that although 50 nM rotenone does not alter basal synaptic transmission, it impairs synaptic plasticity in rat hippocampal slices.
Figure 2.
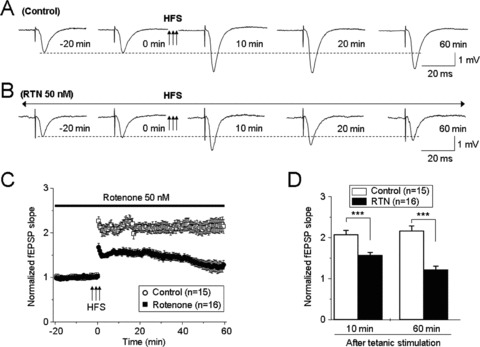
The effects of 50 nM rotenone on LTP initiation and maintenance. Typical high‐frequency stimulation (HFS)‐induced fEPSPs in control (A) and rotenone‐treated (B) slices. (C) Summary of the effects of 50 nM rotenone on LTP (n = 16 slices from 10 rats). Compared to responses from control slices (n = 15 from 10 rats), rotenone impaired HFS‐induced LTP (D). ***P < 0.001 when comparing normalized slopes of fEPSPs under control conditions versus LTP induction at 10 and 60 min.
Effects of Ketones on Rotenone‐Induced Impairment of LTP in Hippocampal Slices
Rotenone blocks mitochondrial complex I and consequently causes oxidation and energy crisis [1, 3]. We hypothesized that these mechanisms may underlie rotenone's effects on hippocampal synaptic functions. To test this, we co‐incubated hippocampal slices with ketones (1 mM lithium acetoacetate plus 1 mM DL‐β‐hydroxybutyric acid sodium salt) and rotenone (50 nM) together for the recording period (total of 80 min). As shown in Figure 3, co‐treatment with ketones plus rotenone (n = 7 from 5 rats) significantly prevented rotenone‐induced impairment of LTP (n = 6 from 4 rats, ***P < 0.001, when comparing rotenone plus ketones verses rotenone alone at 60 min of LTP). However, ketones alone (i.e., without co‐application of rotenone) did not alter synaptic plasticity (n = 7 from 4 rats). These results suggest that ketones significantly prevent acute exposure of rotenone‐induced impairment of hippocampal synaptic plasticity, which is likely mediated via dysfunction of mitochondrial complex I.
Figure 3.
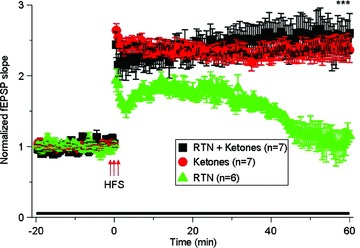
The effects of ketones on rotenone‐induced LTP impairment. This figure represents high frequency stimulation (HFS)‐induced LTP under the following conditions: (1) 50 nM rotenone (RTN) plus ketones (red symbols, n = 7 from 5 rats; 1 mM lithium acetoacetate plus 1 mM DL‐β‐hydroxybutyric acid sodium salt dissolved in incubation solution); (2) ketones alone (black symbols, n = 7 from 4 rats), and (3) rotenone alone (green symbols, n = 6 from 4 rats). These data showed that ketones significantly prevented rotenone‐induced LTP impairment.
Effects of Rotenone and Ketones on Complex I‐Driven Mitochondrial Respiration
Micromolar concentrations of rotenone have previously been shown to inhibit mitochondrial complex I [17]. Incubation of isolated mitochondria with 50 nM rotenone for 10 min partially decreased oxygen consumption—a direct measure of mitochondrial respiration—in the presence of the complex I activators glutamate and malate (combined with ADP) whereas 500 nM rotenone completely eliminated oxygen consumption (Figure 4A). In contrast, preincubation of mitochondria with ketones (1 mM lithium acetoacetate plus 1 mM DL‐β–hydroxybutyrate acid sodium salt) for 10 min enhanced complex I‐driven mitochondrial respiration and prevented the inhibitory effects of rotenone. The differences were statistically significant (n = 5 animals in each experimental group; ANOVA, P < 0.05). However, ketones did not affect mitochondrial respiration driven by the complex II substrate succinate (10 mM) in the presence of 1 mM ADP and 1 μM rotenone (n = 4 animals; Figure 4B).
