

Summary
Alzheimer's disease (AD) has a devastating impact on aged people worldwide. Although sophisticated and advanced molecular methods have been developed for its diagnosis since early phases, pharmacological treatment still represents an unresolved topic. The more the disease progresses, the more the uneffectiveness of antidementia drugs emerges. New and encouraging results from experimental works indicate that glutamate pathway may play a substantial role in the pathogenesis since early stages of the disease. Several experimental data together with the clinical use of the uncompetitive N‐methyl‐d‐aspartate (NMDA) antagonist memantine strengthen this idea. Unfortunately, definitive data on the glutamatergic transmission involvement in AD are still incomplete. Moreover, clinical results indicate only temporarily limited effects of memantine. Currently, memantine is indicated for moderate‐to‐severe cases of AD, an indication that may limit its efficacy and impact on Alzheimer's dementia. The association of memantine with the acetylcholinesterase inhibitor drugs used to treat dementia symptoms appears to be beneficial, in both experimental and clinical studies. Because cholinergic and glutamatergic dysfunction occurs early in AD, the coadministration of appropriate treatment in early stages of the disease might represent a valid option from the beginning of cognitive decline. Moreover, to better evaluate drug efficacy, the association of the recently introduced biomarkers with a clinical AD profile should be considered an aim to pursue.
Keywords: Alzheimer's disease, Aβ42, Clinical profile, Glutamate, Nicotinic receptor
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder leading to progressive cognitive decline and dementia. Pathological hallmarks of AD are neuritic plaques and neurofibrillary tangles, which are responsible for the degeneration of hippocampal, cortical, and subcortical neurons. Several biochemical, molecular, genetic, and clinical studies indicate that Aβ oligomers have a pivotal role in the mechanisms of synaptic impairment and neuronal degeneration in AD 1. Based on this hypothesis (amyloid hypothesis of AD), the formation of soluble species of Aβ peptides (oligomers) can directly interfere with memory formation mechanisms, inducing synaptic degradation, neurofibrillary tangles, excitotoxicity, cell death 2, 3, 4. Together with senile plaques and neurofibrillary tangles formation, the progressive disarrangement of neurotransmission 5 is also responsible for cognitive decline. Overall, cholinergic (and to a lesser extent also aminergic and glutamatergic) neurons appear to be particularly sensitive to Aβ toxicity. Several experimental and pathologic articles focused on the ‘glutamate‐mediated’ toxicity as one of the main processes responsible for memory impairment and cell death in AD 6, 7, 8, 9, 10.
Glutamate and Synaptic Plasticity
Although several receptor proteins, such as α7‐nAchR, insulin receptors, cellular prion protein, ephrin receptor 11, have been proposed to be able to bind Aβ peptides, inducing its toxic effects, the N‐methyl‐d‐aspartate (NMDA) receptors attracted much interest because of their involvement in neurodegenerative mechanisms. Glutamate is one of the main excitatory neurotransmitters in central nervous system and is particularly involved in synaptic plasticity, memory, and learning. There are two families of glutamate receptors located in the plasmalemma on neurons: the ionotropic and the metabotropic receptors. The ionotropic receptor family is further divided into three classes: N‐methyl‐d‐aspartate (NMDA) receptor NR1, NR2A‐D, and NR3A‐B, permeable to Ca2+ ions, α‐amino‐3‐hydroxyl‐5‐methyl‐4‐isoxazole‐propionate (AMPA), GluR1‐4, and kainate GluR5‐7; KA1‐2 receptors permeable to Na+ and K+ ions. Metabotropic receptors are G‐protein‐coupled glutamate receptors divided into three subgroups on the basis of their function and structure: group 1 (mGluRs1 and 5), group 2 (mGluRs 2 and 3), and group 3 (mGluR4 and mGluR6‐8). Ionotropic glutamate receptors are predominantly located in postsynaptic sites and mediate fast excitatory transmission, while metabotropic ones can be found in various compartments, in both neurons and glial cells in the brain. Following synapse activation, glial cells take up the glutamate excess, via their transporters EAAT1 and 2 (excitatory amino acids transporters). Here, glutamate is transformed into glutamine that is transported back to presynaptic terminals where it is transformed by the enzyme glutaminase to glutamate that returns to vesicles thanks to the activity of the vesicular transporters, VGLUT1 and 2. Glutamate is essential for establishing new neural networks and forms of memory and learning. NMDA receptor activation has a central role in the regulation of long‐term potentiation (LTP) and long‐term depression (LTD) mechanisms. The process is generated by high‐frequency stimulation of the presynaptic site with an increased release of glutamate in the synaptic cleft. The AMPA and mGluRs are involved in the first phase, whereas NMDA receptors become active after the continuous and synchronized activation of the AMPA and mGluRs. The activation of synaptic NR2A‐containing NMDA receptors induces a large increases in calcium [Ca2+]i in postsynaptic sites, which triggers additional events involving calcium calmodulin‐dependent kinase II (CaMKII), extracellular signal‐regulated kinase (ERK) activation, and phosphorylation of cyclic AMP response element binding protein (CREB), which is crucial for increasing protein synthesis and synaptic density. All these changes are involved in LTP induction. Several other protein kinases, like extracellular signal‐related kinase (ERK) 12, p38 mitogen‐activated protein kinase (MAPK), and glycogen synthase kinase 3‐beta (GSK3β) 13, have been shown to modulate LTP induction in the brain. In particular, the activation of ERK and the inactivation of MAPK and GSK3β have enhancing effects on LTP (see Figure 1 for summary). On the contrary, the decreased stimulation of excitatory synapses induces internalization of synaptic NMDA receptors and activation of perisynaptic NR2B‐containing NMDA receptors, with an ultimate lower increase in [Ca2+]i. This condition is responsible for LTD, a mechanism associated with neuronal remodeling with spine shrinkage and synapse rearrangement, and that in pathologic conditions is associated with synaptic collapse and failure 14.
