

Summary
Aims
To study the contribution of epidermal growth factor receptor variant III (EGFRvIII) to glioblastoma multiforme (GBM) stemness and gefitinib resistance.
Methods
CD133+ and CD133− cells were separated from EGFRvIII + clinical specimens of three patients with newly diagnosed GBM. Then, RT‐PCR was performed to evaluate EGFRvIII and EGFR expression in CD133+ and CD133− cells. The tumorigenicity and stemness of CD133+ cells was verified by intracranial implantation of 5 × 103 cells into immunodeficient NOD/SCID mice. Finally, cells were evaluated for their sensitivity to EGFR tyrosine kinase inhibition by gefitinib.
Results
RT‐PCR results showed that the sorted CD133+ cells expressed EGFRvIII exclusively, while the CD133− cells expressed both EGFRvIII and EGFR. At 6–8 weeks postimplantation, CD133+/EGFRvIII +/EGFR − cells formed intracranial tumors. Cell counting kit‐8 results showed that the IC 50 values of the three isolated EGFRvIII + cell lines treated with gefitinib were 14.44, 16.00, and 14.66 μM, respectively, whereas the IC 50 value of an isolated EGFRvIII − cell line was 8.57 μM.
Conclusions
EGFRvIII contributes to the stemness of cancer stem cells through coexpression with CD133 in GBMs. Furthermore, CD133+/EGFRvIII +/EGFR − cells have the ability to initiate tumor formation and may contribute to gefitinib resistance.
Keywords: CD133, EGFRvIII, Gefitinib resistance, Stemness
Introduction
Glioblastoma multiforme (GBM) is the most common and malignant type of primary brain tumor. Overexpression of the epidermal growth factor receptor (EGFR) has been found in more than 50% of GBMs 1, 2, 3 and is often associated with the genomic deletion mutation of exons 2–7 (EGFRvIII) 4, 5, 6, 7, the most common gain‐of‐function mutation. EGFRvIII, a truncated receptor with weak but constitutive oncogenic activation 8, 9, usually correlates with a poor prognosis for GBM patients 10. Numerous data have demonstrated that EGFRvIII expression can be regarded as a marker of poor prognosis because EGFRvIII+ gliomas are always far more malignant than gliomas with EGFR expression only 8, 11, 12. Many studies have confirmed that EGFRvIII+ cells are more radioresistant and chemoresistant to EGFR inhibitors 13, 14, 15, 16, 17, 18. Recently, the concept of glioma stem cells (GSCs), which have been identified in human brain tumors, has gained more attention in research on GBM malignancy. This minority cell population has shown potent tumorigenicity in several in vivo studies of immunodeficient animals and has a high degree of similarity to normal neural stem cells (NSCs) because they express NSC markers and possess the properties of self‐renewal and differentiation 19, 20, 21. Therefore, considering the properties of EGFRvIII, a recent study demonstrated that EGFRvIII contributes to cancer stem cell (CSC) phenotypes, suggesting that EGFRvIII may serve as a CSC marker 22.
CD133 was first reported as a NSC marker and is now considered as a GSC marker 20. Many clinical studies have also demonstrated that CD133 promotes GSC resistance against chemotherapeutics, resulting in poor clinical outcomes 23, 24, 25. In this study, we show that EGFRvIII indeed contributes to stemness, and that EGFRvIII and CD133 can synergistically lead to stemness and increase malignancy, tumorigenicity, and drug resistance in GBMs.
Materials and Methods
GBM Specimens, Cell Lines, and Tissue Culture
Ten clinical specimens from GBM patients diagnosed by magnetic resonance imaging (MRI) were provided by Peking Union Medical College Hospital, Navy General Hospital of PLA China, and The Military General Hospital of Beijing PLA. Permission to use human tissue was granted by the ethical committees of these hospitals. Details of the each patient specimen are summarized in Table 1. After testing for EGFRvIII/EGFR expression, we selected three EGFRvIII+ specimens (Patient No. 1–3) from Peking Union Medical College Hospital for further study.
Table 1.
Characteristics of study patients and their tumors according to CD133 and EGFRvIII status
| Patient No. | Age (years) | Gender | Pathologic diagnosis (WHO classification grade) |
|---|---|---|---|
| 1 | 18 | M | Astrocytoma (grade III) |
| 2 | 31 | F | Astrocytoma (mixed type, grade III) |
| 3 | 53 | M | Gliosarcoma (grade IV) |
| 4 | 21 | F | Gliosarcoma (grade IV) |
| 5 | 32 | M | Astrocytoma (grade III‐IV) |
| 6 | 58 | F | Astrocytoma (poorly differentiated,grade III) |
| 7 | 34 | F | Glioblastoma (grade IV) |
| 8 | 58 | F | Glioblastoma (grade IV) |
| 9 | 45 | M | Astrocytoma (grade III‐IV) |
| 10 | 63 | M | Glioblastoma (grade IV) |
M, male; F, female.
Cells from these three specimens were isolated, purified, and primary cultured in Dulbecco's modified Eagle's medium/F12 (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Hyclone, Logan City, UT, USA), epidermal growth factor (EGF; 20 ng/mL; PeproTech Inc., Rocky Hill, NJ, USA), basic fibroblast growth factor (20 ng/mL, PeproTech), and B27 (50×; Invitrogen, Carlsbad, CA, USA). At 95% confluence, cells were transferred to a new flask and cultured continuously to form a monolayer.
