

Summary
Aims
Programmed Cell Death 5 (PDCD5) is a protein that accelerates apoptosis in different types of cells in response to various stimuli and is down‐regulated in many cancer tissues. We hypothesized in this study that down‐regulating PDCD5 can protect the brain from ischemic damage by inhibiting PDCD5‐induced apoptotic pathway.
Methods
One hundred and sixty male Sprague‐Dawley rats were randomly assigned to five groups: Sham surgery (n = 25), MCAO (n = 45), MCAO+rhPDCD5 (RhPDCD5) (n = 30), MCAO+control siRNA (n = 30), and MCAO+PDCD5 siRNA (n = 30). At 24 h following MCAO, immunohistochemistry and Western blot were performed.
Results
PDCD5 siRNA reduced the infarct volume, improved neurological deficits, improved cerebral blood flow (CBF), and reduced Evans blue extravasation. Meanwhile, over‐expression of PDCD5 protein with recombinant human PDCD5 (rhPDCD5) had an opposite effect. Immunohistochemistry and Western blot demonstrated PDCD5 siRNA decreased the expressions of key proapoptotic proteins such as p53, Bax/Bcl‐2, and cleaved caspase‐3 in the penumbra areas, whereas rhPDCD5 increased cell apoptosis. Double fluorescence labeling showed the positive immunoreactive materials of PDCD5 were partly colocalized with MAP2, GFAP, CD34, p53, and caspase‐3 in the penumbra areas in brain.
Conclusions
PDCD5‐induced apoptosis and over‐expression of PDCD5 are harmful to the ischemic neurons in vivo. Meanwhile, the inhibition of PDCD5 may be protective via reducing the apoptotic‐related protein such as p53, Bax, and caspase‐3. This observation may have potential for the treatment of ischemic cerebral stroke.
Keywords: Apoptosis, MCAO, PDCD5, siRNA in vivo
Introduction
Ischemic stroke is a substantial public health problem that leads to long‐term disability worldwide 1. Most researches have focused on rescuing injured brain tissue that is facing apoptosis in the penumbra area.
Programmed cell death 5 (PDCD5), also designated TFAR19 (TF‐1 cell apoptosis‐related gene‐19) by International Human Gene Nomination Committee, is a gene cloned during the apoptotic process of TF‐1 cells induced by cytokine withdrawal and widely expressed in a variety of tissues 2. The wide expression pattern of PDCD5 and its general function in various cell lines indicate that PDCD5 is a critical component in the regulation of programmed cell death. Previous reports have shown that over‐expressed PDCD5 facilitates apoptosis in cancer cell lines such as HeLa and MGC‐803 2. The decreased expression of PDCD5 has been characterized in multiple human tumors, such as astrocytic gliomas 3, breast cancer 4, gastric cancer 5, hepatocellular carcinoma 6, and chronic myelogenous leukemia 7. Functional studies indicate that PDCD5 can accelerate apoptosis of different cell types in response to various stimuli and can also induce different types of cell death, including paraptosis‐like cell death 8. PDCD5 protein is over‐expressed in cells undergoing apoptosis where it translocates rapidly from the cytoplasm to the nucleus of cells 9. The administration of the anti‐PDCD5 antibody or short interfering RNA against PDCD5 can suppress its apoptotic effects in HeLa cells 10, 11. Recently, PDCD5 has been shown to interact with Tip60 in vitro and function as a coactivator to promote apoptosis through the Tip60‐p53 signaling pathway after UV irradiation 12.
However, the effect of PDCD5 and the molecular mechanism in brain ischemia/reperfusion injury are currently unknown. Our hypothesis is that PDCD5 is involved in the apoptosis process during the neuronal injury, and the inhibition of PDCD5 can protect the brain from ischemic damage by inhibiting PDCD5‐induced apoptotic pathway. To test this hypothesis, at first we tested the distribution of PDCD5 expression in the brain after ischemia and detected its changing pattern with time points. Because the application of protein or siRNA of some genes in research and clinical studies has become popular and our previous studies have shown that siRNA could be successfully transfected into the neurons following intracerebroventricular injection 13, 14, 15, 16. Then, an upregulated recombinant human PDCD5 protein (rhPDCD5) and a down‐regulated PDCD5 level with small interference RNA (PDCD5 siRNA) were administrated in vivo via intracerebroventricular (i.c.v.) injection; the potential effect of PDCD5 in focal ischemia rat model was evaluated.
The mortality rate, brain edema, blood–brain barrier (BBB) disruption, cerebral blood flow, and neurobehavioral deficits were observed in different groups. Additionally, to clarify the mechanisms of PDCD5 in the neuronal cell death after cerebral ischemia, we evaluated the expression of proapoptotic proteins such as PDCD5, p53, cleaved caspase‐3, and Bax/Bcl‐2 (for details, see Figure 1).
Figure 1.
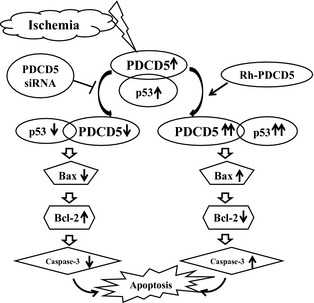
Hypothesis of molecular cascade after brain ischemia: Ischemia induced apoptosis via the upregulation of PDCD5 together with p53 and cooperating with other p53‐downstream apoptotic signals, such as Bax, Bcl‐2, and caspase‐3. After using different interventions for PDCD5 gene expression, p53 and other downstream genes were affected, respectively, followed with the apoptotic cell death process enhanced (following rhPDCD5 intervention) or inhibited (following PDCD5 siRNA intervention).
