

Summary
Background
The activation of nuclear factor‐kappa B (NF‐κB) and NLRP3 inflammasome is involved in neuroinflammation, which is closely linked to Alzheimer's disease (AD). In vivo and in vitro studies have suggested that artemisinin shows antiinflammatory effects in inflammation‐related diseases. However, the impacts of artemisinin on AD have not been investigated.
Aims
In this study, 5‐month‐old APPswe/PS1dE9 transgenic mice were treated daily with 40 mg/kg artemisinin for 30 days by intraperitoneal injection to evaluate the effects of artemisinin on AD.
Results
We found that artemisinin treatment (1) decreased neuritic plaque burden; (2) did not alter Aβ transport across the blood–brain barrier; (3) regulated APP processing via inhibiting β‐secretase activity; (4) inhibited NF‐κB activity and NALP3 inflammasome activation in APPswe/PS1dE9 double transgenic mice.
Conclusions
The in vivo study clearly demonstrates that artemisinin has protective effects on AD pathology due to its effects on suppressing NF‐κB activity and NALP3 inflammasome activation. Our study suggests that targeting NF‐κB activity and NALP3 inflammasome activation offers a valuable intervention for AD.
Keywords: Amyloid beta, Artemisinin, BACE1, NALP3 inflammasome, NF‐κB
Introduction
Alzheimer's disease (AD) is the most common cause of dementia in the elderly, being characterized clinically by progressive cognitive decline and pathologically by the formation of amyloid plaques and neurofibrillary tangles in the brain 1. Amyloid β‐peptide (Aβ) is the central component of amyloid plaques. Aβ is released from amyloid precursor protein by the action of β‐secretase and γ‐secretase, which have been regarded as therapeutic targets for AD 2.
Over the past several years, neuroinflammation has attracted considerable attention because of its pivotal role in the pathogenesis of AD. Biochemical and neuropathological studies of brains from patients with AD provided strong evidence for an activation of inflammatory pathways. Epidemiology studies have shown that long‐term use of antiinflammatory drugs is linked with reduced risk to develop AD 3. Previous studies have shown that inhibition of inflammatory pathway could rescue behavioral deficits and reduce neuropathology in AD mice models 4, 5. Moreover, activation of nuclear factor‐kappa B (NF‐κB) pathway has been proven to be involved in AD 6. Interestingly, NF‐κB signal is associated with BACE1 expression 7, 8, 9. Recent publications have suggested that NF‐κB activation is necessary for NALP3 inflammasome activation 10. In the past 10 years, NALP3 inflammasome has been identified as a sensor of Aβ in a process involving the phagocytosis of Aβ and subsequent lysosomal damage. Furthermore, the NALP3 inflammasome, caspase‐1, and IL‐1β are critical for the recruitment of microglia to exogenous Aβ in the brain 11. Together, it suggests that activation of NF‐κB and NALP3 inflammasome is important for inflammation and tissue damage in AD 11, 12.
Artemisinin is isolated from the plant Artemisia annua, sweet wormwood, an herb employed in Chinese traditional medicine. In addition to its well‐known use as antimalarial drug, it has been a topic of research in cancer treatment 13. Further, several studies have demonstrated that it has potent antiinflammatory effects due to its inhibiting NF‐κB pathway 14, 15, 16. Although artemisinin has been proven to be protective in models of postinfarct myocardial remodeling 17, lupus nephritis 18, and experimental autoimmune encephalomyelitis 19, its effects on AD, a common neurodegenerative disease associated with inflammation, have not been investigated.
In this study, we tested the hypothesis that artemisinin can reduce amyloid plaques in 5‐month‐old APPswe/PS1dE9 transgenic mice and demonstrated that it decreased BACE1 expression and suppressed NF‐κB activity and NALP3 inflammasome activation.
Materials and Methods
Transgenic Mice and Drug Treatment
Animal experiment protocols were approved by the Nanjing Medical University Experimental Animal Care and Use Committee. Ten APPswe/PS1dE9 transgenic mice (5 months old) were obtained from the Institute of Zoology, Chinese Academy of Sciences, and were housed in an air‐conditioned room under a 12‐h/12‐h light/dark cycle (lights on, 8:00 am through 8:00 pm). Food and water were provided ad libitum. Five mice were injected with 40 mg/kg artemisinin (in DMSO) at the same time once daily for 30 days by intraperitoneal injection. The other five mice were injected with DMSO as control. Artemisinin (98% purity) was purchased from Sigma‐Aldrich (St. Louis, MO, USA).
