

Summary
Background and purpose
Beta‐amyloid (Aβ)‐mediated inflammation contributes to the progression and chronicity of Alzheimer's disease (AD), although the exact mechanism remains unclear. This study aimed to investigate whether diammonium glycyrrhizinate (DG) could inhibit Aβ‐induced inflammation in vitro and in vivo and to explore the underlying mechanisms.
Methods
Aβ1–42 was injected to bilateral hippocampus of mice to make the AD models in vivo. The levels of mRNA and protein of inflammatory cytokines were measured by real‐time PCR and Western blotting, respectively. The viability of SH‐SY5Y and HT‐22 cells was determined by MTT. NF‐κB p65 translocation was analyzed by Western blotting and immunostaining. Phosphorylation of ERK, p38, and JNK was tested by Western blotting.
Results
DG suppressed Aβ1–42‐induced activation of microglia and inflammation in vitro and in vivo. The media from Aβ1–42‐activated microglia decreased the viability of SH‐SY5Y and HT‐22 cells, but it was rescued when pretreated with DG. DG could inhibit the activation of MAPK and NF‐κB signaling pathways and attenuate the memory deficits in Aβ1–42‐induced AD mice.
Conclusions
DG protects Aβ1–42‐induced AD models in vitro and in vivo through reducing activation of microglia and inflammation, which may be involved in MAPK and NF‐κB pathways.
Keywords: Alzheimer's disease, Beta‐amyloid, Diammonium glycyrrhizinate, Inflammation, MAPK, NF‐κB pathway
Introduction
Alzheimer's disease (AD), an age‐related disorder with progressive cognitive decline and dementia, afflicts estimated 30 million people worldwide. AD is pathologically characterized by the senile plaques mainly composed of beta‐amyloid (Aβ) and neurofibrillary tangles (NFTs) 1. An increasing number of basic and clinical studies demonstrate that Aβ‐mediated inflammation is a key pathological hallmark of AD, which may contribute to disease progression and chronicity 2, 3. In addition, epidemiological studies show that treatment with nonsteroidal antiinflammatory drugs (NSAIDs) could decrease the risk for developing AD, although more clinical evidences are needed 4.
Inflammatory components related to AD neuroinflammation include brain cells such as microglia, astrocytes, and cytokines. Once activated by Aβ, microglia and astrocytes release inflammatory factors such as interleukin‐1 (IL‐1), IL‐6, inducible nitric oxide synthase (iNOS), cyclooxygenase‐2 (COX‐2), and tumor necrosis factor‐α (TNF‐α), which results in neuron death 3. Mitogen‐activated protein kinase (MAPK) family includes three subtypes: p38, extracellular signal‐regulated kinase (ERK), and c‐Jun N‐terminal kinase (JNK). Reports have indicated that MAPK signaling pathways are excessively activated in AD. ERK and/or p38 inhibitors could reduce the neurotoxicity by Aβ 5. The NF‐κB pathway is also involved in the brain proinflammatory process of AD. Proinflammatory mediators TNF‐α and IL‐1β, which are regulated by NF‐κB, are increased in the brains of AD patients 6, 7, and the activation of NF‐κB signaling pathway also occurs in Aβ‐induced neuron death 7. Suppression of NF‐κB pathway in microglia can ameliorate neurotoxicity 8. COX‐2, mainly regulated by NF‐κB, is notably upregulated in the brains of AD patients, which may be associated with the formation of Aβ plaque 9. These results suggest that suppression of neuroinflammatory molecules could be developed as a therapeutic method of AD. However, no drugs have been clinically proven effective in the treatment of neuroinflammation of AD.
Diammonium glycyrrhizinate (DG), the salt form of glycyrrhizin acid (GA), has more stable, more significant bioactivities than GA. GA has a series of biological functions including antitumor, antivirus, and antiinflammatory effects 10, 11, 12, 13. DG was used for treatment of hepatitis in Asian countries mainly due to its antiinflammatory and antioxidative effects 14, 15. In addition, DG decreases the NF‐κB pathway and inflammatory injury in a rat model of ulcerative colitis induced by acetic acid 16. Previous studies suggest that DG could improve focal cerebral ischemic–reperfusion injury in mice due to its antiinflammatory effect 17. In this study, evidence was provided that DG played a critical role in suppressing Aβ1–42‐induced inflammation through regulating MAPK and NF‐κB pathways, suggesting that DG might be developed into a potential drug for treatment of inflammation‐related neurodegeneration diseases, including AD.