Materials and Methods
Animal Modeling
This protocol was evaluated and approved by the Animal and Ethics Review Committee at Peking University Health Science Center in Beijing, China. Every effort was made to minimize animal suffering and to reduce the number of animals used. One hundred and sixty Sprague‐Dawley male rats weighing 280–300 g were randomly assigned to the following five groups: Sham surgery (n = 25), Middle Cerebral Artery Occlusion/Reperfusion (MCAO) (n = 45), MCAO treated with recombinant human PDCD5 (Rh PDCD5) (n = 30), MCAO treated with control siRNA (n = 30), and MCAO treated with PDCD5 siRNA (n = 30). The animals that died in the experiment were not included. Focal cerebral ischemia was induced by intraluminal middle cerebral artery blockade with a nylon suture, as previously described by Longa et al. 17 and modified by Kawamura et al. 18. Briefly, animals were anesthetized using 4% isoflurane with a mixture of 70% medical air and 30% oxygen; anesthesia was maintained with 2% isoflurane. Under an operating microscope, the right femoral artery was dissected and cannulated using polyethylene‐50 tubing to allow continuous monitoring for mean blood pressure and sampling for analysis of blood gases. The heart rate and blood glucose levels before, during, and after ischemia were also analyzed. The right common carotid artery, including its bifurcation, was dissected and the external carotid artery was divided, leaving a stump of 3–4 mm. The internal carotid artery was isolated and clamped with a small vascular clip. The stump of the external carotid artery was reopened, and a 4.0 monofilament nylon suture with a slightly enlarged and round tip was inserted up 18–20 mm through the internal carotid artery. After occlusion for 2 h, the suture was withdrawn, followed by reperfusion. A similar procedure was performed in the Sham‐operated group except for nylon suture occlusion and reperfusion. All animals had free access to food and water.
In vivo rhPDCD5 Protein and siRNA Transfer
We performed in vivo transfer according to the method described previously 13. The stereotaxic coordinates were 0.8 mm posterior, 1.5 mm lateral to the bregma, and 4.5 mm ventral to the surface of the skull. 5 μL PDCD5 siRNA (Santa Cruz, sc‐270337) or control siRNA‐A (Santa Cruz, sc‐37007) was diluted with the same volume of in vivo transfection reagent (Entranster™‐in vivo; Engreen, Beijing, China). Mix gently by pipetting the solution up and down and injected intracerebroventricularly using a Hamilton microsyringe under a guidance of stereotaxy instrument (Kent Scientific Co., Torrington, CT, USA) under anesthetized, within 60 min after the reperfusion of MCAO. RhPDCD5 protein (70 μg/kg; Beijing Biosea Biotechnology Co., China) was injected into the right lateral ventricle in the same way.
Infarct Volume Measurement
2,3,7‐Triphenyltetrazolium chloride (TTC) (Sigma Inc., St. Louis, MO, USA) staining (n = 5) was performed at 24 h after MCAO as described previously 19. Coronal sections of the brain (2 mm thick) were cut and immersed in 2% solution of TTC for 30 min at 37°C. The normal tissue stains red, and infarct tissue with loss of mitochondrial enzyme activity does not stain and appears white. The infarction area and hemisphere area of each section were traced and measured using an image analysis system (Imaging‐Pro‐Plus [OLYMPUS], Silver Spring, MD, USA). The calculation of infarct volume was performed with infarcted area of the ipsilateral hemisphere/total ipsilateral hemisphere.
Cerebral Blood Flow Measurement
Cerebral blood flow (CBF) was measured using Laser Doppler perfusion image system (PeriScan PIM3 System; PERIMED, Stockholm, Sweden) (n = 5). An incision was made through the scalp, and the skin was retracted to expose the skull. The periosteal connective tissue adherent to the skull was removed with a sterile cotton swab. A parietal bone window was opened on the right side, 1 mm behind the coronal suture, and 1 mm lateral to sagittal suture, with an area of 4 × 6 mm. A computer‐controlled optical scanner directed a low‐powered He‐Ne laser beam over the exposed right cortex. The scanner head was positioned in parallel to the cerebral cortex at a distance of 15.8 cm. The scanning procedure took 4 second for a measurement covering an area of 80 pixels. At each measuring site, the beam illuminated the tissue to a depth of 0.5 mm. A color‐coded image to denote specific relative perfusion levels was displayed on a video monitor 20.
Mortality and Neurological Deficits
Mortality was calculated at 24 h after MCAO. The neurological scores were performed in a blinded fashion at 24 h, which were based on the scoring system of Garcia et al. 21.
Evans Blue Extravasation
Blood–brain barrier permeability was assessed by measuring Evans blue extravasation 22. Evans blue (Sigma, 0.1 mL of 4% solution) was injected into the femoral vein at reperfusion (2 h after occlusion). Rats (n = 5) were transcardially perfused with 100 mL of heparinized saline solution (10 IU/mL) 24 h after reperfusion. Brains were removed, and hemispheres were separated. Each hemisphere was well homogenized in 1 mL 0.1 M PBS and then centrifuged at 1000 × g for 15 min; 0.7 mL of 100% trichloroacetic acid was added into 0.7 mL of supernatant. The mixture was incubated at 14°C for 18 h and then centrifuged at 1000 × g for 30 min. The amount of Evans blue in supernatant was measured spectrophotometrically at 610 nm wavelength by comparison with readings obtained from standard solutions. Data were expressed as μg Evans blue/g tissue.
Brain Water Content
Brain water content was measured as previously described 23. Briefly, animals were decapitated under deep anesthesia. Brains were immediately removed and cut into four parts: both ipsilateral and contralateral hemisphere, cerebellum, and brain stem. Tissue samples were weighed on an electronic analytical balance (APX‐60; Denver Instrument, Denver, CO, USA) to obtain the wet weight (WW). The tissue was then dried at 100°C for 24 h to determine the dry weight (DW). Brain water content (%) was calculated as ([WW−DW]/WW) × 100%.
BBB Leakage using IgG Staining
To measure BBB leakage, immunolocalization of IgG was used as previously described 24. Sections were incubated overnight with goat antirat IgG biotin‐conjugated antibody 1:100 (Santa Cruz Inc., Santa Cruz, CA, USA) at 4°C and subsequently treated with an ABC staining kit.
Western Blot
The brain samples were prepared for Western blot from two parts. One part contained animals after MCAO at 5 different time points (killed at 12, 24, 36, 72 h, and 7 days) to detect the location and changing pattern of PDCD5 expression; another part contained five experimental groups (Sham, MCAO, rhPDCD5, control siRNA, and PDCD5 siRNA). Tissues from brain cortex including the penumbra area were homogenized, and aliquots of each fraction were used to determine the protein concentration of each sample using a detergent compatible assay. Protein samples (50 μg) were loaded onto polyacrylamide gel, electrophoresed, and transferred to a nitrocellulose membrane. The nitrocellulose membranes were then blocked followed by incubation with the primary antibodies overnight at 4°C. The following primary antibodies were purchased from Santa Cruz Biotechnology: (1) rabbit anti‐PDCD5, (2) rabbit anti‐p53, (3) rabbit anti‐Bax and Bcl‐2, and (4) mouse anticleaved caspase‐3. Immunoblots were processed with secondary antibodies (Santa Cruz Biotechnology) for 1 h at room temperature. Immunoblots were probed and then exposed to X‐ray film. The X‐ray films were scanned, and the optical density was determined by Bio‐Rad image analysis (Bio‐Rad, Hercules, CA, USA).