Figure 1.
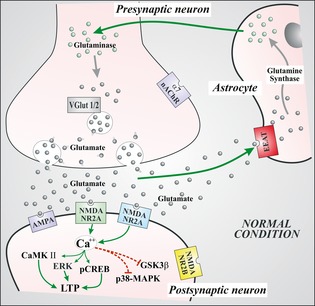
This figure represents the physiological glutamate‐mediated transmission at synaptic level. Glutamate release from presynaptic terminal acts through the activation of ionotropic glutamate receptors located in postsynaptic terminal. In the figure is in particular described the N‐methyl‐d‐aspartate (NMDA) signaling. The activated NMDA NR2A induces increase in calcium, which in turn favors the activation of several metabolic pathways (CaMK, ERK, and CREB) responsible for anabolic activation with subsequent activation of long‐term potentiation (LTP) mechanisms. Conversely, the calcium increase inhibits metabolic pathways (GSK3β and MAPK) responsible for long‐term depression and synaptic remodeling. Glutamate excess is transported via the EAAT into astrocytes where it is transformed to glutamine via the synthase activity which then again returns to synaptic terminals where the enzyme glutaminase produces again glutamate. The produced glutamate is subsequently filled in vesicles through a specific transporter (VGlut). VGlut (vesicular glutamate transporter); EAAT (excitatory amino acid transporter); α7‐nAchR (alpha‐7 nicotinic acetylcholine receptor); NMDANR2A (N‐methyl‐d‐aspartate NR2A subunit); NMDANR2B (N‐methyl‐d‐aspartate NR2B subunit); ERK (extracellular signal‐related kinase); CaMKII (calcium calmodulin‐dependent kinase II); pCREB (phosphorylated cyclic AMP response element binding protein); GSK3β (glycogen synthase kinase 3β); p38‐MAPK (p38 mitogen‐activated protein kinase).
Synaptic Plasticity in Alzheimer's Disease
In physiological conditions, Aβ peptides are able to induce synaptic plasticity mechanisms by stimulating at presynaptic level the α7 nicotinic receptor for acetylcholine. In pathologic conditions, Aβ oligomerizes through unknown mechanisms and forms several different soluble dimers, trimers, till higher‐order oligomers, protofibrils and fibrils. In particular, dimers and trimers have been found to have toxic effects on synapses. In fact, experimental evidences showed that pathologically elevated Aβ oligomers have been shown to inhibit LTP by interfering with the activity of the above‐mentioned LTP‐related molecular pathways 14, 15. The pathologically elevated levels of Aβ are able to block neuronal glutamate uptake at the synaptic cleft, leading to increased glutamate levels 16, 17. A rise in glutamate would activate synaptic NMDA receptors, subsequently inducing a desensitization of the receptors with ultimately synaptic depression. A second effect of increased levels of glutamate would be a spillover and an activation of extra‐synaptic NR2B‐enriched NMDA receptors 18, 19. The excessive activation of the NR2B receptors has been demonstrated to be responsible for synaptic plasticity impairment 13, 20, 21. In particular, the Aβ induces a partial blockade of NMDA receptor's currents, reducing the calcium influx into the spines 14, 15. In turn, the reduced calcium influx inactivates CAMKII and Akt/protein kinase B. Such change would inactivate prosurvival pathways like ERK, CREB, or BDNF, favoring MAPK‐p38, JNK, GSK3β pathways involved in hyperphosphorylation of tau (see Figure 2) and cell death signaling 13, 15, 22, 23. Furthermore, Aβ is able to induce both synaptic depression by activating mGluRs, which trigger a series of molecular events leading to the internalization of AMPA receptors and synapse collapse, and to down‐regulate the levels of postsynaptic density 95 protein (PSD‐95) and synaptophysin, with subsequent suppression of NR2A subunit function and activation of NR2B subunit 15, 24. The activation of NR2B subunits is able to induce caspase‐8 and caspase‐3 apoptotic pathways. These mechanisms are considered responsible for synaptic disarrangement, cell loss, and neuronal death in AD.
Figure 2.