RT‐PCR Assay
RT‐PCR was performed to determine wild‐type EGFR and EGFRvIII expression. Total RNA was extracted from the original clinical specimens, and RT‐PCR was performed using the following primers: sense, 5′‐CTTCGGGGAGCAGCGATGCGAC‐3′; and antisense, 5′‐ACCAATACCTATTCCGTTACAC‐3′.
RT‐PCR products were separated by agarose gel electrophoresis. A 1044‐bp fragment indicated wild‐type EGFR expression, and a 243‐bp (deletion product) fragment indicated EGFRvIII expression.
Flow Cytometric Analysis
Cells isolated from each of the three clinical specimens were trypsinized, and a single cell suspension was prepared containing 1 × 106 cells in 100 μL of 1% bovine serum albumin (BSA)/phosphate‐buffered saline (PBS) (w/v) and immediately stained for 30 min at room temperature with a PE‐labeled anti‐CD133 antibody (Miltenyi Biotech, Auburn, CA, USA). A mouse IgG1‐PE (Becton‐Dickinson, Rutherford, NJ, USA) was used as an isotype control. The Caco2 cell line with CD133 overexpression was used as the positive control. Stained cells were analyzed by a FACSCalibur flow cytometer (Becton‐Dickinson).
Immunofluorescence Assay
Cells were subcultured on LabTek chamber slides overnight. The attached cells were then fixed with 2% paraformaldehyde/PBS (v/v) for 15 min on ice. After blocking for 30 min with 2% BSA and 1% normal goat serum, the cells were incubated with the relevant primary antibodies at 4°C overnight and then incubated with the corresponding fluorescence‐conjugated secondary antibodies. Mouse or rabbit IgG was used as isotype controls. Primary polyclonal antibodies were a rabbit anti‐human CD133 (Cell Signaling Technology, Beverly, MA, USA), rabbit anti‐EGFRvIII (Abcam, Cambridge, MA, USA), rabbit anti‐nestin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and rabbit anti‐glial fibrillary acidic protein (GFAP; Chemicon, Billerica, MA, USA). DAPI was used for nuclear staining. Fluorescence signals were detected using a confocal microscope (TCS SP5; Leica, solms, Germany).
Magnetic‐Activated Cell Sorting for CD133
CD133+ cells from the three patients were purified by magnetic‐activated cell sorting (MACS) using a CD133 MicroBead Kit (Miltenyi Biotech), followed by flow cytometric analysis using CD133/2‐PE (Miltenyi Biotech).
Intracranial Tumorigenic Implantation and Immunohistochemistry
Twenty NOD/SCID mice were equally and randomly divided into four experimental groups. Group 1, 2 and 3 corresponded to sorted CD133+ cell lines derived from Patient No. 1–3, respectively. The forth group of mice was inoculated with CD133− cells from Patient No. 1. A total of 5 × 103 cells of each sorted cell population were intracranially injected into the area between the cerebral cortex and dorsal hippocampus CA3 of experimental mice. When symptoms of hydrocephalus were observed, the mice immediately underwent micro‐CT scanning or micro‐PET imaging after 37 MBq 11C‐choline in 0.1 mL PBS was injected via the tail vein. Then, the intact brains were removed and fixed with 10% neutral buffered formalin and then embedded in paraffin for hematoxylin and eosin (H&E) staining and immunofluorescence analysis. Animal procedures and care were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Drug‐Resistance Assay
A cell counting kit‐8 (CCK‐8; Dojindo Laboratories, Kumamoto, Japan) assay was performed to evaluate the tolerance of the three cell lines for gefitinib, an EGFR tyrosine kinase inhibitor. Cells were seeded in 96‐well plates at 5 × 103 cells per well. After incubation overnight at 37°C, complete medium containing 0.5, 1, 2, 5, 10, 20, 30, 40, 50, 60, 70, 80, or 90 μM gefitinib or a vehicle control was added to the cells. After incubation for 48 h at 37°C, 10 μL of CCK‐8 solution was added to each well, and the cells were incubated for a further 4 h at 37°C. The optical density was measured at 450 nm with a Delta Soft ELISA analysis program interfaced with a Bio‐Tek Microplate Reader (EL‐340; Bio‐Metallics, Princeton, NJ, USA). Each reaction was performed in four replicate wells for each drug concentration and carried out independently three or four times. The IC50 value was defined as the concentration needed for a 50% reduction in the absorbance based on the survival curves.