Materials and Methods
Aβ1–42‐mediated AD Mice Model
Aβ1–42 (Millipore, CA, USA) was dissolved in 1% NH3·H2O at a concentration of 100 μM and incubated at 37°C for 5 days to allow for fibril formation. DG was purchased from Jiangsu Chia‐Tai Tianqing Pharmacy Company (Lianyungang, China). As described previously 18, Aβ1–42 (4 ug) was injected to the bilateral hippocampus (anterior–posterior position −2.0 mm, medial–lateral position ±1.6 mm, dorsoventral −1.5 mm from Bregma) of male ICR mice by infusion cannulae once. DG (10 mg/kg per day, i.p.) was injected intraperitoneally for 14 days. Control mice were injected with saline. Mice were divided into four groups, that is, Aβ1–42‐mediated AD mice with or without DG, and control mice with or without DG. All animal experiments were approved by the Animal Care Committee of Nanjing University and performed according to institutional guidelines. Every effort was made to minimize the number of mice used and their suffering.
Cell Culture
BV‐2 (a murine microglia cell line) and SH‐SY5Y (a neuroblastoma cell line) cells were obtained from American Type Culture Collection (ATCC). HT‐22, a murine hippocampus neural cell line, was a kind gift of Dr. Jian Chen (Suzhou University, China). BV‐2, SH‐SY5Y, and HT‐22 cells were maintained in DMEM containing 10% heat‐inactivated fetal bovine serum (FBS, Gibco), 2 mM of l‐glutamine, 100 U/mL of penicillin, and 100 μg/mL of streptomycin at 37°C in a humidified 5% CO2 incubator.
Real‐time PCR
Real‐time PCR was performed as described previously 18. Briefly, total RNA was extracted by using the Trizol reagent (Takara, Dalian, China) and was reverse‐transcribed into cDNA using a PrimeScript RT reagent kit (Takara) for Quantitative PCR (ABI 7500, Foster City, CA, USA) in the presence of a fluorescent dye (SYBR Green I; Takara). The primers are as follows:
iNOS: Forward, CAGCTGGGCTGTACAAACCTT, Reverse, CATTGGAAGTGAAGCGTTTCG;
COX‐2: Forward, GATGACTGCCCAACTCCC, Reverse, AACCCAGGTCCTCGCTTA;
IL‐1β: Forward, AAGCCTCGTGCTGTCGGACC, Reverse, TGAGGCCCAAGGCCACAGGT;
TNF‐α: Forward, CAAGGGACAAGGCTGCCCCG, Reverse, GCAGGGGCTCTTGACGGCAG;
IL‐10: Forward, GGCATGAGGATCAGCAGGGGC, Reverse, TGGCTGAAGGCAGTCCGCAG;
GAPDH: Forward, GCCAAGGCTGTGGGCAAGGT, Reverse, TCTCCAGGCGGCACGTCAGA.
Western Blotting
Equal amounts of protein were separated by SDS‐PAGE, electrophoretically transferred onto polyvinylidene fluoride membranes, and incubated overnight at 4°C with anti‐COX‐2 (1: 500, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐iNOS (1: 1000, Cell Signaling, Danvers, MA, USA), anti‐p‐ERK (1: 1000, Cell Signaling), anti‐ERK (1: 1000, Cell Signaling), anti‐p‐JNK (1: 1000, Cell Signaling), anti‐JNK (1: 1000, Cell Signaling), anti‐p‐p38 (1: 1000, Cell Signaling), anti‐p38 (1: 1000, Cell Signaling), anti‐p65 (1: 500, Santa Cruz Biotechnology), and anti‐β‐actin (1: 5000, Bioworld, Louis Park, MN, USA) antibodies. Subsequently, the membranes were incubated with the corresponding secondary antibodies and visualized with chemiluminescence reagents provided with an ECL kit (Bioworld).
MTT Assay
BV‐2 microglial cells were treated with Aβ1–42 (5 μM) for 6 and 24 h in the presence and absence of DG (0.001 mg/mL). The culture media of BV‐2 cells were collected as conditioned media. SH‐SY5Y and HT‐22 cells were treated with different groups of conditional media for 6 and 24 h, and cell viabilities were measured by MTT assay as described previously 18.
Nuclear Protein Extraction
The nuclear protein was extracted using the Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Nantong, China) following the manufacturer's instruction. Hippocampal tissue was homogenized and resuspended in the cytoplasmic protein extraction reagent, followed by centrifugation at 14,000 g for 10 min at 4°C. The supernatant was the cytoplasmic protein, and the pellet was resuspended in the nuclear protein extraction reagent followed by centrifugation at 14,000 g for 15 min at 4°C. The supernatant was the nuclear protein. The cytoplasmic and nuclear protein was then subjected to Western blotting.