Immunohistochemistry, TUNEL Staining, and Double Fluorescence Labeling
For the histological analysis, each animal (n = 5) at 24 h after operation was anesthetized. After perfusion with 250 mL of 4% paraformaldehyde in 0.1 M PB (pH 7.4), their brains were removed and postfixed with formalin, and cryoprotected in 30% sucrose in PBS for over 48 h at 4°C. Coronal brain sections (20 μm thick) were cut on a cryostat (Leica CM3050 S). Immunohistochemistry was performed as the way described previously 25 using the same antibodies as in Western blot. The methods for double fluorescence labeling have been described previously 25. The fragmentation of nuclear DNA in cells has been identified extensively with TUNEL staining 26. They were observed under an OLYMPUS BX51 microscope.
Data Analysis
Data were expressed as mean ± SE. Statistical significance was assured by ANOVA performed in 1‐way ANOVA followed by the Tukey test for multiple comparisons. The clinical behavior scores were compared by Kruskal–Wallis 1‐way ANOVA followed by multiple comparison procedures by Dunn method. For the mortality statistical analysis, the Fisher exact test was used in two groups' comparisons 27. A probability value of P < 0.05 was considered statistically significant.
Results
Physiological Data Showed No Statistical Differences
No statistical differences were observed in the PDCD5 siRNA/control siRNA/MCAO/rh PDCD5/Sham groups with regard to mean arterial blood pressure, heart rate, arterial blood gases, or glucose levels before, during, or after ischemia (data not shown).
PDCD5 Upregulated Following Ischemia/Reperfusion Injury
The expression of PDCD5 after ischemia at 5 different time points was detected by Western blot analysis at 12, 24, 36, 72 h, and 7 days following MCAO (Figure 2A). The protein slightly increased as early as 12 h following MCAO and increased to the highest level at 24 h, and then reduced continuously at both 36 and 72 h (P < 0.05) until no significant difference compared with Sham by 7 days (P > 0.05) (Figure 2B). The 24 h was chosen as the time point for further study because of its peak expression.
Figure 2.
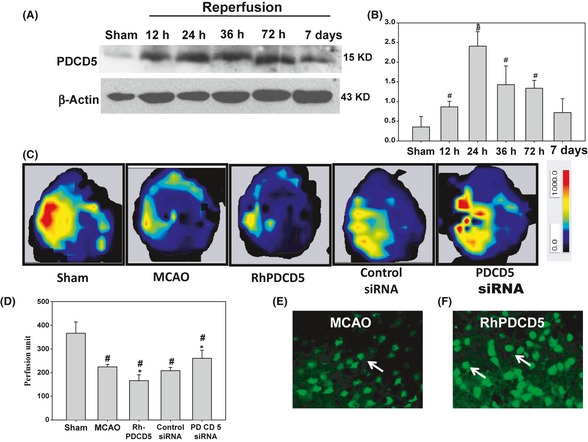
(A&B) The expression of PDCD5 postischemia at 12, 24, 36, 72 h, and 7 days. Western blot analysis of PDCD5 expression postischemia at 12, 24, 36, 72 h, and 7 days. PDCD5 significantly increased at 12 h, and the peak of expression was at 24 h. Then, it reduced continuously at both 36 and 72 h until no significant difference compared with Sham by 7 days (P > 0.05). # P < 0.05 versus Sham; *P < 0.05 versus other groups. (C&D) cerebral blood flow in the ipsilateral cerebral cortex of rat at 24 h following MCAO. (C) The representative images of cerebral blood flow (CBF) of ipsilateral cortex in different groups. The magnitude of CBF is represented by different colors, with blue to red denoting low to high. (D) Quantitative analysis of CBF in different groups. # P < 0.05 versus Sham; *P < 0.05 versus MCAO/control siRNA. (E&F) PDCD5 distribution after the rhPDCD5 protein injection. PDCD5 fluorescence staining using FITC‐conjugated PDCD5 antibody was detected in the penumbra area in the MCAO rats (E) and in the rhPDCD5 rats (F). We found that the expression of PDCD5 in the nucleus is stronger after the injection of rhPDCD5. The arrows showed the PDCD5‐positive cells.
To observe the PDCD5 tissue distribution after the rhPDCD5 protein injection, we detected PDCD5 fluorescence staining using FITC‐conjugated PDCD5 antibody in the penumbra area in the MCAO rats (Figure 2E) and in the rhPDCD5 rats (Figure 2F). We found that the expression of PDCD5 in the nucleus is stronger after the injection of rhPDCD5, which was consistent with previous study about the cellular uptake of exogenous PDCD5 protein 28. The study showed that PDCD5 protein could make use of clathrin‐independent endocytosis to enter the cells and promote programmed cell death. Unfortunately, we could not distinguish the endogenously expressed and exogenously delivered PDCD5.
PDCD5 siRNA Improved the Cerebral Blood Flow
Cerebral blood flow was determined by a laser Doppler perfusion image system in different groups as shown in Figure 2C, and the quantification of the result is presented in Figure 2D. Impressively, MCAO injury for 2 h evoked a significant reduction in CBF compared with Sham group (231.83 ± 4.17 vs. 366.33 ± 47.21), and this reduction remained for 24 h (224.31 ± 10.57) after reperfusion. PDCD5 siRNA and rhPDCD5 significantly attenuated and aggravated the ischemia evoked decrease in CBF (260.41 ± 34.05 in PDCD5 siRNA group and 166.03 ± 24.84 in rhPDCD5 group).