Immunohistochemical Staining
All the 10 mice (five mice in each group) were anesthetized and sacrificed. Then, the brains were removed and cut into halves. In each group, there were 10 cerebral hemispheres. In 10 cerebral hemispheres, 4 hemispheres were used for immunohistochemistry staining, 3 hemispheres for ELISA, and 3 hemispheres for Western blotting. The cerebral hemispheres used for immunohistochemistry were fixed in 4% paraformaldehyde for 20 h. After being dehydrated in alcohol, the brains were embedded in paraffin and cut into 3‐ to 4‐μm sections. Sections were deparaffinized, hydrated in distilled water, treated with 3% H2O2 for 10 min to remove residual peroxidase activity, and rinsed again with PBS. Sections were permeabilized with 1% NP‐40 and 0.1% Triton X‐100 for 10 min, rinsed in PBS, blocked with 10% normal goat serum, and incubated with primary antibody (rabbit anti‐β‐amyloid1–42, 1:600; Abcam (Cambridge, MA, USA); rabbit anti‐NF‐κB p65 antibody, 1:600; Bioworld Technology, Minneapolis, MN, USA) overnight. After being rinsed in PBST (PBS containing 0.05% Tween 20), sections were further incubated with secondary antibody. The immunoreactivity was developed using DAB for 3–10 min. Plaques were counted under a microscope at 200× magnification. Plaques were quantified, and the mean plaque count per slice was recorded for each mouse.
Western Blotting
The following primary antibodies were used: mouse anti‐LRP1 antibody [5A6] (1:3000; Merck, Darmstadt, Germany), rabbit anti‐RAGE antibody (1:1000; Cell Signaling, Denver, MA, USA), rabbit anti‐APP carboxyl terminal antibody (1:4000; Sigma), rabbit anti‐BACE1 antibody (1:1000; Cell Signaling), rabbit anti‐PS1 antibody (1:1000; Cell Signaling), rabbit anti‐Aph‐1a antibody (1:3000; Invitrogen, Camarillo, CA, USA), rabbit polyclonal antibody against serine 536 phosphorylated NF‐κB p65 (1: 800; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit monoclonal antibody against NF‐κB p65 (1:500; Bioworld Technology), rabbit anti‐IκB‐α antibody (1:1000; Bioworld Technology), rabbit polyclonal antibody against serine 32 phosphorylated IκB‐α (1:1000; Bioworld Technology), rabbit anti‐NALP3 antibody (1:800; Santa Cruz), rabbit anticleaved caspase‐1 (Asp297) (D57A2) antibody (1:1000; Cell Signaling), and mouse anti‐β‐actin antibody (1:1000; Santa Cruz).
Western blotting was performed as previously described 20. Brain tissue was lysed in RIPA lysis buffer supplemented with protease inhibitors (Complete; Roche, Indianapolis, IN, USA). The lysates were resolved by SDS‐PAGE. Supernatants and the final pellets from each sample were heat blocked for 5 min in loading buffer (125 mM Tris–HCl, 20% glycerol, 10% 2‐mercaptoethanol, 4% SDS, 0.02% bromophenol blue, pH 6.8) and then subjected to electrophoresis on 10–20% Tris–glycine SDS‐PAGE gels. Proteins were then electrically transferred to a transfer membrane (Bio‐Rad, Hercules, CA, USA) and blocked for 1 h in Tris–HCl‐buffered saline containing 5% skim milk and 0.1% Tween. Membranes were incubated in primary antibodies at 4°C overnight in TBS buffer containing 5% bovine albumin. Membranes were then rinsed with TBS buffer containing 0.1% Tween 20, incubated with HRP‐labeled secondary antibody for 2 h and then stained with detection reagents. Finally, membranes were developed using the enhanced chemiluminescence (ECL) system. The signal intensity of primary antibody binding was analyzed using Image J software (Rasband, W.S., ImageJ v1.44p, U. S. National Institutes of Health, Bethesda, MD, USA, http://rsb.info.nih.gov/ij/, 1997‐2011) and normalized to a loading control β‐actin.
ELISA
Tissue homogenates from transgenic mouse hippocampal and neocortical regions were obtained and centrifuged at 1000 g in 4°C for 15 min to remove cellular debris. The supernatant was collected and stored at −80°C until use. The concentrations of IL‐6, TNF‐α, and IL‐1β were measured by specific ELISA kits (R&D, Minneapolis, MN, USA).
The levels of soluble and insoluble Aβ42 in brain samples were quantified using ELISA, as previously described 20. Briefly, brain tissue was homogenized in extraction buffer consisting of 50 mM Tris (pH 7.4), 2 mM EDTA, 400 mM NaCl, and complete protease inhibitor cocktail (Roche). The homogenates were centrifuged at 20,000 × g for 5 min at 4°C. The resulting supernatants were analyzed for soluble Aβ. The pellets were homogenized in 70% formic acid and centrifuged at 44,000 × g for 5 min at 4°C. The resulting supernatants were neutralized with 1 M Tris and then diluted in ELISA buffer for the measurement of insoluble Aβ. Samples were prepared from five animals in each group. All samples were analyzed in triplicate. Standard curves were made using human Aβ42 standards provided in the ELISA kit (Invitrogen).
Statistical Analysis
Statistical analyses were performed by an individual blinded to the groups. All results are expressed as means ± SD and were examined for the homogeneity of variance. Statistical analysis was performed using SPSS 13.0 software. Comparisons between two groups were performed using Student's t‐test. A value of P < 0.05 was considered to be statistically significant.