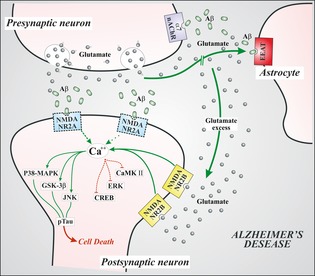
This figure represents the glutamate‐mediated transmission at synaptic level in Alzheimer's disease. Aβ oligomers interfere with NMDA signaling, inducing an internalization of postsynaptic NMDANR2A subunits (dotted). Due to interference of Aβ with EAAT, glutamate concentration in synaptic cleft increases. Moreover, Abeta oligomers form complexes with alpha7‐nicotinic receptors at presynaptic sites. This interaction induces increased levels of glutamate release. Glutamate spillover would activate extrasynaptic NMDANR2B receptors, with increased calcium levels and activation of metabolic pathways (green path) responsible for neuronal shrinkage and synaptic loss, associated with inhibition of prosurvival pathways (red path). The more this condition persists, the more the activated pathologic pathways lead to hyperphosphorylation of cytoskeletal tau protein, with neuronal degeneration and cell death. VGlut (vesicular glutamate transporter); EAAT (excitatory amino acid transporter); α7‐nAchR (alpha‐7 nicotinic acetylcholine receptor); NMDANR2A (N‐methyl‐d‐aspartate NR2A subunit); NMDANR2B (N‐methyl‐d‐aspartate NR2B subunit); ERK (extracellular signal‐related kinase); CaMKII (calcium calmodulin‐dependent kinase II); CREB (cyclic AMP response element binding protein); GSK3β (glycogen synthase kinase 3β); p38‐MAPK (p38 mitogen‐activated protein kinase), JNK (c‐Jun N‐terminal kinase), pTau (hyperphosphorylated tau protein).
Glutamate Hypothesis and Alzheimer's Disease
A clear direct involvement of glutamate in memory loss and in mechanisms of neurodegeneration observed in AD is far from being demonstrated in humans. However, these recent pathological observations showed that, to some extent, glutamatergic transmission could be attenuated since early stages of AD. In this view, the lower levels of VGLUTs and EAAT observed in the prefrontal and parietal cortex of AD brains 6, 7, 25 would be indicative of reduced glutamate metabolism. Similarly, impaired LTP mechanisms shown in recent electrophysiological studies made on patients with AD 26, 27 would be the consequence of NMDA signaling impairment, as predicted by experimental models 14. It is noteworthy that LTD was normal, a finding that is different from what experimentally observed 26 and does not confirm the supposed pathologic glutamate‐mediated synaptic depression. Thus, the glutamatergic dysfunction (impaired LTP) as an early event does not appear to be related to degeneration and cell death processes as suggested in experimental models 14. Because LTP and LTD impairments are also related to the aging process, changes in glutamatergic transmission observed in early phases of AD could probably be related to a physiological memory neuronal network impairment rather than to a pathological one 28 (Burke and Barnes 2011). On the contrary, neurodegeneration could be related to the Aβ‐mediated process that would induce toxic effects of glutamate in later stages of the disease, long after the memory impairment 4, 14. Therefore, to link the glutamatergic dysfunction to cognitive decline symptoms, it would be necessary to consider it as the consequence of the cholinergic dysfunction, an event that is closely related to memory loss 29, 30 and that occurs since early stages of the disease 30 and prior to the glutamatergic dysfunction. Particularly interesting is the role played by the α‐7‐nAchR, the nicotinic receptor for acetylcholine, which is extremely altered in patients with AD 31, 32. This receptor is located in presynaptic terminals of hippocampus and cerebral cortex and facilitates the release of a variety of neurotransmitters (both excitatory and inhibitory) throughout the brain 33. Its activation results in an enhancement of glutamatergic transmission 34, and the receptor is involved in the regulation of synaptic plasticity mechanisms 33, 35. In AD cases, it has been demonstrated that α‐7‐nAchR can bind with high affinity to Aβ peptides 35, 36. The interaction between these two structures would lead to the formation of complexes that are able to interfere with synaptic transmission with a pathogenic mechanism 37. This could be the trigger of the Aβ‐induced changes in glutamate transmission. Excessive and diffuse activation of glutamatergic receptors could be responsible for a further degeneration, a condition that could be more evident in later stages of the disease 37. This hypothesis could explain the reason why memantine is more effective in later stages of AD rather than in early stages.
Memantine in Alzheimer's Disease
Memantine currently represents the rationale for the “glutamate hypothesis” 15, 38, 39 in AD cases, and it is the only NMDA receptor antagonist in use in human clinical studies approved by the European Agency for the Evaluation of Medicinal Products (EMEA) (2002) and by the US Food and Drug Administration (FDA) (2003) for the treatment of moderate‐to‐severe AD (see drug summary Box). Memantine is a low‐to‐moderate affinity, uncompetitive NMDA receptor antagonist. Memantine preferentially blocks NMDA receptor channels when excessively activated. Because NMDA channels during normal synaptic activity are opened for few milliseconds, memantine is unable to act, thus sparing normal synaptic activity. Instead, during prolonged receptor activation, as in excitotoxic conditions, memantine becomes effective, blocking receptor activity. This particular mechanism
Box 1. Drug Summary Box.