PDCD5 Increased the Cerebral Infarction
The cerebral infarction at 24 h after MCAO was shown in Figure 3A. The white colored area represented the infarction regions in these sections. The ratios of cerebral infarction in five groups were shown in Figure 3B. Severe infarction was observed in all rats of MCAO and MCAO+ control siRNA groups (P < 0.05 vs. Sham), and the infarct ratios were significantly increased by about 14.35% and decreased by about 8.5% in rats with the i.c.v. injection of rhPDCD5 and PDCD5 siRNA (P < 0.05 vs. MCAO/control siRNA, respectively).
Figure 3.
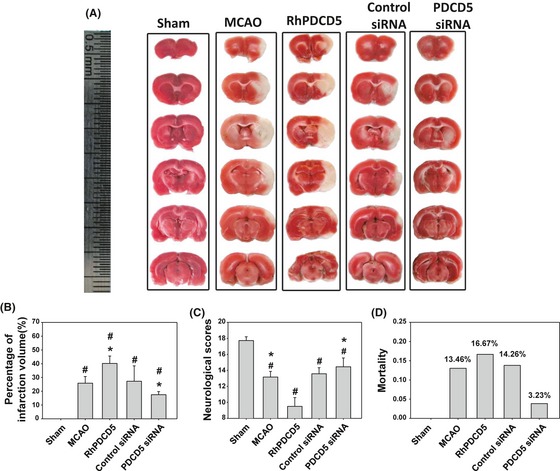
2,3,7‐Triphenyltetrazolium chloride (TTC) staining, infarction ratios of rat brains, neurobehavioral scores, and mortality rate. (A) Representative samples of TTC‐stained brain sections from rats killed at 24 h following ischemia. Severe infarction was shown in MCAO and control siRNA rats. The white areas represented the infarction regions in these sections. RhPDCD5 and PDCD5 siRNA increased and reduced the infarction volume, respectively, especially in the cortex. No ischemic lesion was found in the Sham group. (B) Statistical analysis of the infarction ratio, which was calculated by taking the infarcted area of the ipsilateral hemisphere divided by the total infarcted area. RhPDCD5 and PDCD5 siRNA significantly changed the infarction area. # P < 0.05 versus Sham; *P < 0.05 versus MCAO/control siRNA. (C) Grades of 3–18 were used for neurological scores. PDCD5 siRNA significantly improved the postischemic neurological deficit. However, rhPDCD5 protein made the rats take a turn for the worse state. # P < 0.05 versus Sham; *P < 0.05 versus MCAO/control siRNA. (D) There was no significant difference among the MCAO, rhPDCD5, control siRNA, and PDCD5 siRNA groups with regard to mortality (P > 0.05) by the Fisher exact test. A probability value of P < 0.05 was considered statistically significant.
PDCD5 Worsened Neurological Scores and Increased Mortality
The neurological score was 17.7 ± 0.47 in the Sham group, 13.2 ± 0.7 in the MCAO group, 9.5 ± 1.1 in the rhPDCD5 group, and 13.5 ± 0.76 and 14.45 ± 1.1 in the control siRNA and PDCD5 siRNA groups, respectively (Figure 3C). PDCD5 siRNA was found to increase the neurological scores significantly; however, the rhPDCD5 protein worsened the neurological deficits (P < 0.05).
The mortality rate (Figure 3D) in MCAO rats was 13.46% (7/52 rats); in Rh‐PDCD5 rats, 16.67% (6/36 rats); in MCAO+control siRNA rats, 14.26% (5/35 rats); in MCAO+PDCD5 siRNA rats, 3.23% (1/31 rats); and 0% (0/25 rats) in Sham rats. Statistical analysis revealed no significant difference between MCAO+PDCD5 siRNA versus MCAO, and rhPDCD5 versus MCAO using the Fisher exact test.
PDCD5 Deteriorated the BBB Disruption and Brain Water Content
Evans blue extravasation was reduced greatly with the use of PDCD5 siRNA and increased by the rhPDCD5 protein injection as seen from Figure 4A (blue‐stained area), which was in accordance with the TTC staining. Evans blue content in brain tissue in Sham‐operated was 0.34 ± 0.15 μg/g tissue. At 24 h after MCAO, Evans blue content in the area of infarction was markedly increased in MCAO group and MCAO+control siRNA group (3.36 ± 0.79 μg/g tissue, and 3.6 ± 0.69 μg/g tissue, P < 0.05 vs. Sham). Rats treated with rhPDCD5 and PDCD5 siRNA; Evans blue content was increased by about 74% and reduced by 41% (5.26 ± 1.11 μg/g tissue and 1.87 ± 0.43 μg/g tissue, respectively, P < 0.05 versus MCAO and MCAO+control siRNA group) (Figure 4B).
Figure 4.
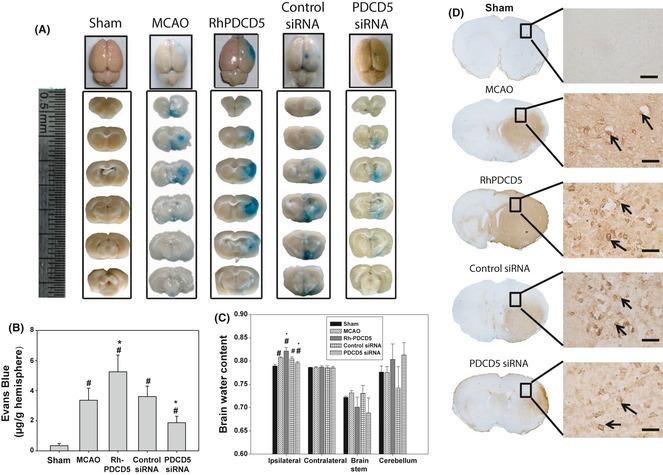
Evans blue extravasation, brain water content, and blood–brain barrier leakage IgG staining. (A) Representative samples of Evans blue‐stained brain sections from rats killed at 24 h following MCAO. Negative dye leakage was observed in the Sham group while notable staining was seen in the ischemic areas in the MCAO group and control siRNA group. RhPDCD5 protein group showed larger Evans blue extravasation, but PDCD5 siRNA group displayed weaker Evans blue staining in the infarcted areas. (B) Vascular leakage was determined by measuring the amount of brain‐extracted Evans blue by spectrophotometry at 620 nm and expressed as μg/g of brain tissue. RhPDCD5 and PDCD5 siRNA group demonstrated an increased and reduced EB content compared with MCAO/control siRNA groups (P < 0.05). Values were expressed as means±SEM. (C) The brain water content of the ipsilateral ischemic hemisphere, the contralateral hemisphere, brain stem, and cerebellum. The ipsilateral hemisphere of rhPDCD5 and PDCD5 siRNA group had significantly increased and decreased water content compared with the MCAO and control siRNA groups (P < 0.05, ANOVA). The results of the contralateral hemisphere, brain stem, and cerebellum from all groups showed no difference. # P < 0.05 versus Sham; *P < 0.05 versus MCAO/control siRNA. (D) No staining of IgG was observed in the cerebral cortex in the Sham group. Strong staining was seen in the MCAO and control siRNA groups in the penumbra area. Numerous positive granules were visible around the blood vessels and also in some cells, which had phagocytized these granules. Extensive stronger staining and more positive cells were seen in rhPDCD5 group. However, PDCD5 siRNA group demonstrated slighter staining area and less cell number. Black arrows indicate examples of the positive cells.