Results
Artemisinin Decreases Neuritic Plaque Burden in APPswe/PS1dE9 Transgenic Mice
We first examined whether artemisinin could inhibit amyloid plaque deposition in APPswe/PS1dE9 transgenic mice. Brain tissues from vehicle‐ and artemisinin‐treated mice were subjected to Aβ immunohistochemistry. Artemisinin reduced amyloid plaque deposition in the cortex and hippocampus compared with vehicle treatment (Figure 1A). Semi‐quantitative analysis revealed that artemisinin treatment reduced plaque number by 47.86% in the cortex (6.08 ± 1.51 vs. 3.17 ± 1.27, P < 0.05; Figure 1B) and by 60.85% in hippocampus (2.35 ± 0.89 vs. 0.92 ± 0.67, P < 0.05; Figure 1B).
Figure 1.
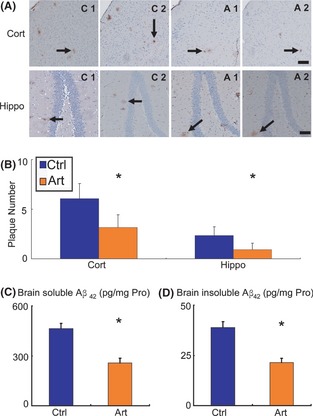
Effect of artemisinin on neuritic plaque formation in APPswe/PS1dE9 double transgenic mice. APPswe/PS1dE9 transgenic mice aged 5 months were treated with 40 mg/kg artemisinin (Art) for 30 days, whereas age‐matched control APPswe/PS1dE9 mice received vehicle solution (Ctrl) as control. The mice were killed, and the brains were dissected, fixed, and sectioned. (A) Neuritic plaques were detected by immunohistochemistry using an Aβ1‐42 antibody. The plaques were visualized by microscopy with 200× magnification. Artemisinin significantly reduced the numbers of neuritic plaques in the cortex (Cort) and hippocampus (Hippo) of mice compared with controls. Black arrows point to plaques. Bars: 100 μm. (B) Quantification of neuritic plaques in APPswe/PS1dE9 mice in each group. An ELISA was conducted to measure brain‐soluble Aβ42 (C) and brain‐insoluble Aβ42 (D) levels in transgenic mice in each group. The numbers represent the mean ± SEM. n = 4 mice each (for immunohistochemistry). n = 3 mice each (for ELISA). *P < 0.05 by Student's t‐test.
To confirm the above results, we investigated the effect of artemisinin on Aβ42 concentration by detecting buffer‐soluble and buffer‐insoluble (extracted with 70% formic acid) Aβ42. As expected, artemisinin reduced the brain‐soluble Aβ42 concentration by 44.35% (464.53 ± 29.68 vs. 258.53 ± 26.49, P < 0.05; Figure 1C). Meanwhile, artemisinin reduced brain‐insoluble Aβ42 concentration by 44.94% (38.87 ± 2.85 vs. 21.4 ± 2.06, P < 0.05; Figure 1D).
Artemisinin does not Alter Aβ Transport Across The Blood–Brain Barrier in APPswe/PS1dE9 Double Transgenic Mice
Aβ transport across the blood–brain barrier, which represents an important way in clearance of Aβ from brain, is mainly mediated by LRP1 and RAGE. To clarify the mechanisms by which artemisinin inhibited plaque deposition, the effects of artemisinin on Aβ transport across the blood–brain barrier were evaluated by determining Aβ concentration in peripheral blood and the expression levels of LRP1 and RAGE in mice brains.
As shown in Figure 2A, artemisinin did not cause a significant change in peripheral blood Aβ42 concentration (22.63 ± 3.68 vs. 20.10 ± 3.00, P > 0.05; Figure 2A). Furthermore, there were no significant differences in LRP1 and RAGE levels between the drug‐treated groups and the vehicle group (P > 0.05) (Figure 2B C). These data clearly indicated that artemisinin‐inhibited Aβ deposition was not mediated by its regulation on transport across blood–brain barrier.
Figure 2.
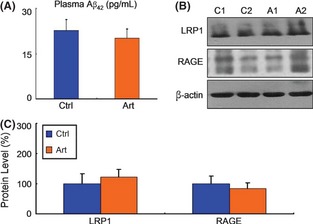
Effect of Artemisinin on the transport of Aβ across the blood–brain barrier in APPswe/PS1dE9 double transgenic mice. An ELISA was conducted to measure peripheral blood Aβ42 (A) levels in transgenic mice in each group. Artemisinin did not alter the peripheral blood Aβ42 level. n = 3. *P < 0.05 by Student's t‐test. (B) Half brains from mice in each group were lysed in RIPA buffer. LRP1 and RAGE were detected by Western blotting using β‐actin as a loading control. (C) Quantification of LRP1 and RAGE in brains. Artemisinin had no influence on LRP1 and RAGE expression. n = 3. *, P < 0.05 by Student's t‐test.