Figure 4.
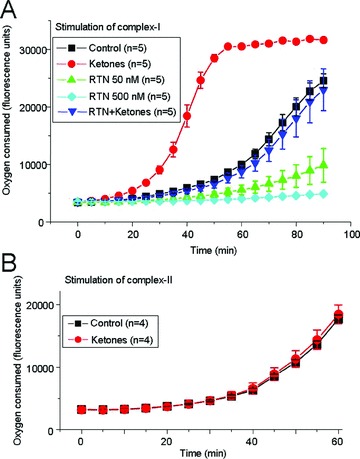
The effects of rotenone and ketones on complex I‐driven mitochondrial respiration. (A) The amount of oxygen consumed by isolated mitochondria was estimated using the Oxygen Biosensory System (BD Biosciences, San Jose, CA, USA) in the presence of rotenone alone or in combination with ketones (1 mM acetoacetate plus 1 mM DL‐β‐hydroxybutyrate). Complex I was activated with 10 mM glutamate, 10 mM malate and 1 mM ADP. Oxygen consumption was clearly decreased by 50 nM rotenone (green), and 500 nM rotenone completely blocked mitochondrial respiration (cyan). In contrast, ketones enhanced complex I‐driven mitochondrial respiration and prevented the inhibitory effects of rotenone. The differences between the rotenone and rotenone plus ketones groups were statistically significant (n = 5 animals in each experimental group; ANOVA, P < 0.05). (B) Ketones did not affect mitochondrial respiration driven by the complex II substrate succinate (10 mM) in the presence of ADP (1 mM) and rotenone (1 μM).
Discussion
The main findings of this study are that acute exposure to rotenone at low nanomolar concentrations (50 nM) did not affect basal synaptic transmission but significantly impaired LTP in hippocampal slices. Rotenone toxicity was prevented by enhancing complex I activity with ketones. Direct measurement of mitochondrial function showed that 50 nM rotenone partially impaired oxygen consumption. Our findings suggest that complex I is likely a sensitive target for hippocampal synaptic plasticity and might potentially represent a therapeutic target for treatment of cognitive deficits in patients with PD and possibly other disorders characterized by hippocampal damage or cognitive decline such as Alzheimer's disease.
Rotenone causes highly‐selective degeneration of nigrostriatal neurons and can reproduce many features of PD [18]. Previous studies have suggested that the hippocampus is much less sensitive than the striatum to complex I inhibition by rotenone [10], but growing evidence supports our conclusion that rotenone, even at low concentrations, exerts significant toxicity within the hippocampus. For instance, studies have shown that brief exposure (1 h) of cultured hippocampal slices to 1–2 μM rotenone induced CA1 neuron death [8], and chronic exposure (24 h) of cultured hippocampal slices to 200–300 nM rotenone significantly worsened cellular degeneration [9]. In addition, a recent report has shown that acute exposure of rat hippocampal slices to 0.1–1 μM rotenone for 30 min dramatically reduced the amplitude of evoked population spikes [11]. Collectively, these reports suggest that inhibition of mitochondrial respiratory chain complex I can produce cellular and synaptic changes within the hippocampus.
In the present study, we examined the acute effects of 50 nM rotenone, a concentration lower than that described in previous reports but sufficient to significantly decrease complex I activity. We found that although 50 nM rotenone did not acutely alter the slope of evoked baseline fEPSPs, it significantly increased both neuronal excitability and short‐term synaptic plasticity but inhibited CA3‐CA1 LTP maintenance. Pretreatment with the ketones acetoacetate and DL‐β‐hydroxybutyrate—a combination that enhanced complex I‐driven mitochondrial respiration—prevented rotenone toxicity. Our data, therefore, suggest that synaptic plasticity is more susceptible to acute mitochondrial dysfunction than basal synaptic transmission.