Western Blot Analysis
As indicated in Figure 4, all cell lines were mock‐treated with DMSO or treated with gefitinib and EGF for up to 3 days. Briefly, at the time of harvest, the cells were washed once in 0.01 M PBS, centrifuged, and then lysed. Protein concentrations in lysates were determined using BCA Protein Assay Reagent (Pierce Chemical, Rockford, IL, USA). Total protein (60 μg) was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred onto nitrocellulose membranes at 250 V for 2 h. Membranes were blocked for 1 h with 5% nonfat milk powder in 1× TBS/0.1% Tween 20 and then incubated with a rabbit anti‐phospho‐Tyr‐1048 EGFR polyclonal antibody (Santa Cruz Biotechnology), rabbit anti‐EGFR polyclonal antibody (Santa Cruz Biotechnology), rabbit anti‐CD133 polyclonal antibody (Cell Signaling Technology), or rabbit anti‐nestin polyclonal antibody (Santa Cruz Biotechnology) at 4°C overnight. The membranes were washed and incubated with a secondary antibody conjugated with Dylight 800 (EarthOx, San Francisco, CA, USA). After the membranes were washed, immunoblotted proteins were detected using an Odyssey Western Blotting Detection System.
Figure 4.
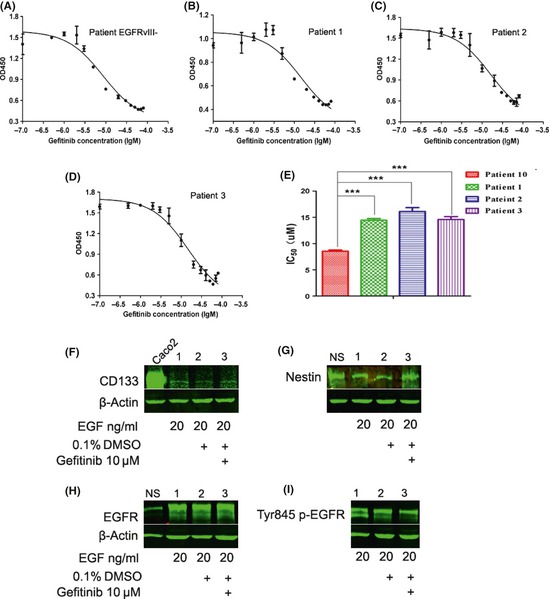
The growth inhibition curves of cells treated with gefitinib were determined by a CCK‐8 assay. (A) Patient No. 10 without EGFRvIII expression. (B, C, and D) Patient No. 1–3, respectively. (E) Column diagram showing each IC 50 value. Western blot results showed that the expression levels of CD133 (F), nestin (G), EGFR (H) in cells from Patient No. 1 were essentially stable, while phosphorylation of EGFR tyrosine 845 was slightly reduced at the indicated concentrations of EGF and gefitinib treatment (I). Caco2 cells and neurospheres were used as positive controls. Lanes 1, 2, and 3 represent the indicated treatments of EGF, DMSO, or gefitinib, respectively. Western blot results of cells from Patient No. 2 and 3 are not shown because similar results were obtained.
Results
Status of EGFRvIII and CD133 Expression in Clinical GBM Specimens
RT‐PCR was used to detect EGFR and EGFRvIII expression in 10 clinical specimens of newly diagnosed WHO Grade III–IV GBM. The results showed that all 10 GBM clinical specimens were wild‐type EGFR+, and eight of the 10 were EGFRvIII+ (Figure 1A). We chose three EGFRvIII+ specimens (No. 1, 2, and 3) to study further. MRIs of these clinical specimens all showed that the intracranial space was occupied with lesions (Figure 1B). Immunophenotyping demonstrated that CD133‐expressing cells accounted for an extremely low percentage of cells among the isolated and cultured GBM cells: 1.1, 3.6, and 0.3%, respectively (Figure 1C). To evaluate stemness, CD133, nestin, and EGFRvIII were detected by immunofluorescence staining (Figure 2A). Next, we evaluated the relationship between EGFRvIII and CD133 coexpression in GBMs.
Figure 1.
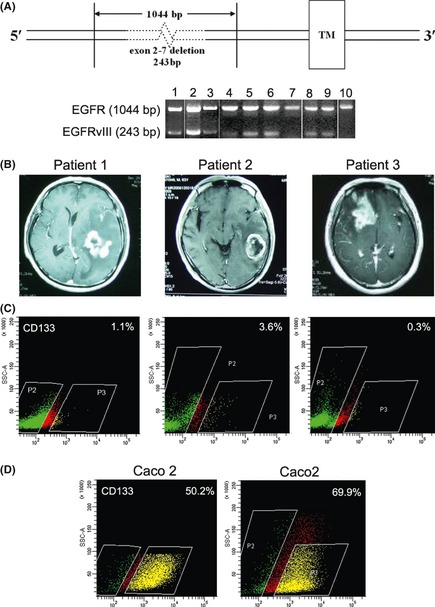
RT‐PCR results showed that all 10 GBM clinical specimens were wild‐type EGFR +, and eight of the 10 were EGFRvIII + (A). Lanes correspond to the patient number. DNA fragments of 1044 bp and 243 bp in size represented EGFR and EGFRvIII, respectively. MRI showed space‐occupying lesions in all three patients (B). Patient No. 1. Space‐occupying lesions were located in the left temporal lobe rear, and the midline structure shifted right. Patient No. 2 showed a class round abnormal signal shadow, and rich blood supply space‐occupying lesions were located in the left temporal lobe rear. Patient No. 3 showed space‐occupying lesions in the right frontal lobe, the right lateral ventricle was compressed by the tumor, and the midline structure shifted left. Flow cytometric analysis showed that the percentages of CD133+ cells from each patient were 1.1%, 3.6%, and 0.3% (C). The Caco2 cell line with CD133 overexpression was used as a positive control (D).