Artemisinin Regulates APP Processing via Inhibiting β‐Secretase Activity in APPswe/PS1dE9 Double Transgenic Mice
We have shown that artemisinin decreases amyloid plaque deposition in APPswe/PS1dE9 transgenic mice, which was not mediated by its regulation on Aβ transport across blood–brain barrier. To investigate the underlying mechanism, the levels of some key proteins in the APP processing pathway were determined by Western blotting, including APP‐full length (FL), BACE1 (β‐secretase enzyme), PS1, Aph‐1a (the key subunits of γ‐secretase enzyme), and APP C‐terminal fragments (CTFs) (Figure 3A). As shown in Figure 3B, artemisinin had no influence on APP‐FL expression in animal (P > 0.05). Significantly, artemisinin decreased BACE1 level in transgenic mice brains by 33% (P < 0.05) (Figure 3B). Furthermore, we failed to detect any significant differences in the levels of PS1/Aph‐1a/APP‐CTFs between artemisinin‐treated group and vehicle group (P > 0.05) (Figure 3B). Together, these results suggested that artemisinin inhibited the APP amyloidogenic pathway through downregulating β‐secretase cleavage of APP.
Figure 3.

Effect of artemisinin on APP processing pathways in APPswe/PS1dE9 double transgenic mice. (A) Brain tissues from APPswe/PS1dE9 mice were subjected to Western blotting to determine the levels of APP‐full length (FL), BACE1, PS1, Aph‐1a, and APP‐CTFs, with β‐actin as a loading control. (B) Quantification of APP‐FL, BACE1, PS1, Aph‐1a, and APP‐CTFs in brain tissues. Artemisinin significantly decreased BACE1 expression and had no influence on the levels of APP‐FL, PS1, Aph‐1a, and APP‐CTFs. n = 3. *P < 0.05 by Student's t‐test.
Artemisinin Inhibits NF‐κB Activity in APPswe/PS1dE9 Double Transgenic Mice
We have shown that artemisinin decreases Aβ production by inhibiting β‐secretase cleavage of APP. To clarify the mechanisms, we determined the activity of NF‐κB, which had been proven to play a pivotal role in BACE1 expression. The nuclear translocation of NF‐κB p65 was determined by immunohistochemistry analysis, which showed that the ratio of NF‐κB nuclear–positive cells was significantly reduced by 65.84% (P < 0.05) (Figure 4A–C).
Figure 4.
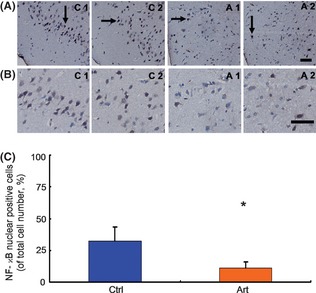
Effects of artemisinin on the nuclear translocation of nuclear factor‐kappa B (NF‐κB) p65 in APPswe/PS1dE9 double transgenic mice. Brain tissues from APPswe/PS1dE9 mice were subjected to immunohistochemistry to determine the nuclear translocation of NF‐κB p65. (A) Representative photographs of immunohistochemistry staining with anti‐NF‐κB p65 antibody (with 200× magnification). (B) Representative photographs of immunohistochemistry staining with anti‐NF‐κB p65 antibody (with 400× magnification). Artemisinin significantly reduced the ratio of NF‐κB p65 nuclear–positive cells. Black arrows point to NF‐κB p65 nuclear–positive cells. Bars: 50 μm. (C) Quantification of the ratio of NF‐κB p65 nuclear–positive cells in APPswe/PS1dE9 mice in each group; the numbers represent the mean ± SEM. n = 3 mice each. *P < 0.05 by Student's t‐test.
In addition, the protein levels of total NF‐κB p65, NF‐κB p65 phosphorylated at serine 536, I‐κBα, and I‐κBα phosphorylated at serine 32 were detected by Western blot (Figure 5A). Consistent with immunohistochemistry results, artemisinin increased the level of I‐κBα by 105% and decreased the levels of NF‐κB p65, p‐NF‐κB p65, and p‐I‐κBα by 56%, 27%, and 45%, respectively (Figure 5B). These results indicated that artemisinin treatment reduced the nuclear translocation of NF‐κB p65 and increased I‐κBα expression, which led to the inhibition of NF‐κB activity in APPswe/PS1dE9 transgenic mice brains.
Figure 5.

Effect of artemisinin on nuclear factor‐kappa B (NF‐κB) activity in APPswe/PS1dE9 double transgenic mice. (A) Brain tissues from APPswe/PS1dE9 mice were subjected to Western blotting to determine the levels of p‐NF‐κB p65, NF‐κB p65, I‐κBα, and p‐I‐κBα, with β‐actin as a loading control. (B) Quantification of p‐NF‐κB p65, NF‐κB p65, I‐κBα, and p‐I‐κBα. Artemisinin significantly decreased the levels of p‐NF‐κB p65, NF‐κB p65, and p‐I‐κBα and increased the level of I‐κBα. n = 3. *, P < 0.05 by Student's t‐test. (C) An ELISA was conducted to measure brain IL‐6 and TNF‐α level in the brains of mice in each group. Artemisinin significantly reduced IL‐6 and TNF‐α level in brains. n = 3. *, P < 0.05 by Student's t‐test.