Immunostaining
BV‐2 cells were seeded on cover slips after treatment cells were fixed in 4% paraformaldehyde for 20 min at room temperature. The cells were incubated overnight with anti‐p65 (1: 100, Santa Cruz) at 4°C and then incubated with the secondary antibody (1: 200, Invitrogen, Carlsbad, CA, USA) for 45 min at room temperature. Fluorescent images were taken using a fluorescent microscope (Olympus, Japan).
Mice were anesthetized and transcardially perfused with 0.9% saline followed by 4% paraformaldehyde (PFA). The brains were cut into consecutive frozen sections and incubated overnight with antiglial fibrillary acidic protein (GFAP) (1: 100, BD) and antiionized calcium‐binding adaptor molecule 1 (Iba1) (1: 400, Abcam, Cambridge, MA, USA) at 4°C and then incubated with the secondary antibody (1: 200, Invitrogen) for 45 min at room temperature. The immunoreactive cells to GFAP and Iba1 in about 100 areas of 100 × 100 μm in 5 slices per brain were counted. All counting procedures were made by an expert in morphology blinded to the experiment.
Morris Water Maze Test
The Morris water maze test was performed as described previously 18. Briefly, four training trials per day were conducted for four consecutive days. In each trial, the latency to escape onto the platform was recorded up to 60 s. If the mouse found the platform, it was allowed to remain on the platform for 5 s, and then returned to the home cage. If the mouse was unable to find the platform within 60 s, it was gently guided to the platform and allowed to remain on the platform for 10 s and the latency was recorded for 60 s. On the fifth day, the probe trial was conducted for memory retention by removing the platform and each mouse was allowed to swim freely for 60 s. The time spent in each quadrant was recorded and analyzed. All data were recorded with a computerized video system.
Statistical Analysis
The data were expressed as means ± SEM and analyzed by SPSS 12.0 statistical analytical software (SPSS, Chicago, IL, USA). Group differences in the escape latency and searching distance during the MWM test were analyzed using two‐way analysis of variance (ANOVA) with repeated measures followed by Bonferroni multiple comparison test with day and treatment as the sources of variation. Otherwise, statistical analysis among groups was performed with one‐way ANOVA followed by Bonferroni's post hoc test. Values of P < 0.05 were considered statistically significant.
Results
DG Inhibits the Inflammatory Response in Aβ1–42‐treated BV‐2 Cells
To investigate the effect of DG on the inflammatory response induced by Aβ1–42 in vitro, BV‐2 cells were pretreated with DG (0.001 mg/mL) for 1 h and then with Aβ1–42 (5 μM) for 3, 6, 12, and 24 h. As shown in Figure 1A–C, and 1D, the mRNA level of TNF‐α, COX‐2, iNOS, and IL‐1β was significantly increased at 3, 6, 12, and 24 h after Aβ1–42 treatment (P < 0.01), while it was greatly decreased in the presence of DG. In addition, Aβ1–42 treatment could significantly downregulate the expression of antiinflammatory factor IL‐10 (P < 0.01), while DG treatment could significantly increase the expression of IL‐10 at 6 h (P < 0.01, Figure 1E). DG also attenuated the protein expression of COX‐2 (96.92 ± 4.85% at 3 h, 72.73 ± 4.31% at 6 h, and 76.78 ± 6.16% at 24 h) and iNOS (63.34 ± 2.77% at 3 h, 56.82 ± 3.08% at 6 h, and 82.32 ± 6.58% at 24 h) in Aβ1–42‐treated BV‐2 cells (Figure 1F).
Figure 1.
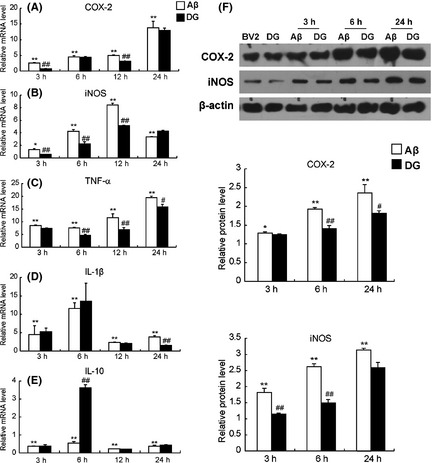
Diammonium glycyrrhizinate (DG) suppresses the inflammatory response in Aβ1–42‐treated BV‐2 cells. BV‐2 cells were pretreated with DG (0.001 mg/mL) for 1 h and Aβ1–42 (5 μM) for 3, 6, and 24 h. The mRNA levels of TNF‐α (A), COX‐2 (B), iNOS (C), IL‐1β (D), and IL‐10 (E) were determined by real‐time PCR. (F)The protein levels of COX‐2 and iNOS were determined by Western blotting. The relative mRNA and protein levels of BV‐2 cells were set as 1. *: P < 0.05 versus BV‐2 cells; **: P < 0.01 versus BV‐2 cells; #: P < 0.05 versus Aβ1–42‐treated BV‐2 cells; ##: P < 0.01 versus Aβ1–42‐treated BV‐2 cells.