Brain water content was shown in Figure 4C. There was no significant difference among all the groups for the contralateral hemisphere, brain stem, and cerebellum. Compared to Sham‐operated animals, the brain water content in the other groups significantly increased in ipsilateral hemisphere at 24 h post‐MCAO injury (P < 0.05). Subsequent PDCD5 siRNA injection showed significant reduction statistically in brain water content (P < 0.05 vs. MCAO and control siRNA). However, the rhPDCD5 protein group showed the highest brain water content (P < 0.05 vs. MCAO and control siRNA) in the ipsilateral hemisphere.
Ischemic brain coronal sections were administered with IgG to assess BBB permeability further. The results showed an increase of IgG staining after injection of rhPDCD5 compared with MCAO and control siRNA groups, while PDCD5 siRNA group demonstrated less staining area and cell number (Figure 4D).
PDCD5 Upregulated the Apoptotic Signaling
Immunochemistry analysis of the penumbra area of cerebral cortex showed a strong upregulation of PDCD5, p53, and caspase‐3 following MCAO as well as in the control siRNA group, but they were markedly inhibited by PDCD5 siRNA and enhanced by rhPDCD5 (Figure 5A–C). Western blot analysis of the infarcted brain tissue in each group showed that the expression levels of PDCD5 (Figure 6A), p53 (Figure 6B), cleaved caspase‐3 (Figure 6C), and Bax/Bcl‐2 (Figure 6D) were significantly increased in the MCAO and control siRNA groups compared to those of the Sham group (P < 0.05). However, administration of rhPDCD5 and PDCD5 siRNA enhanced and reduced the levels of PDCD5, p53, cleaved caspase‐3, and Bax/Bcl‐2 proteins.
Figure 5.
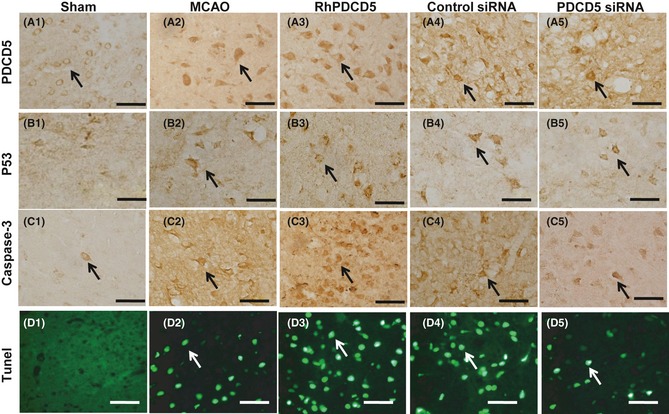
Immunohistochemistry and TUNEL staining in the penumbra area. Immunoreactivity staining of PDCD5 in Sham group mainly localized in the cytoplasm or cytomembrane (A1). After MCAO, it showed the transposition to the nuclei in the infarction regions (A2). The rhPDCD5 (A3) and PDCD5 siRNA (A5) group demonstrated increased and decreased number of positive staining cell. Almost no p53 or caspase‐3 in the infarcted areas was observed in the Sham‐operated rats (B1, C1). Massive immunoreactivity of p53 was localized in the infarction regions at 24 h in MCAO and control group. However, the extent of p53 immunostaining was enhanced in the rhPDCD5 group and lessened in PDCD5 siRNA group (B2, B5). Similar results were found for caspase‐3 (C1–C5). The apoptosis is marked by the terminal deoxynucleotidyl transferase (TdT)‐mediated biotinylated UTP nick end labeling (TUNEL) assay (green). No detectable TUNEL‐positive cells were found in the Sham‐operated animals (D1). In samples collected from the MCAO (D2) and control group (D4), the damaged cells were characterized by a round and shrunken morphology. The processus disappeared and the neuronal body became rounded with strong TUNEL staining in the nucleus. In rhPDCD5 and PDCD5 siRNA group, the number of positive cells observed in the cortex had increased and decreased dramatically by 24 h (D2, D5). Bar = 50 μm. The arrows showed the positive immunostaining cell.
Figure 6.
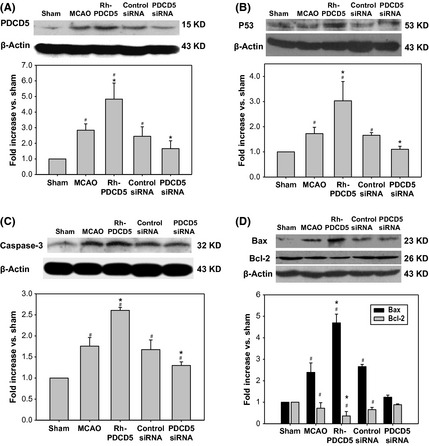
Western blot analysis of the penumbra area in each group. The expression levels of PDCD5 (A), p53 (B), cleaved caspase‐3 (C), and Bax/Bcl‐2 (D) were significantly increased in the MCAO and control siRNA groups compared to those of the Sham group (P < 0.05). However, administration of rhPDCD5 and PDCD5 siRNA enhanced and reduced the levels of PDCD5, p53, cleaved caspase‐3, and Bax/Bcl‐2 proteins. # P < 0.05 versus Sham; *P < 0.05 versus MCAO/control siRNA.