Furthermore, downstream cytokines' (IL‐6, TNF‐α) production was detected to further determine the activity of NF‐κB. As shown in Figure 5C, artemisinin treatment reduced IL‐6 and TNF‐α production by 37.16% (1631.90 ± 76.31 vs. 1025.50 ± 253.53, P < 0.05) and 34.46% (2500.60 ± 232.90 vs. 1639.00 ± 304.47, P < 0.05) in transgenic mice brains.
Artemisinin Inhibits NALP3 Inflammasome Activation in APPswe/PS1dE9 Double Transgenic Mice
We have shown that artemisinin reduces Aβ production via downregulation the level of BACE1, which is mediated by the inhibition of NF‐κB activity. Recent publications have suggested NF‐κB activation is necessary for NALP3 inflammasome activation, which is important for inflammation and tissue damage in AD. Hence, the activation of NALP3 inflammasome was determined in vehicle‐ and artemisinin‐treated groups. In artemisinin‐treated mice, the level of NALP3 was significantly reduced by 37% than that in control group (P < 0.05, Figure 6A, B). Meanwhile, artemisinin treatment decreased the level of caspase‐1 p20 subunit by 50% (P < 0.05, Figure 6A,B). Further, artemisinin treatment decreased the production of IL‐1β by 29.73% (1967.50 ± 68.84 vs. 1382.50 ± 80.79, P < 0.05, Figure 6C). Together, these results suggested that artemisinin inhibited NALP3 inflammasome activation in APPswe/PS1dE9 double transgenic mice.
Figure 6.
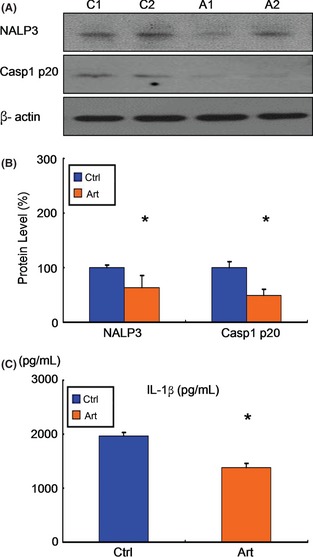
Effects of artemisinin on NALP3 inflammasome activation in APPswe/PS1dE9 double transgenic mice. (A) Brain tissues from APPswe/ PS1dE9 mice were subjected to Western blotting to determine the levels of NALP3 and caspase‐1 p20 subunit, with b‐actin as a loading control. (B) Quantification of NALP3 and caspase‐1 p20 subunit. Artemisinin significantly decreased the levels of NALP3 and caspase‐1 p20 subunit. n = 3. *, P < 0.05 by Student's t‐test. (C) An ELISA was conducted to measure brain IL‐1b level in the brains of mice in each group. Artemisinin significantly reduced IL‐1b level in brains. n = 3. *, P < 0.05 by Student's t‐test.
Discussion
Antimalarial drug artemisinin showed protective effects against AD in this study. First, artemisinin treatment decreased neuritic plaque burden. Second, artemisinin did not alter Aβ transport across the blood–brain barrier. Third, artemisinin regulated APP processing via inhibiting β‐secretase activity. Fourth, artemisinin inhibited NF‐κB activity and NALP3 inflammasome activation in APPswe/PS1dE9 double transgenic mice. Hence, our study suggests that targeting NF‐κB activity and NALP3 inflammasome activation offers a valuable intervention for AD.
Transgenic mice models have played an important role in AD research community. APPswe/PS1dE9 transgenic mouse model has been widely used in scientific research, which was first described by Jankowsky et al. in 2004 21. The mice overexpress the Swedish mutation of APP, together with PS1 deleted in exon 9. The mice shows significant behavioral deficits in Morris water maze at 12 months of age. At 6 months of age, only memory deficits could be seen in radial arm water maze 22. Usually, behavioral tests are performed in 7‐ to 12‐month‐old mice. To observe the effects of artemisinin by early intervention, 5‐month‐old transgenic mice were used in the present study. Considering that behavioral deficits of 5‐month‐old transgenic mice might be unremarkable in Morris water maze, behavioral tests were not included in the study. In previously published studies on AD, the days of drugs were often selected between 7 and 60 days 23, 24, 25. Hence, the mice were injected once daily for 30 days by intraperitoneal injection in the study. The LD50 of artemisinin by intraperitoneal injection in mice is 1558 mg/kg. Usually, we can choose the dose between 1/5 LD50 and 1/50 LD50. Hence, we chose 40 mg/kg in the present study according to published study 26.