DG Inhibits the Inflammatory Factors in Aβ1–42‐induced AD Mice
To further confirm inhibitory inflammation of DG in vivo, the relative mRNA levels of COX‐2, iNOS, TNF‐α, IL‐1β, and IL‐10 and the relative protein levels of COX‐2 and iNOS in the hippocampus of mice were determined. As shown in Figure 2A, the mRNA level of COX‐2 was increased 1.49 ± 0.07‐fold in Aβ1–42‐induced AD mice, while DG could significantly decrease the mRNA level of COX‐2 (77.43 ± 3.07%, P < 0.01). Meanwhile, DG significantly decreased the mRNA level of TNF‐α, iNOS, and IL‐1β (66.43 ± 4.44%, 24.05 ± 5.93%, and 58.79 ± 5.90%, P < 0.01, Figure 2B–D). In addition, the mRNA level of IL‐10 was increased by 1.53 ± 0.07‐fold in Aβ1–42‐induced AD mice treated with DG (Figure 2E). The protein levels of COX‐2 and iNOS in the hippocampus of AD mice were also attenuated after the treatment of DG (52.89 ± 4.15% and 56.75 ± 6.36%, Figure 2F).
Figure 2.
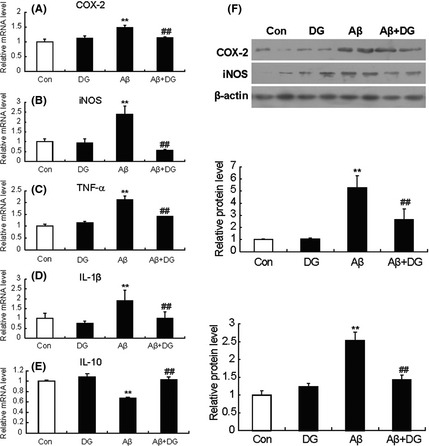
Diammonium glycyrrhizinate (DG) reduces the inflammatory factors in Aβ1–42‐induced AD mice. The mRNA levels of TNF‐α (A), COX‐2 (B), iNOS (C), IL‐1b (D), and IL‐10 (E) were determined by real‐time PCR. (F)The protein levels of COX‐2 and iNOS were determined by Western blotting. The relative mRNA and protein levels of normal mice were set as 1. Con: normal mice; DG: normal mice with DG (10 mg/kg per day, i.p. for 14 days); Aβ: Aβ1–42‐induced AD mice; Aβ+DG: Aβ1–42‐induced AD mice with DG (10 mg/kg per day, i.p. for 14 days). n = 4 mice per group. *: P < 0.05 versus Control; **: P < 0.01 versus Control; #: P < 0.05 versus Aβ1–42‐induced AD mice; ##: P < 0.01 versus Aβ1–42‐induced AD mice.
DG Ameliorates the Microglia Activation and Attenuates Memory Deficits in Aβ1–42‐mediated AD Mice
Next, to reveal whether the inflammation was due to the activation of microglia and astrocyte by Aβ1–42, Iba‐1 and GFAP staining was performed. Iba‐1 has specific affinity for microglial Ca2+‐binding proteins, which are highly expressed in activated microglia, and GFAP is a marker of astrocyte activation. The results indicated that Iba‐1 expression in the hippocampus of AD mice was significantly increased compared with that in control mice (P < 0.01), whereas it was significantly reduced to 57.14 ± 7.41% after the treatment of DG (P < 0.01). Likewise, the magnitude of astrocytosis was also significantly reduced to 52.17 ± 4.55% in the hippocampus of DG‐treated AD mice (P < 0.01, Figure 3A).
Figure 3.