The apoptotic activity shown by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was a common method for detecting DNA fragmentation that results from apoptotic signaling cascades. No detectable TUNEL‐positive cells were found in the Sham‐operated animals (Figure 5D1). In samples collected from the MCAO (Figure 5D2) and control siRNA group (Figure 5D4), the damaged cells were characterized by a round and shrunken morphology. The axons and dendrites disappeared, and the neuronal body became rounded with strong TUNEL staining in the nucleus. The number and density of TUNEL‐positive staining were increased and decreased in rats treated with rhPDCD5 protein and PDCD5 siRNA (Figure 5D3,5D5).
PDCD5 Colocalized with MAP2, GFAP, CD34, P53, and Caspase‐3
Double fluorescence labeling revealed positive costaining (yellow) of PDCD5 (green) to MAP2 (red, microtubule‐associated protein 2, marker for neurons) (Figure 7A2) and endothelia to CD34 (red, endothelium marker) (Figure 7B2) but rarely with astrocytes to GFAP (red, glial fibrillary acidic protein) (Figure 7C2), indicating that PDCD5 expressed in neurons and also in some endothelia and astrocytes.
Figure 7.
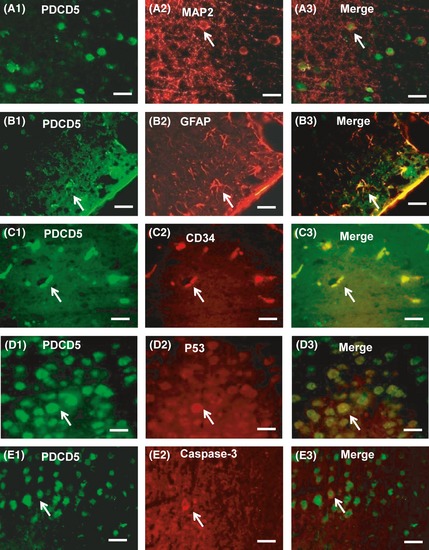
Double immunofluorescence staining. Double immunofluorescence staining showed the colocalization of PDCD5 (A1, B1, C1) with MAP2 (A2) and endothelia shown by CD34 (C2) but rarely with astrocytes shown by GFAP (B2). PDCD5 (D1, E1) also colocalized with p53 (D2), caspase‐3 (E2) shown by the merged images (A3, B3, C3, D3, E3) in the cerebral cortex at 24 h after middle cerebral artery occlusion (MCAO). PDCD5 (green) was expressed by fluorescein isothiocyanate (FITC), and MAP2, GFAP, CD34, p53, and caspase‐3 were expressed by tetramethylrhodamine isothiocyanate (TRITC). Scale bars: 50 um. Small white arrows indicate examples of the positive cells.
Double fluorescence labeling of PDCD5 with p53 and caspase‐3 in the penumbra area after 24 h of MCAO showed that PDCD5 (Figure 7D1,E1) was partly superposition and coexpressed with p53 (Figure 7D2) and caspase‐3 (Figure 7E2) in neuronal tissues.
Discussion
Regulation of apoptosis process is essential to maintain the balance between cell survival and cell death, which is also important in the ischemic injury. PDCD5 has been identified as a proapoptotic gene and a potent enhancer of cellular apoptosis 2. Reduced PDCD5 expression levels are often associated with suppression of the apoptotic process, and PDCD5 enhancement may increase the susceptibility of cells to apoptosis and lead to clinical benefit in the treatment of many cancer 29, 30, 31, 32. The aim of this study was to investigate the roles of PDCD5 protein after ischemic injury in the brain.
The Pattern of PDCD5 Expression in MCAO Model
To determine the effect of PDCD5 in middle cerebral artery occlusion model, we firstly observed the pattern of PDCD5 expression in the injured brain tissue. We found that after ischemic injury, PDCD5 increased significantly compared with Sham rat. Double fluorescence labeling showed that PDCD5‐immunoreactive cells were mainly colocalized with neurons as determined by MAP2 staining and rarely with astrocytes as determined by GFAP staining. In our experiment, we have found CD34 (endothelium marker) in the ischemic zone, and these CD34‐positive endothelial cells coexpressed PDCD5. The endothelium is one important component of the BBB. So the changes of PDCD5 might affect the integrity of endothelium, which might lead to brain edema and Evans blue extravasation as shown in our study. Nuclear translocation of PDCD5 was found after MCAO, which was consistent with previous study by Chen et al. 9. Meanwhile, we chose 24 h after ischemia as the time point for our study because of the peak expression of PDCD5 although it had significantly increased as early as 12 h.
The Role of PDCD5 in the MCAO‐Induced Focal Ischemia
PDCD5 is a novel protein associated with the regulation of cell apoptosis and para‐apoptosis 8. Previous studies have shown that PDCD5 plays a role in accelerating apoptosis rather than being an initiating factor in this process. However, the effect of PDCD5 in the brain ischemia has not been studied. In the present study, for the first time, we investigated the effect of PDCD5 as an apoptosis‐facilitating factor in brain ischemia. We used rhPDCD5 protein and siRNA against PDCD5 to increase or decrease the expression of PDCD5 as measured on protein levels by Western blot and immunohistochemistry. Furthermore, we demonstrated that decreased expression of PDCD5 by siRNA resulted in significant inhibition of p53 and Bax activation, and attenuation of caspase‐3 expression following brain ischemic injury. Administration of PDCD5 siRNA also resulted in an apparent improvement in cerebral blood flow and cerebral infarction, as demonstrated by CBF and TTC staining in addition to the attenuation of brain edema, Evans blue extravasation, and improvement of neurological score. The relationship among these results was still unclear. It is most likely that alleviation in brain edema, which reduces the intracranial pressure, leading to an improvement in the supply of cerebral blood and energy and, finally, the attenuation of infarction and neurological outcome. Further study was needed to clarify this mechanism. Meanwhile, the rhPDCD5 protein injection had the opposite effect. These results implicated that the elevation of PDCD5 may be harmful in cerebral ischemia, and an abatement of PDCD5 could be neuroprotective from brain damage.
The Possible Mechanism of PDCD5 Involved in the Ischemia Injury
In our study, rhPDCD5 protein was injected into the rat brain to induce its upregulation. We found that over‐expression of PDCD5 could enhance the apoptosis‐inducing effects of focal ischemia by TUNEL staining. These results are consistent with the report by Chen et al. 30, in which PDCD5 could enhance apoptosis in chondrosarcomas induced by cisplatin.