In basic and clinical studies, inflammation has been demonstrated to be involved in the pathogenesis and progress of AD 3. Elevated inflammatory biomarkers are observed in patients with AD 27. Clinical trials clearly indicate that NSAIDs show protective effects on the prevention of AD 28, 29. Aβ‐activated glial cells secrete mounts of pro‐inflammation cytokines through NF‐κB activation 30, 31. More interestingly, the signaling from NF‐κB activation is necessary for the activation of NALP3 inflammasome 9. Together, both NF‐κB and NALP3 inflammasome play an important role in the inflammation of AD. In the present study, artemisinin significantly inhibited the activation of NF‐κB and NALP3 inflammasome, thus leading to the decreased production of downstream cytokines (TNFα, IL‐6, IL‐1β). In fact, the antiinflammatory effects of artemisinin from NF‐κB inhibition have been proven in models of lupus nephritis, experimental autoimmune encephalomyelitis, and other disease 14, 15, 17, 18, 19, 32. Our study first demonstrated the antiinflammatory effects of artemisinin in AD animal model.
In our previous study, we have demonstrated that antiinflammation strategy could alleviate the pathology of AD though upregulation of Aβ clearance 20. The present study also confirmed the protective effects of antiinflammation strategy in AD, which was mediated by inhibition of Aβ production. We showed that artemisinin inhibited NF‐κB activity, leading to downregulation of Aβ production through BACE1 inhibition. In fact, the association between BACE1 expression and NF‐κB activity has been clarified in other studies. The age‐ and AD‐associated increases in NF‐kappaB in brain may be significant contributors to activation of BACE1 transcription 7. Furthermore, the levels of BACE1 and NF‐κB p65 were significantly elevated in brains of patients with AD. Overexpression and knockout studies have demonstrated that NF‐κB signaling facilitates BACE1 gene expression 8. Hence, inhibition of NF‐κB‐mediated BACE1 expression may be an effective target for AD therapy. Some drugs attenuated Aβ secretion by reducing BACE1 expression in vitro and in vivo 16, 33, 34, 35, 36, 37, 38. These results are consistent with our study. Further work is needed to explore the mechanisms that artemisinin inhibited NF‐κB activity in AD.
There is a close relationship between NF‐κB and Aβ plaques. Some drugs lowered Aβ plaques through inhibitory effects of NF‐κB 33, 39. A recently published study suggested a role for NALP3 inflammasome in AD. NALP3 or caspase‐1 knockout largely protected AD transgenic model from memory loss and enhanced Aβ clearance. Furthermore, NALP3 inflammasome deficiency decreased Aβ deposition in the APP/PS1 model of AD. Hence, NALP3 inflammasome inhibition is regarded as a therapeutic target for AD 40. The present study showed that artemisinin exerted inhibitory effects on NALP3 inflammasome, which could partly explain its protective effects on Aβ pathology.
AD is characterized by amyloid plaques in brain, which was caused by an imbalance between Aβ generation and Aβ clearance. A recent study suggested that an impairment of Aβ clearance might be a major cause of sporadic AD 41. Brain‐to‐blood transport of Aβ is an important way of Aβ clearance, which is mainly mediated by LRP1 (Aβ transport from brain to blood) and RAGE (Aβ transport from blood to brain) 42, 43. Our results indicated that artemisinin did not alter Aβ transport from brain to blood through the regulation of LRP1 and RAGE expression. Interestingly, binding of RAGE ligands to RAGE activates NF‐κB pathway, thus mediating pro‐inflammatory effects and upregulating BACE1 44. Because artemisinin exerted no effects on RAGE expression in our study, we suspected that the NF‐κB‐inhibiting effects of artemisinin might not be ascribed to regulation of RAGE pathway.
Kim et al. tested the effect of artemisinin on Aβ fibril formation in vitro 45. The results showed that artemisinin exerted no impact on Aβ fibril formation in vitro. The present study showed that artemisinin exerted inhibitory effects on Aβ generation in vivo. However, there are many differences between process of Aβ generation and process of Aβ fibril formation. Hence, there is a possibility that a compound exerts different impacts on Aβ generation and Aβ fibril formation.
In conclusion, the current study reveals the neuroprotective effects of artemisinin against the neuropathology in APPswe/PS1dE9 transgenic mice. Artemisinin decreased neuritic plaque burden in APPswe/PS1dE9 transgenic mice. Artemisinin decreased Aβ production via inhibition of NF‐κB activity and NALP3 inflammasome activation in APPswe/PS1dE9 double transgenic mice. On the other hand, artemisinin did not alter Aβ transport across the blood–brain barrier. These data suggest that artemisinin might be an effective drug for the treatment of AD. However, its efficacy and safety should be further investigated in clinical trials.
Conflict of Interest
The authors declare no conflict of interests.
Acknowledgments
This work was supported by National Natural Science Foundation of China grant (30700248; 81271211). We thank Laboratory Animal Center of Nanjing First Hospital for breeding the animals and providing animal experimental facility.
The first two authors contributed equally to this work.