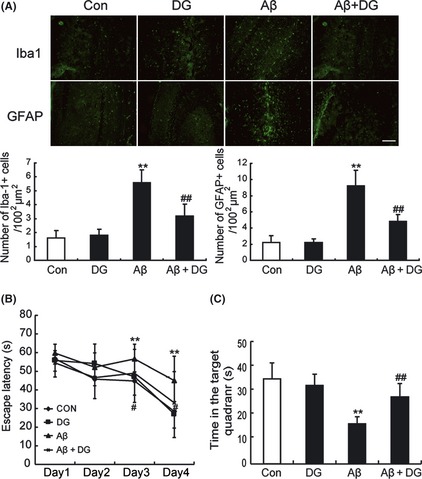
Diammonium glycyrrhizinate (DG) ameliorates the microglia activation and attenuates memory deficits in Aβ1–42‐induced AD mice. (A) Immunostaining for Iba‐1 and GFAP in the hippocampus of normal mice and Aβ1–42‐induced AD mice treated with or without DG (10 mg/kg per day, i.p. for 14 days). n = 3 mice per group. **: P < 0.01 versus Control; ##: P < 0.01 versus Aβ1–42‐induced AD mice. (B) Escape latency for escape to a submerged platform in the training trials. (C) Time spent in the target quadrant during the probe trail. **: P < 0.01 compared to control mice, #: P < 0.05 compared to Aβ1–42‐induced AD mice. n = 10 mice per group. Con: normal mice; DG: normal mice with DG (10 mg/kg per day, i.p. for 14 days); Aβ: Aβ1–42‐induced AD mice; Aβ+DG: Aβ1–42‐induced AD mice with DG (10 mg/kg per day, i.p. for 14 days).
To confirm that DG could attenuate memory impairment in Aβ1–42‐induced AD mice, Morris water maze test was performed. As shown in Figure 3B, the mean escape latency of AD mice was significantly increased compared with control mice (P < 0.01), while DG‐treated AD mice showed significant improvements compared with AD mice after the training periods (P < 0.05). During the probe trial, Aβ1–42‐induced AD mice spent significantly less time in the target quadrant than control mice and DG treatment could reverse such effect (Figure 3C). These results demonstrated that DG treatment could ameliorate microglia activation and attenuate memory deficits in Aβ1–42‐induced AD mice.
Neuroprotective Effects of DG against Neuronal Cell Death by Aβ1–42‐activated Microglia in Conditioned Media System
To validate whether inhibition of the inflammatory response by DG in microglia could protect the neurons, conditional media of Aβ1–42‐treated BV‐2 cells with or without DG were collected to treat HT22 and SH‐SY5Y cells. As shown in Figure 4A, treatment of conditional media of Aβ1–42‐treated BV‐2 cells without DG decreased cell viability of HT‐22 cells to 41.18 ± 2.76% and 31.40 ± 2.05% at 6 and 24 h. However, cell viability, when treated with conditional media with DG, was markedly increased by 1.05 ± 0.11‐fold and 1.58 ± 0.10‐fold at 6 and 24 h. Similarly, results appeared in SH‐SY5Y cells (Figure 4B), suggesting that neuroprotection of DG against Aβ1–42‐mediated toxicity may be partially related to inhibition of activated microglia.
Figure 4.
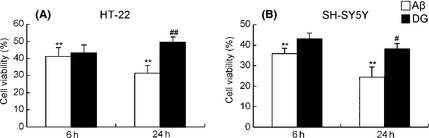
Neuroprotective effects of diammonium glycyrrhizinate (DG) against neuronal cell death by Aβ‐activated microglia in conditioned media system. Conditional media of Aβ1–42‐treated BV‐2 cells with or without DG were collected to treat HT22 and SH‐SY5Y cells for 6 and 24 h. (A) Cell viability of SH‐SY5Y cells was determined by MTT. (B) Cell viability of HT‐22 cells was determined by MTT. *: P < 0.05 versus conditional media of Aβ1–42‐treated BV‐2 cells, **: P < 0.01 versus conditional media of Aβ1–42‐treated BV‐2 cells.
DG Inhibits Aβ1–42‐induced Activation of NF‐κB p65 and MAPK Signaling Pathways In Vivo and In Vitro
To elucidate the mechanism of DG in suppression of inflammation by Aβ1–42, NF‐κB p65 and MAPK signaling pathways, which contributed to the microglial activation and inflammation, were studied. As shown in Figure 5A,B, there was a low basal level of p65 in the nuclei of normal mice and Aβ1–42‐induced a translocation of p65 from the cytoplasm to the nucleus, while DG significantly attenuated the p65 translocation. These results were also confirmed by immunostaining of p65 in BV‐2 cells (Figure 5C), which indicated that DG could attenuate Aβ1–42‐induced NF‐κB nuclear translocation.
Figure 5.