Figure 2.
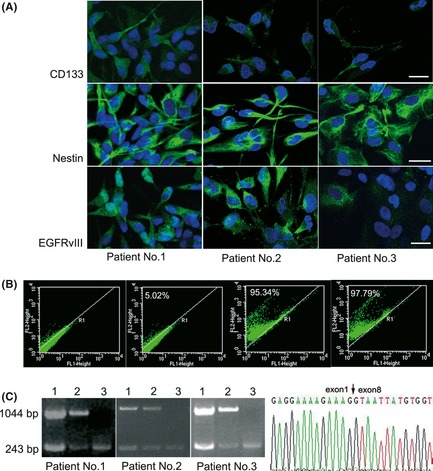
The results of immunofluorescence assays showed that CD133, nestin, and EGFRvIII were all expressed in cells derived from the three patient specimens (A). For cells from Patient No. 1, the percentages of CD133+ cells were 95.34% and 97.79% after initial and secondary MACS was performed, respectively. (B). The first left panel is the blank control, and the second left panel is the isotype control using IgG instead of the anti‐CD133 antibody as the primary antibody. The sorting results of cells from Patient No. 2 and 3 are not shown here. RT‐PCR results showed that CD133+ cells were EGFRvIII + but EGFR − (C). Lanes 1 and 2, corresponding to primary nonsorted cells and sorted CD133− cells, respectively, showed two bands including EGFR (1044 bp) and EGFRvIII (243 bp), while CD133+ cells in lane 3 only showed the 243‐bp band. Sequencing results confirmed expression of the EGFRvIII gene.
CD133‐positive GBM Cells Express the EGFRvIII Mutant Isoform But Not EGFR
To confirm the specificity of the relationship between CD133 and EGFRvIII expression in GBM cells, the cells were separated into two subpopulation of CD133+ and CD133− cells by MACS. To obtain a high purity of isolated CD133+ cells, MACS was performed twice with a similar method. Then, flow cytometry was used to test the purity of isolated CD133+ cells from Patient No. 1, which showed that the cells were more than 97% CD133+ after MACS was performed twice (Figure 2B). Results of flow cytometric analyses for Patient No. 2 and 3 are shown in Supplemental Figure 1A, 1B. RT‐PCR and sequence analysis showed that CD133− cells were both wild‐type EGFR+ and EGFRvIII+. Surprisingly, CD133+ cells were only EGFRvIII+ and EGFR− (CD133+/EGFRvIII+/EGFR−) (Figure 2C), suggesting that the CD133+/EGFRvIII+ phenotype may have great potential as a CSC‐like marker.
CD133+/EGFRvIII+/EGFR− GBM Cells Show Stem Cell‐like Features and Initiate Intracranial Tumor Formation
To test the in vivo tumorigenicity of CD133+/EGFRvIII+/EGFR− and CD133−/EGFRvIII−/EGFR+ GBM cells, immunodeficient (NOD/SCID) mice were used. All 15 mice that received intracranial implantation of CD133+/EGFRvIII+/EGFR− cells showed fleshless, hunched backs and were unresponsive after 6–8 weeks. Results of micro‐CT scanning could not detect any tumor formation probably because of the low sensitivity of the equipment and/or the tumor volume was too small in the mouse brain. However, micro‐PET imaging verified tumor formation in mouse brains at 4 weeks postinoculation. (Supplemental Figure S1C). Histologically, these neoplastic cells invaded the surrounding normal nervous tissues. H&E staining revealed that these tumor cells had large, oval nuclei with some prominent nucleoli, suggesting that these tumor cells could proliferate quickly (Figure 3A). Figure 3B shows that the transplanted cells grew along with the needle track. H&E staining results from Patient No. 2 and 3 are shown in Supplemental Figure S1A,B. After intracranial implantation, immunofluorescence staining demonstrated that these cell lines expressed GSC markers CD133 (Figure 3C) and nestin (Figure 3D), the glial cell marker GFAP (Figure 3E), and EGFRvIII (Figure 3F). Mice in group four that received CD133−/EGFRvIII−/EGFR+ cells showed no neurological symptoms at 4 weeks postimplantation. Both micro‐CT scanning and micro‐PET imaging could not detect any tumor formation in these mice. However, tumor formation occurred in two mice up to 4–6 months postimplantation. Taken together, these data revealed that CD133+/EGFRvIII+/EGFR− GMB cells can initiate tumor formation in the brain and acquire the features of malignancy and stemness.
Figure 3.