References
- 1. Cummings JL, Cole G. Alzheimer disease. JAMA 2002;287:2335–2338. [DOI] [PubMed] [Google Scholar]
- 2. O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci 2011;34:185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wyss‐Coray T, Rogers J. Inflammation in Alzheimer disease‐a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med 2012;2:a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. He P, Zhong Z, Lindholm K, et al. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol 2007;178:829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lim JE, Kou J, Song M, et al. MyD88 deficiency ameliorates β‐amyloidosis in an animal model of Alzheimer's disease. Am J Pathol 2011;179:1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Granic I, Dolga AM, Nijholt IM, van Dijk G, Eisel UL. Inflammation and NF‐kappaB in Alzheimer's disease and diabetes. J Alzheimers Dis 2009;16:809–821. [DOI] [PubMed] [Google Scholar]
- 7. Bourne KZ, Ferrari DC, Lange‐Dohna C, Rossner S, Wood TG, Perez‐Polo JR. Differential regulation of BACE1 promoter activity by nuclear factor‐kappaB in neurons and glia upon exposure to beta‐amyloid peptides. J Neurosci Res 2007;85:1194–1204. [DOI] [PubMed] [Google Scholar]
- 8. Chen CH, Zhou W, Liu S, et al. Increased NF‐κB signalling up‐regulates BACE1 expression and its therapeutic potential in Alzheimer's disease. Int J Neuropsychopharmacol 2012;15:77–90. [DOI] [PubMed] [Google Scholar]
- 9. Paris D, Patel N, Quadros A, et al. Inhibition of Abeta production by NF‐kappaB inhibitors. Neurosci Lett 2007;415:11–16. [DOI] [PubMed] [Google Scholar]
- 10. Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF‐kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009;183:787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid‐beta. Nat Immunol 2008;9:857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Salminen A, Ojala J, Suuronen T, Kaarniranta K, Kauppinen A. Amyloid‐beta oligomers set fire to inflammasomes and induce Alzheimer's pathology. J Cell Mol Med 2008;12:2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Crespo‐Ortiz MP, Wei MQ. Antitumor activity of artemisinin and its derivatives: From a well‐known antimalarial agent to a potential anticancer drug. J Biomed Biotechnol 2012;2012:247597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xiong Z, Sun G, Zhu C, et al. Artemisinin, an anti‐malarial agent, inhibits rat cardiac hypertrophy via inhibition of NF‐κB signaling. Eur J Pharmacol 2010;649:277–284. [DOI] [PubMed] [Google Scholar]
- 15. Wang Y, Huang Z, Wang L, et al. The anti‐malarial artemisinin inhibits pro‐inflammatory cytokines via the NF‐κB canonical signaling pathway in PMA‐induced THP‐1 monocytes. Int J Mol Med 2011;27:233–241. [DOI] [PubMed] [Google Scholar]
- 16. Zhu C, Xiong Z, Chen X, et al. Artemisinin attenuates lipopolysaccharide‐stimulated proinflammatory responses by inhibiting NF‐κB pathway in microglia cells. PLoS One 2012;7:e35125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gu Y, Wang X, Wang X, et al. Artemisinin attenuates post‐infarct myocardial remodeling by down‐regulating the NF‐κB pathway. Tohoku J Exp Med 2012;227:161–170. [DOI] [PubMed] [Google Scholar]
- 18. Wu X, Zhang W, Shi X, An P, Sun W, Wang Z. Therapeutic effect of artemisinin on lupus nephritis mice and its mechanisms. Acta Biochim Biophys Sin (Shanghai) 2010;42:916–923. [DOI] [PubMed] [Google Scholar]
- 19. Wang Z, Qiu J, Guo TB, et al. Anti‐inflammatory properties and regulatory mechanism of a novel derivative of artemisinin in experimental autoimmune encephalomyelitis. J Immunol 2007;179:5958–5965. [DOI] [PubMed] [Google Scholar]
- 20. Shi JQ, Shen W, Chen J, et al. Anti‐TNF‐α reduces amyloid plaques and tau phosphorylation and induces CD11c‐positive dendritic‐like cell in the APP/PS1 transgenic mouse brains. Brain Res 2011;1368:239–247. [DOI] [PubMed] [Google Scholar]
- 21. Jankowsky JL, Fadale DJ, Anderson J, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta‐amyloid peptide in vivo: Evidence for augmentation of a 42‐specific gamma secretase. Hum Mol Genet 2004;13:159–170. [DOI] [PubMed] [Google Scholar]