Drug name: Axura and Akatinol (Merz), Namenda (Forest Laboratories), Ebixa and Abixa (Lundbeck), and Memox (Unipharm)
Indication: Moderate‐to‐severe Alzheimer's dementia
Mechanism of action: Uncompetitive NMDA (N‐methyl‐D‐aspartate) antagonist
Route of Administration: Oral administration 20 mg/day
Chemical structure: Memantine hydrochloride
Pivotal trial (S): Winblad et al., (1999), Reisberg et al., (2003), Tariot et al., (2004) and Howard et al., (2012)
makes memantine a potential neuroprotective agent against glutamate‐mediated neurotoxicity on the one hand and an enhancer of synaptic transmission on the other 40, 41. Memantine has recently been demonstrated to selectively target glutamate NR2B‐containing NMDA receptors, which are located in extrasynaptic sites and are linked to several molecular pathways related to cell death 42, 43 that are considered relevant to AD pathophysiology. Several experimental studies demonstrated neuroprotective effects of memantine against Aβ1‐42 effects 44. Memantine is able to reduce apoptotic Aβ‐induced effects, by reducing caspase‐3 and Bcl‐2 activation 43. In addition, memantine was demonstrated to be able to reverse Aβ‐induced LTP deficits 43, to reduce Aβ burden, and to increase synaptic density in the hippocampus of mice 45, 46. Moreover, neuroprotective effects of memantine have been shown to be increased by the coadministration with galantamine, an acetylcholine esterase inhibitor approved for AD treatment 47, 48, 49, 50. Galantamine is a reversible competitive cholinesterase inhibitor and allosteric modulator of acetylcholine receptors, particularly potentiates the activity of nicotinic receptors, increasing in turn NMDA receptors' activity. The coadministration of galantamine and memantine would potentiate the NMDA synaptic transmission, protecting from excitotoxicity and cell death 51, 52. These data can partially explain the beneficial effect obtained by the coadministration of memantine with acetylcholine inhibitors. To date, most studies showed positive effects of the association of donepezil with memantine. Unfortunately, data on the association of memantine with acetylcholinesterase inhibitor like galantamine (and/or rivastigmine, pseudo‐irreversible cholinesterase inhibitor) are not available so far. Because the mechanisms of action of these drugs are different, further studies are encouraged.
Therapeutic Perspectives for Alzheimer's Disease
“Amyloid and glutamate hypothesis”‐based treatment for cognitive symptoms represents the most recently adopted treatment option of AD. In recent years, several studies have been performed to evaluate the clinical efficacy of memantine in patients with AD. Earlier studies evaluated the efficacy of memantine alone and also in combination with acetylcholinesterase inhibitors (ACheIs). Each available study evaluated memantine efficacy considering two different endpoints. Primary variables of efficacy (primary endpoint) were the global rating (CIBIC‐plus) and the functional rating (modified ADCS‐ADL). Secondary variables (secondary endpoint) included the SIB (Severe Impairment Battery), MMSE, FAST (Functional Assessment Staging), GDS (Global deterioration Scale), NPI, and RUD (Resource Utilization and caregiver burden). In general, benefits of memantine emerge from all these studies (see Table 1). Two of these studies were randomized double‐blinded placebo‐controlled trials that evaluated memantine efficacy in a group of AD patients with moderate‐to‐severe dementia. Both obtained statistically significant results on primary endpoints, in particular on ADCS‐ADL score scale; however, they found no effects on secondary variables, in particular on behavioral symptoms 56, 57. These studies showed modest results, although encouraging, in severe cases. Other three studies evaluated memantine efficacy associated with the reversible cholinesterase inhibitor donepezil 10 mg/day 54, 55, 56. Results of these studies showed that memantine in combination with ACheIs was well tolerated. Memantine and ACheIs have different pharmacokinetics and dynamics 57. The association revealed better results with respect to monotherapy with ACheIs alone 58, 59 in both cognitive and functional domains. Memantine was also administered to mild cases of AD; in particular, two studies were performed and reported, however, contradictory results on both cognition (evaluated by the use of ADAS‐Cog) and functional domains 60, 61. The Cochrane analysis 62 showed the main results that memantine has beneficial effects on cognitive and functional decline, although small, and at six months, in moderate‐to‐severe AD, while in mild‐to‐moderate AD these effects were not detectable. In general, it can be concluded that all available drugs for AD are indicated for the treatment of cognitive decline symptoms. ACheIs are indicated for memory and attention deficits, but from all the studies examined, however, it does not emerge a clinical profile of the AD patient who deserves memantine's treatment. This may limit the correct evaluation and might also account for modest results of clinical evaluation. This is true for both ACheIs and memantine. As ACheIs are indicated for earlier phases of cognitive decline, it is supposed that in these phases cholinergic deficit prevails overall; similarly, memantine is indicated for moderate‐to‐severe cases, and thus for more advanced forms of cognitive decline, during which the dysfunction of glutamatergic transmission occurs, it is supposed to prevail. Of note is the evidence that the association of the two drugs represents the best available option, indicating, as above mentioned, that cognitive decline might be the result of more complex mechanisms, involving more than one or two neurotransmitters at the same time. More importantly, the evaluation of cognitive status and its pharmacological approach would often take into account the disease years and the rate of disease progression. The treatment of patients with rapidly progressive cognitive decline often unravels an unsuspected drug treatment refractoriness. Moreover, in all of the studies on memantine efficacy—that is, counteracting the excitotoxic effects at glutamate synapses and also slowing down the amyloid cascade effects on cortical neurons, or for the treatment of severe dementia symptoms (in this case, it would be useful to know which symptom has to be treated with memantine)—whether memantine was used because of the drug anti‐NMDA profile remains unclear. Reconsidering the evaluation of memantine's efficacy would need to enroll patients considering years of disease and, as a consequence, the rate of disease progression 63, 64, 65, 66. Given the high diagnostic and even prognostic value of CSF biomarkers (Aβ1‐42, t‐tau, and p‐tau) 67, 68, 69, 70 and PIB studies 71, 72, 73, their association with cognitive and functional assessment would make more reliable the evaluation of drug efficacy and also the evaluation for a more precise classification of patients (even the simple distinction between responders and nonresponders would be helpful). This would make more adherent to the reality, that is, the evaluation of treatment. In particular, because memantine has the great potential to counteract the amyloid cascade and subsequent glutamatergic toxicity, it is likely that its introduction since early phases of cognitive decline as mentioned above, possibly using reduced dosage, could represent a new and interesting therapeutic option to treat AD and other neurodegenerative disorders. Therefore, memantine has beneficial effects on AD evolution, although temporarily limited. This could be the effect of a delayed drug administration, which could limit its efficacy. Because amyloid pathology glutamate‐based hypothesis accounts for mechanisms of neurodegeneration, its administration since early phases of disease is reasonable. Use of different dosages in relation to the degree of cognitive decline could be considered as a therapeutic option. Association of memantine with donepezil, generally well tolerated, seems to be the most fruitful. Of note is the lack of data on either rivastigmine or galantamine (ACheIs with a different pharmacological profile than donepezil) combination therapy in these cases. It is likely to suppose that the pharmacological profile of these drugs could represent a valid therapeutic option. Thus, to reduce discrepancies observed in previous studies and to test real potential of memantine, more studies are needed. To obtain better results, it would be useful to test its efficacy in a population of patients with AD defined both by neuropsychological and by instrumental/biochemical features (CSF biomarkers, PET, MRI studies), with early‐diagnosed AD. This, on the one hand, would clarify whether memantine is a disease‐modifying drug, and could also help to define clinical profiles of both memantine's responders and nonresponders.