Western blot and immunochemistry analysis were also used to detect the level of protein expression of Bcl‐2 family proteins (Bax/Bcl‐2). Specifically, the Bcl‐2 family of proteins has been shown to have vital roles in several different pathways affected by apoptosis, with the ratio of Bax/Bcl‐2 being critical for the induction of apoptosis 33. In our study, injection with rhPDCD5, the expression of Bax was increased and expression of Bcl‐2 was decreased, which gave us an instinct that PDCD5 were related with apoptosis in the brain ischemia.
Previous study showed that PDCD5 enhancement of caspase‐3 activity is required for DNA‐damage‐induced apoptosis, and rhPDCD5 sensitizes cells to DNA damage by promoting caspase‐3 activity 34. In this study, we found increased activated caspase‐3 expression after rhPDCD5 but reduced level after the inhibition of PDCD5. We speculated that activated caspase‐3 is a vital element of the apoptosis signaling pathway in the PDCD5‐related brain injury but initiating an unclear signal to trigger apoptosis.
Here, we showed that PDCD5 siRNA could attenuate the expression of p53 accompanied by the reduction of Bax after ischemic injury. These findings were consistent with other studies that transfer of anti‐PDCD5 antibody into cells via in situ electroporation could suppress apoptosis induced by etoposide in HeLa cells 10. Our data also showed that suppressed expression of PDCD5 induced the inhibition of caspase‐3 activity. Accordingly, we speculated that PDCD5 may exert its apoptosis‐accelerating role by caspase‐3‐dependent pathway and may act as a general enhancer of programmed cell death.
Furthermore, in our study, the results of p53 level regulated by PDCD5 in the injured brain tissue gave us a clue that PDCD5 might competitively disturb the interaction between p53 and MDM2 or other functional proteins. Previous studies had demonstrated that after UV irradiation, PDCD5 binds to Tip60 and enhances the stability of Tip60 protein, increases Tip60‐dependent acetylation of p53, and promotes the expression of proapoptotic gene Bax, and then consequently accelerates apoptosis 12. The interaction of PDCD5 with p53 may be one of the mechanisms by which PDCD5 performs its function such as apoptosis. In the present study, we have also observed the colocalization of PDCD5 and p53 in the neurons. In neurons, PDCD5 may promote neuronal death involving enhanced p53 transcriptional activity. A direct interaction between p53 and PDCD5 is not detectable in vivo, but PDCD5‐mediated activation of p53 affected the brain injury. Whether these mechanisms play a role in apoptotic signaling after ischemia or other neurodegenerative conditions remains to be established. We supposed that during ischemia, PDCD5 stabilized p53 and activates the expression of various genes including Bax, a proapoptotic member of Bcl‐2 family proteins. Bax translocates to mitochondria and releases cytochrome c to cytosol to interact with Apaf‐1 to activate caspase‐9, which in turn activates downstream caspases, such as caspase‐3.
Apoptosis is a type of programmed cell death that can be induced by various means, and many genes are closely involved in the regulation of apoptosis 35. How PDCD5 interacts with these genes awaits further investigations. Further studies on the interaction between PDCD5 and these proteins will be beneficial for the elucidation of the molecular mechanisms involved in PDCD5 regulation.
In conclusion, PDCD5 plays an important role in the process of cell apoptosis, which would aggravate the brain injury, and the inhibition of PDCD5 may have protective effect. PDCD5 siRNA with its transfection reagent improved neurological deficits and protected blood–brain barrier. It also decreased the expression of p53, Bax, and caspase‐3, which are associated with apoptosis. Thus, the therapeutical effect of PDCD5 inhibition may have potentials in the treatment of acute ischemic cerebral stroke.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81000523 and 31271280). The authors appreciate Prof. Ying‐Yu Chen (Peking University, Beijing) for advice and guidance and Dr. Nikan H. Khatibi (Department of Anesthesiology, Loma Linda University Medical Center, Loma Linda, California 92354) who revised the English version of this article.
References
- 1. Lansberg MG, O'Donnell MJ, Khatri P, et al. Antithrombotic and thrombolytic therapy for ischemic stroke: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence‐Based Clinical Practice Guidelines. Chest 2012;141:e601S–e636S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liu H, Wang Y, Zhang Y, et al. TFAR19, a novel apoptosis‐related gene cloned from human leukemia cell line TF‐1, could enhance apoptosis of some tumor cells induced by growth factor withdrawal. Biochem Biophys Res Commun 1999;254:203–210. [DOI] [PubMed] [Google Scholar]
- 3. Li H, Wang Q, Gao F, et al. Reduced expression of PDCD5 is associated with high‐grade astrocytic gliomas. Oncol Rep 2008;20:573–579. [PubMed] [Google Scholar]
- 4. Hedenfalk I, Duggan D, Chen Y, et al. Gene‐expression profiles in hereditary breast cancer. N Engl J Med 2001;344:539–548. [DOI] [PubMed] [Google Scholar]
- 5. Yang YH, Zhao M, Li WM, et al. Expression of programmed cell death 5 gene involves in regulation of apoptosis in gastric tumor cells. Apoptosis 2006;11:993–1001. [DOI] [PubMed] [Google Scholar]
- 6. Xu XR, Huang J, Xu ZG, et al. Insight into hepatocellular carcinogenesis at transcriptome level by comparing gene expression profiles of hepatocellular carcinoma with those of corresponding noncancerous liver. Proc Natl Acad Sci USA 2001;98:15089–15094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ruan GR, Qin YZ, Chen SS, et al. Abnormal expression of the programmed cell death 5 gene in acute and chronic myeloid leukemia. Leuk Res 2006;30:1159–1165. [DOI] [PubMed] [Google Scholar]