Rotenone‐induced inhibition of mitochondrial complex I has two potential consequences: (1) increased production of reactive oxygen species (ROS) and (2) ATP depletion. Ketones have consistently been shown to prevent oxidative stress and energy failure [19, 20]. In support of the oxidative stress hypothesis, previous studies have shown that the oxidant compound hydrogen peroxide (H2O2) impairs hippocampal LTP [21, 22] possibly by activating protein phosphatases 2A or 2B [23]. In the present study, we found that ketones significantly prevented rotenone‐induced LTP impairment. We also showed that ketones significantly enhanced oxygen consumption in hippocampal mitochondrial preparations. Taken together, these results are consistent with previous findings that ketones prevent oxidative stress and energy failure [19, 20], which may underlie the protective effect of ketones found in the present study.
Although PD is characterized by movement disorders in later stages of the disease, it is now appreciated that there also may be cognitive impairment, including dementia and behavioral changes [24, 25, 26]. Dementia in patients with PD is consequential and has been associated with reduced quality of life, shortened survival and increased caregiver distress. Thus far, there has been considerable progress in regards to defining the scope of cognitive impairment in PD both in terms of epidemiology and risk profiles for those most at risk for its development. Investigators, using multiple approaches, including neuropsychology, imaging and pathology, suggest there exists a common denominator referable to early dopamine deficiency but highlight an increasingly important focus on nondopaminergic pathway involvement [25]. Unfortunately, the mechanisms of cognitive deficits in PD animal models and patients have not been well‐elucidated. The hippocampus is a key brain structure for several forms of learning and memory. It is well‐known that the hippocampus is highly sensitive to oxidative stress, which makes hippocampal synaptic transmission highly dependent on mitochondrial products such as ATP and ROS [27]. Therefore, it is reasonable that changes in hippocampal synaptic integrity under such pathological conditions would be responsible for the observed cognitive deficits in PD. In the present study, we provide direct evidence that rotenone, even at a low concentration (e.g., 50 nM) that does not affect basal synaptic transmission, significantly impairs hippocampal synaptic plasticity, which is an important factor related to synaptic integrity in vitro and thought to be critical in processes governing learning and memory. Based on these findings, we speculate that impairment of hippocampal LTP by rotenone might serve as an underlying mechanism contributing to cognitive deficits in PD. In addition, our data also suggest that antioxidative therapies may serve as useful strategies for the prevention and/or treatment of cognitive deficits in PD.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was partially supported by a Shantou University Seed Fund, China (JW and SJX), and by the Barrow Neurological Foundation (MM).
The first two authors contributed equally to this work.
References
- 1. Schapira AH. Complex I: Inhibitors, inhibition and neurodegeneration. Exp Neurol 2010;224:331–335. [DOI] [PubMed] [Google Scholar]
- 2. Zhu J, Chu CT. Mitochondrial dysfunction in Parkinson's disease. J Alzheimers Dis 2010;20(Suppl 2):S325–S334. [DOI] [PubMed] [Google Scholar]
- 3. Greenamyre JT, Cannon JR, Drolet R, Mastroberardino PG. Lessons from the rotenone model of Parkinson's disease. Trends Pharmacol Sci 2010;31:141–142; author reply 2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jenner P. Parkinson's disease, pesticides and mitochondrial dysfunction Trends Neurosci 2001;24:245–247. [DOI] [PubMed] [Google Scholar]
- 5. Sano K, Kirino T. Ammon's horn sclerosis: Its pathogenesis and clinical significance. Tohoku J Exp Med 1990;161(Suppl):273–295. [PubMed] [Google Scholar]