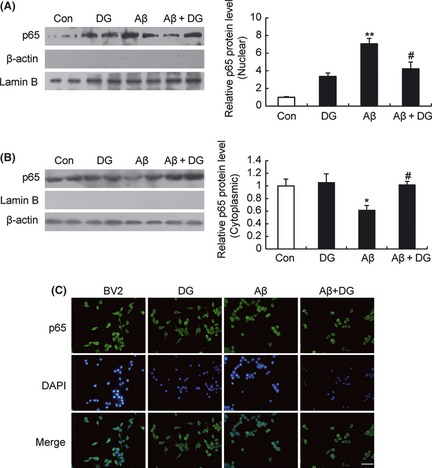
Diammonium glycyrrhizinate (DG) inhibits Aβ1–42‐induced translocation of NF‐κB p65 in vivo and in vitro. The expression of NF‐κB p65 in the nucleus (A) and cytoplasm (B) of hippocampus of normal mice and Aβ1–42‐induced AD mice treated with or without DG (10 mg/kg per day, i.p. for 14 days) was determined by Western blotting. n = 4 mice per group. **: P < 0.01 versus Control; #: P < 0.05 versus Aβ1–42‐induced AD mice. (C) BV‐2 cells were pretreated with DG (0.001 mg/mL) for 1 h and Aβ1–42 (5 μM) for 6 h and immunostaining for NF‐κB p65. Scale bar = 50 μm.
The activation of MAPK pathways in the AD model was also tested. As shown in Figure 6, Aβ1–42‐activated phosphorylation of ERK, JNK, and p38 in the hippocampus of Aβ1–42‐induced AD mice, while DG significantly decreased the phosphorylation levels.
Figure 6.
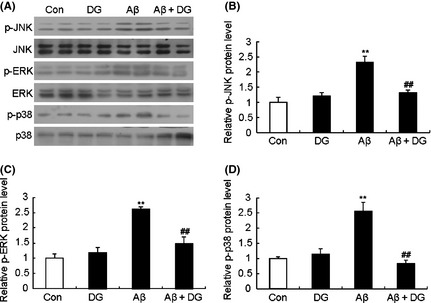
DG inhibits Aβ1–42‐induced activation MAPK signaling pathways in vivo. (A) The expression of JNK, ERK and p38 MAPK signaling of hippocampus of normal mice and Aβ1–42‐induced AD mice treated with or without DG (10 mg/kg per day, i.p. for 14 days) was determined by western blotting. Quantitative analysis of the relative protein levels of p‐JNK (B), p‐ERK (C) and p‐p38 (D) of (A). n = 4 mice per group. **P < 0.01 versus control; ## P < 0.01 versus Aβ1–42‐induced AD mice.
Discussion
This study showed, for the first time, that DG could protect against Aβ1–42‐induced inflammation response in vitro and in vivo, and the underlying mechanism of this neuroprotection may be involved of downregulation of MAPK and NF‐κB pathways.
Aβ is the major component of senile plaques, which contribute to the pathogenesis of AD. Some studies have shown that there is an association between higher inflammatory levels and lower cognitive levels or higher risk of cognitive impairment over time in populations of older adults living in the community. Specific circulating inflammatory markers have been associated with declines in cognitive function and worsening of brain structure 19. Aβ could activate microglia and astrocytes, which produce a wide spectrum of proinflammatory factors such as IL‐1β, TNF‐α, iNOS, COX‐2, and so forth 8, 19. Current study indicated that Aβ1–42 did activate microglia, and the mRNA level of COX‐2, iNOS, TNF‐α, and IL‐1β was increased in vivo and in vitro. DG could reduce the release of Aβ1–42‐induced proinflammatory factors and suppress the activation of microglia. Of great interest were the results showing that media from Aβ1–42‐treated BV‐2 cells could decrease the cell viability of HT22 and SH‐SY5Y cells. However, cell viability of these two cells was significantly increased after adding DG. These findings indicated that DG indeed protected neurons from activated microglia induced by Aβ1–42. Given the fact that DG could directly protect the primary cortical neurons against Aβ1–42‐induced oxidative stress 18, it was likely that DG exerted protective effects by modulating the glial cells and neurons simultaneously. Furthermore, this study demonstrated that DG could ameliorate memory deficits partially through inhibiting brain inflammation in Aβ1–42‐mediated AD mouse models.
However, the mechanism on Aβ‐induced neuroinflammation has not been fully elucidated. Researches have shown that NF‐κB pathway is activated, and both increased proinflammatory cytokines and activated glial cells are frequently around Aβ plaque in the brains of AD patients 20, 21. NF‐κB pathway has been regarded as one of the key mediators of aging and mediates immune and inflammatory responses. Without stimuli, NF‐κB is retained in the cytoplasm by binding to IκB inhibitors. When activated, IκB is phosphorylated by IκB kinases and then degraded by the proteasome, which allows the translocation of p65/p50 from the cytoplasm to the nuclei 22. The SIRT1 agonist resveratrol exerts neuroprotective effects by markedly suppression of NF‐κB signaling in microglia and decreasing of Aβ toxicity 9. NF‐κB pathway also participates in the formation of amyloid‐β precursor (APP) protein 22. Moreover, NF‐κB can regulate the expression of COX‐2 and control the production of proinflammatory mediators such as TNF‐α and IL‐1β. In this study, our results demonstrated that DG significantly attenuated the NF‐κB p65 translocation induced by Aβ1–42 in vivo and in vitro.