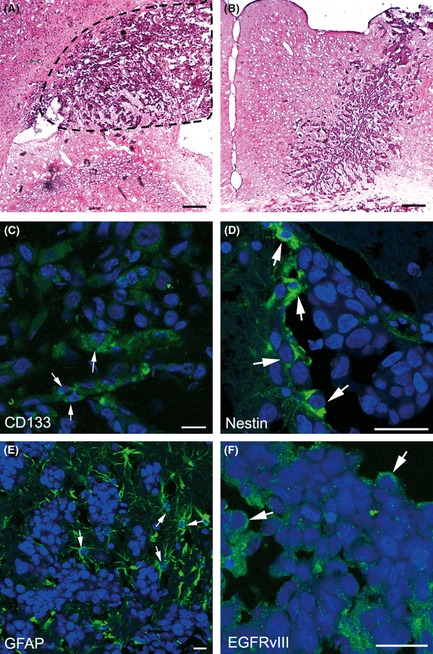
H&E staining of the brain tissues of NOD/SCID mice. Space‐occupying lesions in the area between the cerebral cortex and dorsal hippocampus CA3, as well as many atypical and prominent nuclei, were found in high‐magnification fields (A). The needle track of cell transplantation is shown in (B). Brain tissue sections were also analyzed by immunofluorescence. Some tumor cells were immunoreactive for CD133 (C), nestin (D), GFAP (E), and EGFRvIII (F). Nuclei were counterstained with DAPI. (Scale bars: A, B C, D, and F, 200 μm; E, 40 μm).
CD133+/EGFRvIII+/EGFR− GBM Cells Are More Resistant to Gefitinib
A major possibility raised by the identification of CD133 and EGFRvIII coexpression in GBMs is targeting the EGFR pathway by an inhibitor. For example, gefitinib (trade name Iressa) is an EGFR inhibitor that targets the tyrosine kinase domain. The IC50 values of EGFRvIII+ cells derived from the three GBM clinical specimens were 14.44, 16.00, and 14.66 μM, respectively. In contrast, the IC50 value of EGFRvIII− cells derived from the specimen of Patient No. 10 was 8.57 μM (Figure 4A–E). The P‐values between Patient No. 10 and Patient No. 1–3 were all smaller than 0.0001. The P‐values between Patient No. 1 and 2, Patient No. 1 and 3, and Patient No. 2 and 3 were 0.0226, 0.6940 and 0.0460, respectively. Moreover, western blot results showed that the levels of CD133, nestin, and EGFR were essentially stable after gefitinib treatment, which explicitly illustrates that EGFRvIII+ GBM cells were more resistant to gefitinib compared with that of EGFRvIII− cells (Figure 4F–I). However, phosphorylation of EGFR tyrosine 845, a key regulatory tyrosine residue of the EGFR, and EGFRvIII signal transduction were slightly reduced by daily treatment with 10 μM gefitinib.
Discussion
In 1997, CSCs were isolated by Bonnet and Dick from a subpopulation of leukemic cells that showed a CD34+/CD38− phenotype 26. In recent decades, CSCs have been identified in several solid tumors including brain tumors 20 and breast cancers 27. However, recent studies of CSCs underline many complexities and challenges 28 . The molecular mechanism of the relationship between CSC‐specific markers and the biological characteristics of cancer is still unknown. A number of membrane proteins are considered as markers of multiple types of CSCs. For example, it has been widely accepted that CD133 is a GSC marker 20. In addition, some cell surface proteins associated with signaling pathways are considered as CSC markers, including CD44 29, 30 and CD166 31, as well as high aldehyde dehydrogenase activity 31. Moreover, compelling studies have confirmed that only the CD133+ subset has the ability to maintain tumorigenesis, generate heterogeneity, and initiate tumor formation 32. On the other hand, even a huge number of CD133− cells lack the capacity to form tumors 20, 33. Therefore, CD133 may be a therapeutic target for CSCs 34. However, subsequent studies have shown that CD133 expression is not restricted to CSCs, and both CD133+ and CD133− cells have been detected among brain tumor cells 35, 36. Recently, Stewart et al. 37 found that CD133 expression can change during passaging of some types of stem‐like cells. Taken together, these observations suggest that CD133 as a CSC marker is not convincing. There must be at least some other protein that interacts with CD133 to synergistically promote GSC‐mediated tumorigenesis.
EGFRvIII is a specific variant of the EGFR and is commonly found in glioblastoma 4, 5, 6, 7. Many studies have determined that constitutive activation of EGFRvIII promotes oncogenesis 8, 9. Some recent studies have shown that EGFRvIII expression contributes to CSC phenotypes in some types of cancer, indicating that EGFRvIII may serve as a CSC marker 22. In previous studies, cancers following the hierarchical stem cell model can arise from normal stem cells by acquiring mutations that over‐activate self‐renewal mechanisms 38, 39, or arise from restricted progenitors or differentiated cells as a result of mutations that ectopically activate self‐renewal mechanisms. Therefore, we propose that it is EGFRvIII, a specific genetic deletion mutant, which may synergistically contribute to stemness together with CD133. In our study, we confirmed that CD133+ cells exclusively express EGFRvIII, but not EGFR, and this cell population acquires stemness, suggesting that CD133 possibly has some particular relationship with EGFRvIII, and that EGFRvIII may contribute to the stemness of CSCs. Furthermore, some studies have found that CD133+ only marks CSCs under defined conditions, and CD133+ cells can change along with changes of the niche 37, indicating that CD133+ cells, considered as CSCs, are an unstable and undefined population. Thus, we assume that CD133 expression, when it is regarded as a CSC marker for GBM, would be based on EGFRvIII expression that probably creates a kind of niche for arising CSCs.