- 6. Zarow C, Sitzer TE, Chui HC. Understanding hippocampal sclerosis in the elderly: Epidemiology, characterization, and diagnostic issues. Curr Neurol Neurosci Rep 2008;8:363–370. [DOI] [PubMed] [Google Scholar]
- 7. Jellinger KA. Recent developments in the pathology of Parkinson's disease. J Neural Transm Suppl 2002;62:347–376. [DOI] [PubMed] [Google Scholar]
- 8. Xu G, Perez‐Pinzon MA, Sick TJ. Mitochondrial complex I inhibition produces selective damage to hippocampal subfield CA1 in organotypic slice cultures. Neurotox Res 2003;5:529–538. [DOI] [PubMed] [Google Scholar]
- 9. Schuh RA, Matthews CC, Fishman PS. Interaction of mitochondrial respiratory inhibitors and excitotoxins potentiates cell death in hippocampal slice cultures. J Neurosci Res 2008;86:3306–3313. [DOI] [PubMed] [Google Scholar]
- 10. Costa C, Belcastro V, Tozzi A, et al Electrophysiology and pharmacology of striatal neuronal dysfunction induced by mitochondrial complex I inhibition. J Neurosci 2008;28:8040–8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim do Y, Vallejo J, Rho JM. Ketones prevent synaptic dysfunction induced by mitochondrial respiratory complex inhibitors. J Neurochem 2010;114:130–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004;119:873–887. [DOI] [PubMed] [Google Scholar]
- 13. Wu J, Fisher RS. Hyperthermic spreading depressions in the immature rat hippocampal slice. J Neurophysiol 2000;84:1355–1360. [DOI] [PubMed] [Google Scholar]
- 14. Wu J, Javedan SP, Ellsworth K, Smith K, Fisher RS. Gamma oscillation underlies hyperthermia‐induced epileptiform‐like spikes in immature rat hippocampal slices. BMC Neurosci 2001;2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Song C, Murray TA, Kimura R, et al Role of alpha7‐nicotinic acetylcholine receptors in tetanic stimulation‐induced gamma oscillations in rat hippocampal slices. Neuropharmacology 2005;48:869–880. [DOI] [PubMed] [Google Scholar]
- 16. Wang K, Zheng C, Wu C, et al alpha‐Chloralose diminishes gamma oscillations in rat hippocampal slices. Neurosci Lett 2008;441:66–71. [DOI] [PubMed] [Google Scholar]
- 17. Ayala A, Venero JL, Cano J, Machado A. Mitochondrial toxins and neurodegenerative diseases. Front Biosci 2007;12:986–1007. [DOI] [PubMed] [Google Scholar]
- 18. Betarbet R, Sherer TB, MacKenzie G, Garcia‐Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 2000;3:1301–1306. [DOI] [PubMed] [Google Scholar]
- 19. Sherer TB, Betarbet R, Testa CM, et al Mechanism of toxicity in rotenone models of Parkinson's disease. J Neurosci 2003;23:10756–10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bao L, Avshalumov MV, Rice ME. Partial mitochondrial inhibition causes striatal dopamine release suppression and medium spiny neuron depolarization via H2O2 elevation, not ATP depletion. J Neurosci 2005;25:10029–10040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kamsler A, Segal M. Hydrogen peroxide modulation of synaptic plasticity. J Neurosci 2003;23:269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kamsler A, Segal M. Hydrogen peroxide as a diffusible signal molecule in synaptic plasticity. Mol Neurobiol 2004;29:167–178. [DOI] [PubMed] [Google Scholar]
- 23. Maalouf M, Rho JM. Oxidative impairment of hippocampal long‐term potentiation involves activation of protein phosphatase 2A and is prevented by ketone bodies. J Neurosci Res 2008;86:3322–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leverenz JB, Quinn JF, Zabetian C, Zhang J, Montine KS, Montine TJ. Cognitive impairment and dementia in patients with Parkinson disease. Curr Top Med Chem 2009;9:903–912. [PMC free article] [PubMed] [Google Scholar]
- 25. Kehagia AA, Barker RA, Robbins TW. Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson's disease. Lancet Neurol 2010;9:1200–1213. [DOI] [PubMed] [Google Scholar]
- 26. Shi M, Huber BR, Zhang J. Biomarkers for cognitive impairment in Parkinson disease. Brain Pathol 2010;20:660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Keating DJ. Mitochondrial dysfunction, oxidative stress, regulation of exocytosis and their relevance to neurodegenerative diseases. J Neurochem 2008;104:298–305. [DOI] [PubMed] [Google Scholar]