Table 1.
Summary of the principal trial on memantine's efficacy in AD
| Winblad and Poritis, (1999) “Memantine in severe dementia: results of the 9M‐Best Study. | Benefit and efficacy in severely demented patients during treatment with memantine |
| Reisberg, et al., (2003) Memantine Study Group. Memantine in moderate‐to‐severe Alzheimer's disease. | Memantine vs. placebo showed a better outcome on ACDS‐ADL, CIBIC+, and SIB |
| Tariot et al., (2004) “Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial.” | Memantine plus donepezil showed significant benefits in all measures (ACDS‐ADL, CIBIC+, SIB, NPI), compared with placebo plus donepezil |
| Bakchine et al., (2008) “Memantine treatment in patients with mild to moderate Alzheimer's disease: results of a randomised, double‐blind, placebo‐controlled 6‐month study.” | Memantine showed significant improvement in ADAS‐Cog and CIBIC+ at 12 and 18 weeks, supporting its efficacy in mild‐to‐moderate AD |
| Peskind et al., (2006) “Memantine treatment in mild to moderate Alzheimer disease: a 24‐week randomized, controlled trial.” | Significant improvement in ADAS‐cog, CIBIC+, and NPI in mild‐to‐moderate AD |
| Cummings et al., (2012) “Memantine MEM‐MD‐02 Study Group. Behavioral effects of memantine in Alzheimer disease patients receiving donepezil treatment.” | Memantine reduced agitation/aggression, irritability, and appetite/eating disturbances |
| Howard et al., (2012) Donepezil and memantine for moderate‐to‐severe Alzheimer's disease. | The continued treatment of memantine with donepezil was associated with cognitive benefits that exceeded the minimum clinically important difference and with significant functional benefits over the course of 12 months |
Conclusions
The glutamate receptor family of proteins appears to be involved in neuronal targeting by Aβ oligomers. In particular, a pathologic increase in Aβ levels appears to target the extrasynaptically located subtypes of NR2B‐containing NMDA receptors. These receptors are involved in the modulation of the cognitive functions 74 in the frontal cortex. Moreover, the physiological aging process induces changes in these receptors' localization (reduced) and even in function (decreased) that is related to the physiological memory decline 75, 76, 77. An enhanced activation of these receptors has been suggested to be the possible target for treating normal memory decline 78. Therefore, it is conceivable that, during early phases of AD, these receptors, which are reduced in number and function due to aging, become overactive only in certain regions of the brain (prefrontal cortex, hippocampus), in order to compensate for the memory loss. Then, the continuous activation associated with the pathological stimulus of Aβ oligomers might trigger and spread a glutamatergic cortical overactivation that in advanced stages could even induce an excitotoxic damage of neurons. In this view, an early pharmacological treatment with memantine, or even memantine associated from the beginning with ACheIs, might represent the best option for the treatment of patients with AD.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
We want to express our gratitude to Graziano Bonelli for art‐figures of this work.