Gefitinib (Iressa, ZD1839) is a small molecule inhibitor that specifically binds to and inhibits the EGFR tyrosine kinase, and has been shown to inhibit the growth, proliferation, survival, and invasion of a range of tumor cells overexpressing EGFR 40, 41, 42, 43. However, the clinical response to gefitinib has failed to correlate with EGFR levels and activity, which indicates that there must be other molecular mechanisms involved in the regulation of tumor cell resistance to gefitinib. For this reason, EGFRvIII mutations may be of importance to predict target drug susceptibility and poor prognosis. Many studies have indicated that EGFRvIII expression promotes tumor resistance against gefitinib 44, 45. Moreover, a similar study showed that higher doses and longer exposure to gefitinib decreases EGFRvIII phosphorylation but does not effectively inhibit the biologically relevant processes of DNA synthesis, cellular growth, and invasion 15. Another study reported that gefitinib treatment enhances mitochondrial translocalization of both EGFR and EGFRvIII, and that mitochondrial accumulation of these receptors contributes to tumor drug resistance 18. In addition, a relevant study found that the nuclear EGFRvIII‐STAT5b complex contributes to glioblastoma cell survival by direct activation of the Bcl‐XL promoter 46. Nonetheless, some clinical studies of EGFR inhibitors have still shown inconsistent results 47. Our study verified that EGFRvIII+ GBM shows more resistance to gefitinib compared with that of EGFRvIII− GBM. Exposure to 10 μM gefitinib did not significantly decrease the level of receptor tyrosine phosphorylation. Based on our results, we propose that CD133+/EGFRvIII+/EGFR− cells, as stem‐like cells, can survive gefitinib treatment because of their gefitinib resistance, while CD133−/EGFR+/EGFRvIII+ cells are more sensitive to gefitinib, resulting in a larger number of killed tumor cells (Figure 5). Therefore, individualized treatments for patients with GBMs harboring an active EGFRvIII mutation would be necessary and more reasonable. As new therapeutic programs are developed, it is important to develop a strategy to detect EGFRvIII expression before chemotherapy, so that patients can benefit from the development of a reasonable and effective therapeutic regimen in the clinic.
Figure 5.
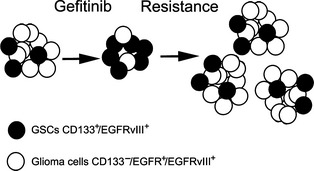
GSC model depicting survival of gefitinib treatment. Glioma following the stem cell model contains different subpopulations of tumorigenic (black) and nontumorigenic (white) cells organized in a hierarchy. In this study, the subpopulation of CD133+/EGFRvIII +/EGFR − cells was confirmed to be tumorigenic and regarded as cancer stem‐like cells. This subpopulation of cells could survive gefitinib treatment and give rise to phenotypically diverse nontumorigenic cells, while the majority of nontumorigenic glioma cells (CD133−/EGFR +/EGFRvIII +) were killed.
Conclusion
In summary, the most significant finding of this study is that cells isolated from clinical specimens express CD133 and are EGFRvIII+/EGFR−. More importantly, this minority subpopulation of cells acquires stemness and the capacity to initiate tumors, which are so‐called “cancer stem‐like cells”. All of these data suggest that coexpression of EGFRvIII and CD133 contributes to the stemness of CSCs in GBMs. Therefore, further research regarding the role of EGFRvIII in GBMs is needed.
Disclosures
The authors certify that this manuscript have not been published or submitted elsewhere.
Conflict of Interest
The authors declare no conflicts of interest.
Supporting information
Figure S1. The middle panels of Figure 1A,B show that the percentages of CD133+ cells from Patient No. 2 and 3 were 89.66% and 85.71%, respectively, by flow cytometric analysis after MACS. The left panels of Figure 1A,B show the results of isotype controls using IgG instead of the anti‐CD133 antibody as the primary antibody. The right panels of Figure 1A,B show H&E staining of the brain tissues of NOD/SCID mice inoculated with cells from Patient No. 2 and 3. We observed space‐occupying lesions in the area between the cerebral cortex and dorsal hippocampus CA3, as well as many atypical and prominent nuclei. Figure 1C shows the micro‐CT scanning (left) and micro‐PET imaging (right) of tumor formation in mouse brains inoculated with CD133+/EGFRvIII+/EGFR− cells (mouse head up position) and CD133−/EGFRvIII−/EGFR+ cells (mouse head down position) from Patient No. 1. Micro‐CT could not detect tumor formation in both mice probably because of the low sensitivity of the equipment and/or the tumor volume was too small. However, micro‐PET could detect tumor formation (at the intersection of the red and yellow lines) in the mouse brain inoculated with CD133+/EGFRvIII+/EGFR− cells, whereas tumor formation was still not detected in the mouse brain inoculated with CD133−/EGFRvIII−/EGFR+ cells.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant number: 81272790) and the Foundation of National Science and Technology Major Project Focusing on Drug Innovation from The Ministry of Science and Technology of the People's Republic of China.