In addition, MAPK pathways are associated with Aβ‐induced inflammation. Aromatic turmerone protects the hippocampus from indirect neuronal toxicity induced by Aβ‐stimulated microglia through inhibiting NF‐κB, JNK, and p38 MAPK signaling pathways 23. Another study suggests that Aβ1–42‐induced activation of MEK/ERK pathway leads to the release of inflammatory factors such as IL‐1β and TNF‐α, which might be toxic to neuroblastoma SK‐N‐SH cells and lead to the onset of AD 24. In Aβ‐stimulated SH‐SY5Y cells, Xylocoside G exerts neuroprotection by inhibition of JNK and NF‐κB signaling and reduction of Aβ‐induced inflammation and apoptosis 25. This study demonstrated that DG significantly decreased the phosphorylation of ERK, JNK, and p38 in the hippocampus of Aβ1–42‐induced AD mice, which was also supported by reports that ERK and p38 inhibitors attenuated memory deficit in Aβ‐treated rats 26, and inhibition of JNK phosphorylation reversed memory deficit in Aβ‐meditated rats 27.
In conclusion, the present study demonstrates that administration of DG provides neuroprotection against Aβ‐induced inflammation in vitro and in vivo, and the mechanisms may be related with downregulation of MAPK and NF‐κB pathways. The findings contribute to the understanding of pathological effects of Aβ‐activated microglia in the inflammatory process of AD brain and point to an unidentified role of DG as a neuroprotective agent for AD.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by the National Nature Science Foundation of China (81200839 and 30971010), the State Key Laboratory of Pharmaceutical Biotechnology of Nanjing University (KF‐GN‐200901), the Medical Leading Talent and Innovation Team Project of Jiangsu Province (LJ201101), the National Natural Science Foundation of Jiangsu Province of China (BL2012013), and Key Project of Nanjing Municipal Bureau of Health (ZKX08027). We thank Brad Peterson for writing modification.
The first two authors contributed equally to this work.
References
- 1. Winner B, Kohl Z, Gage FH. Neurodegenerative disease and adult neurogenesis. Eur J Neurosci 2011;33:1139–1151. [DOI] [PubMed] [Google Scholar]
- 2. Azizi G, Mirshafiey A. The potential role of proinflammatory and antiinflammatory cytokines in Alzheimer disease pathogenesis. Immunopharmacol Immunotoxicol 2012;34:881–895. [DOI] [PubMed] [Google Scholar]
- 3. He FQ, Qiu BY, Zhang XH, et al. Tetrandrine attenuates spatial memory impairment and hippocampal neuroinflammation via inhibiting NF‐kappaB activation in a rat model of Alzheimer's disease induced by amyloid‐beta(1–42). Brain Res 2011;1384:89–96. [DOI] [PubMed] [Google Scholar]
- 4. Rubio‐Perez JM, Morillas‐Ruiz JM. A review: Inflammatory process in Alzheimer's disease, role of cytokines. ScientificWorldJournal 2012;2012:756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Munoz L, Ralay Ranaivo H, Roy SM, et al. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up‐regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer's disease mouse model. J Neuroinflammation 2007;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ashabi G, Alamdary SZ, Ramin M, Khodagholi F. Reduction of Hippocampal Apoptosis by Intracerebroventricular Administration of ERK and/or p38 Inhibitors in Abeta Rat Model of Alzheimer's Disease: Involvement of Nrf2 and NF‐kappaB. Basic Clin Pharmacol Toxicol 2012;doi: 10.1111/bcpt.12000. [DOI] [PubMed] [Google Scholar]
- 7. Steinman L. Nuanced roles of cytokines in three major human brain disorders. J Clin Invest 2008;118:3557–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hull M, Muksch B, Akundi RS, et al. Amyloid beta peptide (25–35) activates protein kinase C leading to cyclooxygenase‐2 induction and prostaglandin E2 release in primary midbrain astrocytes. Neurochem Int 2006;48:663–672. [DOI] [PubMed] [Google Scholar]
- 9. Chen J, Zhou Y, Mueller‐Steiner S, et al. SIRT1 protects against microglia‐dependent amyloid‐beta toxicity through inhibiting NF‐kappaB signaling. J Biol Chem 2005;280:40364–40374. [DOI] [PubMed] [Google Scholar]