- 8. Wang Y, Li X, Wang L, et al. An alternative form of paraptosis‐like cell death, triggered by TAJ/TROY and enhanced by PDCD5 overexpression. J Cell Sci 2004;117:1525–1532. [DOI] [PubMed] [Google Scholar]
- 9. Chen Y, Sun R, Han W, et al. Nuclear translocation of PDCD5 (TFAR19): An early signal for apoptosis? FEBS Lett 2001;509:191–196. [DOI] [PubMed] [Google Scholar]
- 10. Rui M, Chen Y, Zhang Y, Ma D. Transfer of anti‐TFAR19 monoclonal antibody into HeLa cells by in situ electroporation can inhibit the apoptosis. Life Sci 2002;71:1771–1778. [DOI] [PubMed] [Google Scholar]
- 11. Chen LN, Wang Y, Ma DL, Chen YY. Short interfering RNA against the PDCD5 attenuates cell apoptosis and caspase‐3 activity induced by Bax overexpression. Apoptosis 2006;11:101–111. [DOI] [PubMed] [Google Scholar]
- 12. Xu L, Chen Y, Song Q, Xu D, Wang Y, Ma D. PDCD5 interacts with Tip60 and functions as a cooperator in acetyltransferase activity and DNA damage‐induced apoptosis. Neoplasia 2009;11:345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu Q, Chen C, Khatibi NH, et al. Lentivirus‐mediated transfer of MMP‐9 shRNA provides neuroprotection following focal ischemic brain injury in rats. Brain Res 2011;1367:347–359. [DOI] [PubMed] [Google Scholar]
- 14. Hu Q, Chen C, Yan J, et al. Therapeutic application of gene silencing MMP‐9 in a middle cerebral artery occlusion‐induced focal ischemia rat model. Exp Neurol 2009;216:35–46. [DOI] [PubMed] [Google Scholar]
- 15. Li L, Khatibi NH, Hu Q, et al. Transmembrane protein 166 regulates autophagic and apoptotic activities following focal cerebral ischemic injury in rats. Exp Neurol 2012;234:181–190. [DOI] [PubMed] [Google Scholar]
- 16. Chen C, Hu Q, Yan J, et al. Early inhibition of HIF‐1alpha with small interfering RNA reduces ischemic‐reperfused brain injury in rats. Neurobiol Dis 2009;33:509–517. [DOI] [PubMed] [Google Scholar]
- 17. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989;20:84–91. [DOI] [PubMed] [Google Scholar]
- 18. Kawamura S, Yasui N, Shirasawa M, Fukasawa H. Rat middle cerebral artery occlusion using an intraluminal thread technique. Acta Neurochir (Wien) 1991;109:126–132. [DOI] [PubMed] [Google Scholar]
- 19. Yin W, Badr AE, Mychaskiw G, Zhang JH. Down regulation of COX‐2 is involved in hyperbaric oxygen treatment in a rat transient focal cerebral ischemia model. Brain Res 2002;926:165–171. [DOI] [PubMed] [Google Scholar]
- 20. Huang P, Zhou CM, Hu Q, et al. Cerebralcare Granule® attenuates blood–brain barrier disruption after middle cerebral artery occlusion in rats. Exp Neurol 2012;237:453–463. [DOI] [PubMed] [Google Scholar]
- 21. Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke 1995;26:627–634. [DOI] [PubMed] [Google Scholar]
- 22. Gursoy‐Ozdemir Y, Bolay H, Saribas O, Dalkara T. Role of endothelial nitric oxide generation and peroxynitrite formation in reperfusion injury after focal cerebral ischemia. Stroke 2000;31:1974–1980. [DOI] [PubMed] [Google Scholar]
- 23. Xi G, Hua Y, Keep RF, Younger JG, Hoff JT. Brain edema after intracerebral hemorrhage: The effects of systemic complement depletion. Acta Neurochir Suppl 2002;81:253–256. [DOI] [PubMed] [Google Scholar]
- 24. Park S, Sinko PJ. P‐glycoprotein and multidrug resistance‐associated proteins limit the brain uptake of saquinavir in mice. J Pharmacol Exp Ther 2005;312:1249–1256. [DOI] [PubMed] [Google Scholar]
- 25. Li Y, Zhou C, Calvert JW, Colohan AR, Zhang JH. Multiple effects of hyperbaric oxygen on the expression of HIF‐1 alpha and apoptotic genes in a global ischemia‐hypotension rat model. Exp Neurol 2005;191:198–210. [DOI] [PubMed] [Google Scholar]
- 26. Yin D, Zhou C, Kusaka I, et al. Inhibition of apoptosis by hyperbaric oxygen in a rat focal cerebral ischemic model. J Cereb Blood Flow Metab 2003;23:855–864. [DOI] [PubMed] [Google Scholar]
- 27. Ostrowski RP, Colohan AR, Zhang JH. Mechanisms of hyperbaric oxygen‐induced neuroprotection in a rat model of subarachnoid hemorrhage. J Cereb Blood Flow Metab 2005;25:554–571. [DOI] [PubMed] [Google Scholar]
- 28. Wang Y, Li D, Fan H, et al. Cellular uptake of exogenous human PDCD5 protein. J Biol Chem 2006;281:24803–24817. [DOI] [PubMed] [Google Scholar]
- 29. Yin A, Jiang Y, Zhang X, Zhao J, Luo H. Transfection of PDCD5 sensitizes colorectal cancer cells to cisplatin‐induced apoptosis in vitro and in vivo. Eur J Pharmacol 2010;649:120–126. [DOI] [PubMed] [Google Scholar]
- 30. Chen C, Zhou H, Xu L, et al. Recombinant human PDCD5 sensitizes chondrosarcomas to cisplatin chemotherapy in vitro and in vivo. Apoptosis 2010;15:805–813. [DOI] [PubMed] [Google Scholar]
- 31. Gu L, Jiang Y, Wang Y, et al. TFAR19 gene changes the biophysical properties of murine erythroleukemia cells. Cell Biochem Biophys 2005;43:355–363. [DOI] [PubMed] [Google Scholar]
- 32. Wang S, Zeng X, Liu Y, et al. Construction and characterization of a PDCD5 recombinant lentivirus vector and its expression in tumor cells. Oncol Rep 2012;28:91–98. [DOI] [PubMed] [Google Scholar]
- 33. Rosse T, Olivier R, Monney L, et al. Bcl‐2 prolongs cell survival after Bax‐induced release of cytochrome c. Nature 1998;391:496–499. [DOI] [PubMed] [Google Scholar]
- 34. Zhuge C, Chang Y, Li Y, Chen Y, Lei J. PDCD5‐regulated cell fate decision after ultraviolet‐irradiation‐induced DNA damage. Biophys J 2011;101:2582–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol 2007;35:495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]