References
- 1. Ekstrand AJ, James CD, Cavenee WK, Seliger B, Pettersson RF, Collins VP. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res 1991;51:2164–2172. [PubMed] [Google Scholar]
- 2. Libermann TA, Razon N, Bartal AD, Yarden Y, Schlessinger J, Soreq H. Expression of epidermal growth factor receptors in human brain tumors. Cancer Res 1984;44:753–760. [PubMed] [Google Scholar]
- 3. Wong AJ, Ruppert JM, Bigner SH, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci USA 1992;89:2965–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N‐ and/or C‐terminal tails. Proc Natl Acad Sci USA 1992;89:4309–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Malden LT, Novak U, Kaye AH, Burgess AW. Selective amplification of the cytoplasmic domain of the epidermal growth factor receptor gene in glioblastoma multiforme. Cancer Res 1998;48:2711–2714. [PubMed] [Google Scholar]
- 6. Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci USA 1990;87:8602–8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamazaki H, Fukui Y, Ueyama Y, et al. Amplification of the structurally and functionally altered epidermal growth factor receptor gene (c‐erbB) in human brain tumors. Mol Cell Biol 1998;8:1816–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nishikawa R, Ji XD, Harmon RC, et al. Huang, A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA 1994;91:7727–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang HS, Nagane M, Klingbeil CK, et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signalling. J Biol Chem 1997;272:2927–2935. [DOI] [PubMed] [Google Scholar]
- 10. Feldkamp MM, Lala P, Lau N, Roncari L, Guha A. Expression of activated epidermal growth factor receptors, Ras‐guanosine triphosphate, and mitogen‐activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery 1999;45:1442–1453. [DOI] [PubMed] [Google Scholar]
- 11. Tang CK, Gong XQ, Moscatello DK, Wong AJ, Lippman ME. Epidermal growth factor receptor vIII enhances tumorigenicity in human breast cancer. Cancer Res 2000;60:3081–3087. [PubMed] [Google Scholar]
- 12. Nagane M, Coufal F, Lin H, Bögler O, Cavenee WK, Huang HJ. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res 1996;56:5079–5086. [PubMed] [Google Scholar]
- 13. Lammering G, Hewit TH, Valerie K, et al. EGFRvIII‐mediated radioresistance through a strong cytoprotective response. Oncogene 2003;22:5545–5553. [DOI] [PubMed] [Google Scholar]
- 14. Weppler SA, Li Y, Dubois L, et al. Expression of EGFR variant vIII promotes both radiation resistance and hypoxia tolerance. Radiother Oncol 2007;83:333–339. [DOI] [PubMed] [Google Scholar]
- 15. Learn CA, Hartzell TL, Wikstrand CJ, et al. Resistance to tyrosine kinase inhibition by mutant epidermal growth factor receptor variant iii contributes to the neoplastic phenotype of glioblastoma multiforme. Clin Cancer Res 2004;10:3216–3224. [DOI] [PubMed] [Google Scholar]
- 16. Sok JC, Coppelli FM, Thomas SM, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res 2006;12:5064–5073. [DOI] [PubMed] [Google Scholar]
- 17. Pillay V, Allaf L, Wilding AL, et al. The plasticity of Oncogene addiction: Implications for targeted therapies directed to receptor tyrosine kinases. Neoplasia 2009;11:448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cao X, Zhu H, Ali‐Osman F, Lo HW. EGFR and EGFRvIII undergo stress‐ and EGFR kinase inhibitor‐induced mitochondrial translocalization: A potential mechanism of EGFR‐driven antagonism of apoptosis. Mol Cancer 2011;10:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem‐like neural precursors from human glioblastoma. Cancer Res 2004;64:7011–7021. [DOI] [PubMed] [Google Scholar]
- 20. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature 2004;432:396–401. [DOI] [PubMed] [Google Scholar]
- 21. Yuan X, Curtin J, Xiong Y, et al. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene 2004;23(58):9392–9400. [DOI] [PubMed] [Google Scholar]
- 22. Yuan X, Curtin J, Xiong Y, et al. Epidermal growth factor receptor variant III contributes to cancer stem cell phenotypes in invasivebreast carcinoma. Cancer Res 2012;72:2657–2671. [DOI] [PubMed] [Google Scholar]
- 23. Ma HI, Chiou SH, Hueng DY, et al. Celecoxib and radioresistant glioblastoma‐derived CD133+ cells: Improvement in radiotherapeutic effects. Laboratory investigation. J Neurosurg 2011;114:651–662. [DOI] [PubMed] [Google Scholar]
- 24. McCord AM, Jamal M, Williams ES, Camphausen K, Tofilon PJ. CD133 + glioblastoma stem‐like cells are radiosensitive with a defective DNA damage response compared with established cell lines. Clin Cancer Res 2009;15:5145–5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Blazek ER, Foutch JL, Maki G. Daoy medulloblastoma cells that express CD133 are radioresistant relative to CD133‐ cells, and the CD133+ sector is enlarged by hypoxia. Int J Radiat Oncol Biol Phys 2007;67:1–5. [DOI] [PubMed] [Google Scholar]
- 26. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730–737. [DOI] [PubMed] [Google Scholar]