- 10. Michaelis M, Geiler J, Naczk P, et al. Glycyrrhizin exerts antioxidative effects in H5N1 influenza A virus‐infected cells and inhibits virus replication and pro‐inflammatory gene expression. PLoS ONE 2011;6:e19705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chandrasekharan DK, Nair CK. Studies on Silver Nanoparticle‐Glycyrrhizic Acid Complex as a Radioprotector and an Adjuvant in Radiotherapy Under In Vivo Conditions. Cancer Biother Radiopharm 2012;27:642–651. [DOI] [PubMed] [Google Scholar]
- 12. Kim SW, Jin Y, Shin JH, et al. Glycyrrhizic acid affords robust neuroprotection in the postischemic brain via anti‐inflammatory effect by inhibiting HMGB1 phosphorylation and secretion. Neurobiol Dis 2012;46:147–156. [DOI] [PubMed] [Google Scholar]
- 13. Hardy ME, Hendricks JM, Paulson JM, Faunce NR. 18beta‐glycyrrhetinic acid inhibits rotavirus replication in culture. Virol J 2012;9:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feng C, Wang H, Yao C, Zhang J, Tian Z. Diammonium glycyrrhizinate, a component of traditional Chinese medicine Gan‐Cao, prevents murine T‐cell‐mediated fulminant hepatitis in IL‐10‐ and IL‐6‐dependent manners. Int Immunopharmacol 2007;7:1292–1298. [DOI] [PubMed] [Google Scholar]
- 15. Ikeda K, Arase Y, Kobayashi M, et al. A long‐term glycyrrhizin injection therapy reduces hepatocellular carcinogenesis rate in patients with interferon‐resistant active chronic hepatitis C: A cohort study of 1249 patients. Dig Dis Sci 2006;51:603–609. [DOI] [PubMed] [Google Scholar]
- 16. Yuan H, Ji WS, Wu KX, Jiao JX, Sun LH, Feng YT. Anti‐inflammatory effect of Diammonium Glycyrrhizinate in a rat model of ulcerative colitis. World J Gastroenterol 2006;12:4578–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hou SZ, Li Y, Zhu XL, Wang ZY, Wang X, Xu Y. Ameliorative effects of Diammonium Glycyrrhizinate on inflammation in focal cerebral ischemic‐reperfusion injury. Brain Res 2012;1447:20–27. [DOI] [PubMed] [Google Scholar]
- 18. Zhu X, Chen C, Ye D, et al. Diammonium glycyrrhizinate upregulates PGC‐1alpha and protects against Abeta1–42‐induced neurotoxicity. PLoS ONE 2012;7:e35823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rosano C, Marsland AL, Gianaros PJ. Maintaining brain health by monitoring inflammatory processes: A mechanism to promote successful aging. Aging Dis 2012;3:16–33. [PMC free article] [PubMed] [Google Scholar]
- 20. Mattson MP, Camandola S. NF‐kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest 2001;107:247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaltschmidt B, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C. Transcription factor NF‐kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci U S A 1997;94:2642–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tilstra JS, Clauson CL, Niedernhofer LJ, Robbins PD. NF‐kappaB in aging and disease. Aging Dis 2011;2:449–465. [PMC free article] [PubMed] [Google Scholar]
- 23. Park SY, Jin ML, Kim YH, Kim Y, Lee SJ. Anti‐inflammatory effects of aromatic‐turmerone through blocking of NF‐kappaB, JNK, and p38 MAPK signaling pathways in amyloid beta‐stimulated microglia. Int Immunopharmacol 2012;14:13–20. [DOI] [PubMed] [Google Scholar]
- 24. Yin LL, Li W, Chu YQ, Li L. ERK pathway activation is required for amyloid‐beta(1–40)(‐)induced neurotoxicity of THP‐1 human monocytes towards SK‐N‐SH neuroblastoma. Brain Res 2011;1378:9–17. [DOI] [PubMed] [Google Scholar]
- 25. Young KF, Pasternak SH, Rylett RJ. Oligomeric aggregates of amyloid beta peptide 1–42 activate ERK/MAPK in SH‐SY5Y cells via the alpha7 nicotinic receptor. Neurochem Int 2009;55:796–801. [DOI] [PubMed] [Google Scholar]
- 26. Ashabi G, Ramin M, Azizi P, et al. ERK and p38 inhibitors attenuate memory deficits and increase CREB phosphorylation and PGC‐1alpha levels in Abeta‐injected rats. Behav Brain Res 2012;232:165–173. [DOI] [PubMed] [Google Scholar]
- 27. Ramin M, Azizi P, Motamedi F, Haghparast A, Khodagholi F. Inhibition of JNK phosphorylation reverses memory deficit induced by beta‐amyloid (1–42) associated with decrease of apoptotic factors. Behav Brain Res 2011;217:424–431. [DOI] [PubMed] [Google Scholar]