References
- 1. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297:353–356. [DOI] [PubMed] [Google Scholar]
- 2. Selkoe DJ. Soluble oligomers of the amyloid beta‐protein impair synaptic plasticity and behavior. Behav Brain Res 2008;192:106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cheng L, Yin WJ, Zhang JF, Qi JS. Amyloid beta‐protein fragments 25–35 and 31–35 potentiate long‐term depression in hippocampal CA1 region of rats in vivo. Synapse 2009;63:206–214. [DOI] [PubMed] [Google Scholar]
- 4. Ondrejcak T, Klyubin I, Hu NW, Barry AE, Cullen WK, Rowan MJ. Alzheimer's disease amyloid beta‐protein and synaptic function. Neuromolecular Med 2010;12:13–26. [DOI] [PubMed] [Google Scholar]
- 5. Martorana A, Esposito Z, Koch G. Beyond the cholinergic hypothesis: do current drugs work in Alzheimer's disease? CNS Neurosci Ther 2010;16:235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kashani A, Lepicard E, Poirel O, et al. Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol Aging 2008;29:1619–1630. [DOI] [PubMed] [Google Scholar]
- 7. Kirvell SL, Esiri M, Francis PT. Down‐regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer's disease. J Neurochem 2006;98:939–950. [DOI] [PubMed] [Google Scholar]
- 8. Francis PT. Glutamatergic systems in Alzheimer's disease. Int J Geriatr Psychiatry 2003;18(Suppl 1):S15–S21. [DOI] [PubMed] [Google Scholar]
- 9. Gray CW, Patel AJ. Neurodegeneration mediated by glutamate and beta‐amyloid peptide: a comparison and possible interaction. Brain Res 1995;691:169–179. [DOI] [PubMed] [Google Scholar]
- 10. Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. beta‐Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci 1992;12:376–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Selkoe DJ. Resolving controversies on the path to Alzheimer's therapeutics. Nat Med 2011;17:1060–1065. [DOI] [PubMed] [Google Scholar]
- 12. Ivanov A, Pellegrino C, Rama S, et al. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal‐regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Neurophysiol 2006;572:789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble A(beta) oligomers inhibit long term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B‐containing NMDA receptors. J Neurosci 2011;31:6627–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Palop JJ, Mucke L. Amyloid‐beta‐induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci 2010;13:812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Danysz W, Parsons GC. Alzheimer's disease, beta amyloid, glutamate, NMDA receptors and memantine. Br J Pharm 2012;167:324–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koch G, Di Lorenzo F, Bonnì S, Ponzo V, Caltagirone C, Martorana A. Impaired LTP‐ but not LTD‐Like Cortical Plasticity in Alzheimer's Disease Patients. J Alzheimers Dis 2013;33:525–533. [DOI] [PubMed] [Google Scholar]
- 17. Texidó L, Martín‐Satué M, Alberdi E, Solsona C, Matute C. Amyloid β peptide oligomers directly activate NMDA receptors. Cell Calcium 2011;49:184–190. [DOI] [PubMed] [Google Scholar]
- 18. Domingues A, Almeida S, da Cruz e Silva EF, Oliveira CR, Rego AC. Toxicity of beta‐amyloid in HEK293 cells expressing NR1/NR2A or NR1/NR2B N‐methyl‐d‐aspartate receptor subunits. Neurochem Int 2007;50:872–880. [DOI] [PubMed] [Google Scholar]
- 19. Rammes G, Hasenjäger A, Sroka‐Saidi K, Deussing JM, Parsons CG. Therapeutic significance of NR2B‐containing NMDA receptors and mGluR5 metabotropic glutamate receptors in mediating the synaptotoxic effects of β‐amyloid oligomers on long‐term potentiation (LTP) in murine hippocampal slices. Neuropharmacology 2011;60:982–990. [DOI] [PubMed] [Google Scholar]
- 20. Kerven M, Angeli A, Nicole O, et al. Selective impairment of some forms of synaptic plasticity by oligomeric amyloid beta peptide in the mouse hippocampus: implication of extrasynaptic NMDA receptors. J Alzheimer Dis 2012;32:183–196. [DOI] [PubMed] [Google Scholar]
- 21. Bordji K, Becerill‐Ortega J, Nicole O, Buisson A. Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid‐Beta production. J Neurosci 2010;30:15927–15942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long term potentiation by naturally secreted and synthetic amyloid‐beta peptide in hippocampal slices is mediated via activation of the kinases c‐Jun N‐terminal kinase, cyclin‐dependent kinase 5, and p38 mitogen‐activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci 2004;24:3370–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hardingham GE, Fukunaga Y, Banding H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut‐off and cell death pathways. Nat Neurosci 2002;5:405–414. [DOI] [PubMed] [Google Scholar]
- 24. Koffie RM, Hyman BT, Spires‐Jones TL. Alzheimer's disease: synapses gone cold. Mol Neurodeg 2012;6:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parsons CG, Stöffler A, Danysz W. Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system‐too little activation is bad, too much is even worse. Neuropharmacology 2007;53:699–723. [DOI] [PubMed] [Google Scholar]
- 26. Koch G, Di Lorenzo F, Bonnì S, Ponzo V, Caltagirone C, Martorana A. Impaired LTP‐ but not LTD‐like cortical plasticity in Alzheimer's disease patients. J Alzheimers Dis 2012;31:593–599. [DOI] [PubMed] [Google Scholar]