- 27. Al‐Hajj M, Wicha MS, Benito‐Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003;100:3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Visvader JE, Lindeman GJ. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012;10:717–728. [DOI] [PubMed] [Google Scholar]
- 29. Meyer MJ, Fleming JM, Lin AF, Hussnain SA, Ginsburg E, Vonderhaar BK. CD44posCD49fhi CD133/2hi defines xenograft‐initiating cells in estrogen receptor‐negative breast cancer. Cancer Res 2010;70:4624–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang WC, Shyh‐Chang N, Yang H., et al. Glycine decarboxylase activity drives nonsmall cell lung cancer tumor‐initiating cells and tumorigenesis. Cell 2012;148:259–272. [DOI] [PubMed] [Google Scholar]
- 31. Silva IA, Bai S, McLean K, et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells. Cancer Res 2011;71:3991–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lathia JD, Hitomi M, Gallagher J, et al. Distribution of CD133 reveals glioma stem cells self‐renew through symmetric and asymmetric cell divisions. Cell Death Dis 2011;2:e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444:756–760. [DOI] [PubMed] [Google Scholar]
- 34. Rappa G, Fodstad O, Lorico A. The stem cell‐associated antigen CD133 (Prominin‐1) is a molecular therapeutic target for metastatic melanoma. Stem Cells 2008;26:3008–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beier D, Hau P, Proescholdt M, et al. CD133(+) and CD133(‐) glioblastoma‐derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res 2007;67:4010–4015. [DOI] [PubMed] [Google Scholar]
- 36. Chen R, Nishimura MC, Bumbaca SM, et al. A hierarchy of self‐renewing tumor‐initiating cell types in glioblastoma. Cancer Cell 2010;17:362–375. [DOI] [PubMed] [Google Scholar]
- 37. Stewart JM, Shaw PA, Gedye C, Bernardini MQ, Neel BG, Ailles LE. Phenotypic heterogeneity and instability of human ovarian tumorinitiating cells. Proc Natl Acad Sci USA 2010;108:6468–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells‐of‐origin of intestinal cancer. Nature 2009;457:608–611. [DOI] [PubMed] [Google Scholar]
- 39. Yang ZJ, Ellis T, Markant SL, et al. Medulloblastoma can be initiated by deletion of Patched in lineage‐restricted progenitors or stem cells. Cancer Cell 2008;14:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sirotnak FM, Zakowski MF, Miller VA, Scher HI, Kris MG. Efficacy of cytotoxic agents against human tumor xenografts is markedly enhanced by coadministration of ZD1839 (Iressa), and inhibitor of EGFR tyrosine kinase. Clin Cancer Res 2000;6:4885–4892. [PubMed] [Google Scholar]
- 41. Ciardiello F, Caputo R, Bianco R, et al. Tortora inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clin Cancer Res 2001;7:1459–1465. [PubMed] [Google Scholar]
- 42. Lichtner RB, Menrad A, Sommer A, Klar U, Schneider MR. Signaling‐inactive epidermal growth factor receptor/ligand complexes in intact carcinoma cells by quinazoline tyrosine kinase inhibitors. Clin Cancer Res 2001;61:5790–5795. [PubMed] [Google Scholar]
- 43. Baselga J, Averbuch SD. ZD1839 (“Iressa”) as an anticancer agent. Drugs 2000;60:33–40. [DOI] [PubMed] [Google Scholar]
- 44. Heimberger AB, Learn CA, Archer GE, et al. Brain tumors in mice are susceptible to blockade of epidermal growth factor receptor (EGFR) with the oral, specific, EGFR‐tyrosine kinase inhibitor ZD1839 (iressa). Clin Cancer Res 2002;8:3496–3502. [PubMed] [Google Scholar]
- 45. Pedersen MW, Pedersen N, Ottesen LH, Poulsen HS. Differential response to gefitinib of cells expressing normal EGFR and the mutant EGFRvIII. Br J Cancer 2005;93:915–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Latha K, Li M, Chumbalkar V, et al. Nuclear EGFRvIII‐STAT5b complex contributes to glioblastoma cell survival by direct activation of the Bcl‐XL promoter. Int J Cancer 2013;132:509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 2005;353:2012–2024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The middle panels of Figure 1A,B show that the percentages of CD133+ cells from Patient No. 2 and 3 were 89.66% and 85.71%, respectively, by flow cytometric analysis after MACS. The left panels of Figure 1A,B show the results of isotype controls using IgG instead of the anti‐CD133 antibody as the primary antibody. The right panels of Figure 1A,B show H&E staining of the brain tissues of NOD/SCID mice inoculated with cells from Patient No. 2 and 3. We observed space‐occupying lesions in the area between the cerebral cortex and dorsal hippocampus CA3, as well as many atypical and prominent nuclei. Figure 1C shows the micro‐CT scanning (left) and micro‐PET imaging (right) of tumor formation in mouse brains inoculated with CD133+/EGFRvIII+/EGFR− cells (mouse head up position) and CD133−/EGFRvIII−/EGFR+ cells (mouse head down position) from Patient No. 1. Micro‐CT could not detect tumor formation in both mice probably because of the low sensitivity of the equipment and/or the tumor volume was too small. However, micro‐PET could detect tumor formation (at the intersection of the red and yellow lines) in the mouse brain inoculated with CD133+/EGFRvIII+/EGFR− cells, whereas tumor formation was still not detected in the mouse brain inoculated with CD133−/EGFRvIII−/EGFR+ cells.