- 22. Malm T, Koistinaho J, Kanninen K. Utilization of APPswe/PS1dE9 transgenic mice in research of Alzheimer's disease: Focus on gene therapy and cell‐based therapy applications. Int J Alzheimers Dis 2011;2011:517160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gabbita SP, Srivastava MK, Eslami P, et al. Early intervention with a small molecule inhibitor for tumor necrosis factor‐α prevents cognitive deficits in a triple transgenic mouse model of Alzheimer's disease. J Neuroinflammation 2012;9:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sehgal N, Gupta A, Valli RK, et al. Withania somnifera reverses Alzheimer's disease pathology by enhancing low‐density lipoprotein receptor‐related protein in liver. Proc Natl Acad Sci USA 2012;109:3510–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qing H, He G, Ly PT, et al. Valproic acid inhibits Abeta production, neuritic plaque formation, and behavioral deficits in Alzheimer's disease mouse models. J Exp Med 2008;205:2781–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robert A, Benoit‐Vical F, Claparols C, et al. The antimalarial drug artemisinin alkylates heme in infected mice. Proc Natl Acad Sci USA 2005;102:13676–13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reale M, Kamal MA, Velluto L, Gambi D, Di Nicola M, Greig NH. Relationship between inflammatory mediators, Aβ levels and ApoE genotype in Alzheimer disease. Curr Alzheimer Res 2012;9:447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. int'Veld BA, Ruitenberg A, Hofman A, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med 2001;345:1515–1521. [DOI] [PubMed] [Google Scholar]
- 29. Zandi PP, Anthony JC, Hayden KM, et al. Reduced incidence of AD with NSAID but not H2 receptor antagonists: The Cache County Study. Neurology 2002;59:880–886. [DOI] [PubMed] [Google Scholar]
- 30. Bonaiuto C, McDonald PP, Rossi F, Cassatella MA. Activation of nuclear factor‐kappa B by beta‐amyloid peptides and interferon‐gamma in murine microglia. J Neuroimmunol 1997;77:51–56. [DOI] [PubMed] [Google Scholar]
- 31. Kawamoto EM, Lepsch LB, Boaventura MF, et al. Amyloid beta‐peptide activates nuclear factor‐kappaB through an N‐methyl‐D‐aspartate signaling pathway in cultured cerebellar cells. J Neurosci Res 2008;86:845–860. [DOI] [PubMed] [Google Scholar]
- 32. Yi H, Lee SJ, Lee J, et al. Sphingosylphosphorylcholine attenuated β‐amyloid production by reducing BACE1 expression and catalysis in PC12 cells. Neurochem Res 2011;36:2083–2890. [DOI] [PubMed] [Google Scholar]
- 33. Paris D, Beaulieu‐Abdelahad D, Bachmeier C, et al. Anatabine lowers Alzheimer's Aβ production in vitro and in vivo . Eur J Pharmacol 2011;670:384–391. [DOI] [PubMed] [Google Scholar]
- 34. Choi DY, Lee JW, Lin G, et al. Obovatol attenuates LPS‐induced memory impairments in mice via inhibition of NF‐κB signaling pathway. Neurochem Int 2012;60:68–77. [DOI] [PubMed] [Google Scholar]
- 35. Lee YJ, Choi DY, Choi IS, et al. Inhibitory effect of 4‐O‐methylhonokiol on lipopolysaccharide‐induced neuroinflammation, amyloidogenesis and memory impairment via inhibition of nuclear factor‐kappaB in vitro and in vivo models. J Neuroinflammation 2012;9:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Choi DY, Lee JW, Peng J, et al. Obovatol improves cognitive functions in animal models for Alzheimer's disease. J Neurochem 2012;120:1048–1059. [DOI] [PubMed] [Google Scholar]
- 37. Chami L, Buggia‐Prévot V, Duplan E, et al. Nuclear factor‐κB regulates βAPP and β‐ and γ‐secretases differently at physiological and supraphysiological Aβ concentrations. J Biol Chem 2012;287:24573–24584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang LY, Tang SS, Wang XY, et al. PPARγ agonist pioglitazone reverses memory impairment and biochemical changes in a mouse model of Type 2 diabetes mellitus. CNS Neurosci Ther 2012;18:659–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Paris D, Ganey NJ, Laporte V, et al. Reduction of beta‐amyloid pathology by celastrol in a transgenic mouse model of Alzheimer's disease. J Neuroinflammation 2010;7:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 2013;493:674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS beta‐amyloid in Alzheimer's disease. Science 2010;330:1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Deane R, Bell RD, Sagare A, Zlokovic BV. Clearance of amyloid‐beta peptide across the blood‐brain barrier: Implication for therapies in Alzheimer's disease. CNS Neurol Disord Drug Targets 2009;8:16–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta‐peptide clearance through transport across the blood‐brain barrier. Stroke 2004;35(11 Suppl 1):2628–2631. [DOI] [PubMed] [Google Scholar]
- 44. Guglielmotto M, Aragno M, Tamagno E, et al. AGEs/RAGE complex upregulates BACE1 via NF‐κB pathway activation. Neurobiol Aging 2012;33:196.e13‐27. [DOI] [PubMed] [Google Scholar]
- 45. Kim H, Park BS, Lee KG, et al. Effects of naturally occurring compounds on fibril formation and oxidative stress of beta‐amyloid. J Agric Food Chem 2005;53:8537–8541. [DOI] [PubMed] [Google Scholar]