- 27. Battaglia F, Wang HY, Ghilardi MF, et al. Cortical plasticity in Alzheimer's disease in humans and rodents. Biol Psychiatry 2007;62:1405–1412. [DOI] [PubMed] [Google Scholar]
- 28. Burke SN, Barnes CA. Senescent synapses and hippocampal circuit dynamics. Trends Neurosci 2010;33:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Giacobini E. Cholinergic function and Alzheimer's disease. Int J Geriatr Psychiatry 2003;18(Suppl 1):S1–S5. [DOI] [PubMed] [Google Scholar]
- 30. Selkoe DJ. Defining molecular targets to prevent Alzheimer disease. Arch Neurol 2005;62:192–195. [DOI] [PubMed] [Google Scholar]
- 31. Sattelle DB, Buckingham SD, Akamatsu M, et al. Comparative pharmacology and computational modelling yield insights into allosteric modulation of human alpha7 nicotinic acetylcholine receptors. Biochem Pharmacol 2009;78:836–843. [DOI] [PubMed] [Google Scholar]
- 32. Kar S, Slowikowski SP, Westaway D, Mount HT. Interactions between beta‐amyloid and central cholinergic neurons: implications for Alzheimer's disease. J Psychiatry Neurosci 2004;29:427–441. [PMC free article] [PubMed] [Google Scholar]
- 33. McKay BE, Placzek AN, Dani JA. Regulation of synaptic transmission and plasticity by neuronal nicotinic acetylcholine receptors. Biochem Pharmacol 2007;74:1120–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature 1996;383:713–716. [DOI] [PubMed] [Google Scholar]
- 35. Jürgensen S, Ferreira ST. Nicotinic receptors, amyloid‐beta, and synaptic failure in Alzheimer's disease. J Mol Neurosci 2010;40:221–229. [DOI] [PubMed] [Google Scholar]
- 36. Liu Q, Huang Y, Xue F, et al. A novel nicotinic acetylcholine receptor subtype in basal forebrain cholinergic neurons with high sensitivity to amyloid peptides. J Neurosci 2009;29:918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ni R, Marutle A, Nordberg A. Modulation of α7 Nicotinic Acetylcholine Receptor and Fibrillar Amyloid‐β Interactions in Alzheimer's Disease Brain. J Alzheimers Dis 2013;33:841–851. [DOI] [PubMed] [Google Scholar]
- 38. Revett TJ, Baker GB, Jhamandas J, Kar S. Glutamate system, amyloid ß peptides and tau protein: functional interrelationships and relevance to Alzheimer disease pathology. J Psychiatry Neurosci 2013;38:6–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zimmer ER, Kalinine E, Haas CB, et al. Pretreatment with memantine prevents Alzheimer‐like alterations induced by intrahippocampal okadaic acid administration in rats. Curr Alzheimer Res, 2012;9:1182–1190. [DOI] [PubMed] [Google Scholar]
- 40. Nimmrich V, Reymann KG, Strassburger M, et al. Inhibition of calpain prevents NMDA‐induced cell death and beta‐amyloid‐induced synaptic dysfunction in hippocampal slice cultures. Br J Pharmacol 2010;159:1523–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Okamoto S, Pouladi MA, Talantova M, et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med 2009;15:1407–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xia P, Chen HS, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci 2010;30:11246–11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Klyubin I, Wang Q, Reed MN, et al. Protection against Abeta‐mediated rapid disruption of synaptic plasticity and memory by memantine. Neurobiol Aging 2011;32:614–623. [DOI] [PubMed] [Google Scholar]
- 44. Song MS, Rauw G, Baker GB, Kar S. Memantine protects rat cortical cultured neurons against beta‐amyloid‐induced toxicity by attenuating tau phosphorylation. Eur J Neurosci 2008;28:1989–2002. [DOI] [PubMed] [Google Scholar]
- 45. Lacor PN, Buniel MC, Furlow PW, et al. Abeta oligomer‐induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci 2007;27:796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Abeta (1–42) are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 1998;95:6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhao X, Marszalec W, Toth PT, Huang J, Yeh JZ, Narahashi T. In vitro galantamine‐memantine co‐application: mechanism of beneficial action. Neuropharmacology 2006;51:1181–1191. [DOI] [PubMed] [Google Scholar]
- 48. Grossberg GT, Edwards KR, Zhao Q. Rationale for combination therapy with galantamine and memantine in Alzheimer's disease. J Clin Pharmacol 2006;46(Suppl 1):17S–26S. [DOI] [PubMed] [Google Scholar]
- 49. Farrimond LE, Roberts E, McShane R. Memantine and cholinesterase inhibitor combination therapy for Alzheimer's disease: a systematic review. BMJ Open 2012;2:e000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lorrio S, Negredo P, Roda JM, García AG, López MG. Effects of memantine and galantamine given separately or in association, on memory and hippocampal neuronal loss after transient global cerebral ischemia in gerbils. Brain Res 2009;1254:128–137. [DOI] [PubMed] [Google Scholar]
- 51. Csernansky JG, Bardgett ME, Sheline YI, Morris JC, Olney JW. CSF excitatory amino acids and severity of illness in Alzheimer's disease. Neurology 1996;46:1715–1720. [DOI] [PubMed] [Google Scholar]
- 52. Simoni E, Daniele S, Bottegoni G, et al. Combining Galantamine and Memantine in Multitargeted. New Chemical Entities Potentially Useful in Alzheimer's Disease. J Med Chem, 2012;55:9708–9721. [DOI] [PubMed] [Google Scholar]
- 53. Winblad B, Poritis N. Memantine in severe dementia: results of the 9M‐Best Study (Benefit and efficacy in severely demented patients during treatment with memantine). Int J Geriatr Psychiatry 1999;14:135–146. [DOI] [PubMed] [Google Scholar]
- 54. Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ. Memantine in moderate‐to‐severe Alzheimer's disease. N Engl J Med 2003;348:1333–1341. [DOI] [PubMed] [Google Scholar]
- 55. Atri A, Shaughnessy LW, Locascio JJ, Growdon JH. Long‐term course and effectiveness of combination therapy in Alzheimer disease. Alzheimer Dis Assoc Disord 2008;22:209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Porsteinsson AP, Grossberg GT, Mintzer J, Olin JT, Memantine MEM‐MD‐12 Study Group . Memantine treatment in patients with mild to moderate Alzheimer's disease already receiving a cholinesterase inhibitor: a randomized, double‐blind, placebo‐controlled trial. Curr Alzheimer Res 2008;5:83–89. [DOI] [PubMed] [Google Scholar]
- 57. Periclou AP, Ventura D, Sherman T, Rao N, Abramowitz WT. Lack of pharmacokinetic or pharmacodynamic interaction between memantine and donepezil. Ann Pharmacother 2004;38:1389–1394. [DOI] [PubMed] [Google Scholar]
- 58. Howard R, McShane R, Lindesay J, et al. Donepezil and memantine for moderate‐to‐severe Alzheimer's disease. N Engl J Med 2012;366:893–903. [DOI] [PubMed] [Google Scholar]
- 59. Burke D, ACP Journal Club . Donepezil or memantine improved cognitive functioning in moderate‐to‐severe Alzheimer disease. Ann Intern Med 2012;156:JC6–JC10. [DOI] [PubMed] [Google Scholar]
- 60. Peskind ER, Potkin SG, Pomara N, et al. Memantine treatment in mild to moderate Alzheimer disease: a 24‐week randomized, controlled trial. Am J Geriatr Psychiatry 2006;14:704–715. [DOI] [PubMed] [Google Scholar]
- 61. Bakchine S, Loft H. Memantine treatment in patients with mild to moderate Alzheimer's disease: results of a randomised, double‐blind, placebo‐controlled 6‐month study. J Alzheimers Dis 2008;13:97–107. [DOI] [PubMed] [Google Scholar]
- 62. McShane R, Areosa Sastre A, Minakaran N. Memantine for dementia. Cochrane Database Syst Rev 2006;19:CD003154. [DOI] [PubMed] [Google Scholar]
- 63. Cummings JL, Schneider E, Tariot PN, Graham SM, Memantine MEM‐MD‐02 Study Group . Behavioral effects of memantine in Alzheimer disease patients receiving donepezil treatment. Neurology 2006;67:57–63. [DOI] [PubMed] [Google Scholar]
- 64. Martorana A, Semprini R, Koch G. Clinical Profile of Alzheimer's Disease Non‐Responder Patient, Advanced Understanding of Neurodegenerative Diseases, Raymond Chuen‐Chung Chang (Ed.), ISBN: 978‐953‐307‐529‐7, InTech. 2011.
- 65. Wallin AK, Blennow K, Zetterberg H, Londos E, Minthon L, Hansson O. CSF biomarkers predict a more malignant outcome in Alzheimer disease. Neurology 2010;74:1531–1537. [DOI] [PubMed] [Google Scholar]
- 66. Andreasen N, Minthon L, Davidsson P, et al. Evaluation of CSF‐tau and CSF‐Abeta42 as diagnostic markers for Alzheimer disease in clinical practice. Arch Neurol 2001;58:373–379. [DOI] [PubMed] [Google Scholar]
- 67. Van der Vlies AE, Verwey NA, Bouwman FH, et al. CSF biomarkers in relationship to cognitive profiles in Alzheimer disease. Neurology 2009;72:1056–1061. [DOI] [PubMed] [Google Scholar]
- 68. Blennow K, Hampel H. CSF markers for incipient Alzheimer's disease. Lancet Neurol 2003;2:605–613. [DOI] [PubMed] [Google Scholar]
- 69. Hampel H, Broich K. Enrichment of MCI and early Alzheimer's disease treatment trials using neurochemical and imaging candidate biomarkers. J Nutr Health Aging 2009;13:373–375. [DOI] [PubMed] [Google Scholar]
- 70. Vlachos GS, Paraskevas GP, Naoumis D, Kapaki E. Cerebrospinal fluid β‐amyloid 1–42 correlates with rate of progression in Alzheimer's disease. J Neural Transm 2012;119:799–804. [DOI] [PubMed] [Google Scholar]
- 71. Rabinovici GD, Jagust WJ. Amyloid imaging in aging and dementia: testing the amyloid hypothesis in vivo. Behav Neurol 2009;21:117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rabinovici GD, Furst AJ, Alkalay A, et al. Increased metabolic vulnerability in early‐onset Alzheimer's disease is not related to amyloid burden. Brain 2010;133:512–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jack CR Jr, Wiste HJ, Vemuri P, et al. Alzheimer's Disease Neuroimaging Initiative. Brain beta‐amyloid measures and magnetic resonance imaging atrophy both predict time‐to‐progression from mild cognitive impairment to Alzheimer's disease. Brain 2010;133:3336–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tang YP, Shimizu E, Dube GR, et al. Genetic enhancement of learning and memory in mice. Nature 1999;401:63–69. [DOI] [PubMed] [Google Scholar]
- 75. Magnusson KR, Nelson SE, Young AB. Age‐related changes in the protein expression of sub units of the NMDA receptor. Brain Res Mol Brain Res 2002;99:40–45. [DOI] [PubMed] [Google Scholar]
- 76. Magnusson KR, Scruggs B, Zhao X, Hammersmark R. Age‐related declines in a two‐day reference memory task are associated with changes in NMDA receptor subunits in mice. BMC Neurosci 2007;8:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhao X, Rosenke R, Kronemann D, et al. The effects of aging on N‐methyl‐d‐aspartate receptor subunits in the synaptic membrane and relationships to long‐term spatial memory. Neuroscience 2009;162:933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cao X, Cui Z, Feng R, et al. Maintenance of superior learning and memory function in NR2B transgenic mice during ageing. Eur J Neurosci 2007;25:1815–1822. [DOI] [PubMed] [Google Scholar]