

Summary
Aims
The present study evaluated the combined treatment effects of aerobic exercise and antioxidative stress on moderate‐stage Alzheimer's disease (AD).
Methods
Ten‐month‐old APP/PS1 mice were given antioxidative treatment with acetylcysteine, along with aerobic exercise for 6 weeks. Spatial learning and memory were tested using the Morris water maze, and β‐amyloid (Aβ) plaque deposits in the forebrain were quantified by Thioflavin‐S staining. Levels of soluble Aβ1‐42, β‐secretase enzyme, ү‐secretase enzyme, oxidative and antioxidant stress markers nitrotyrosine and peroxiredoxin‐1, glial markers glial fibrillary acidic protein and ionized calcium‐binding adaptor molecule 1, and synaptic protein synaptophysin in the hippocampus were all measured by western blotting and/or immunohistochemistry.
Results
APP/PS1 mice showed severe declines in spatial learning and memory compared with their wild‐type littermates, which were not attenuated by aerobic exercise combined with antioxidative treatment. The pathologic analysis revealed that Aβ deposition and production, oxidative stress, glial inflammation, and synaptic loss were not mitigated in the brain of exercised APP/PS1 mice, compared with the sedentary APP/PS1 animals.
Conclusion
This study reveals that a combined treatment of aerobic exercise plus antioxidative stress does not counteract pathophysiology in the moderate‐ or mid‐stages of AD.
Keywords: Aerobic exercise, Alzheimer's disease, Antioxidative treatment, APP/PS1 mice, Cognitive function, Pathology, β‐amyloid
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disorder, characterized by progressive declines in cognitive and memory function, with the presence of senile plaques and neurofibrillary tangles in the brain 1. The prevalence of AD increases with age, rising from just 3% between ages 65–74 to almost 50% in people over the age of 85 2. By 2030, because of increase in expected life span, the number of people age 65 and older is projected to reach 71 million, thus leading to enormous financial expenditures for families and societies 3. Unfortunately, there is no effective drug treatment for AD currently 4. Therefore, it is urgent to identify other strategies, such as nonpharmacological interventions or combined therapeutic approaches, to fight against this devastating disease.
There is extensive literature demonstrating that aerobic exercise can slow the pathophysiological progress of cardiovascular diseases and type II diabetes 5. Aerobic exercise has also been shown to improve brain function and memory via multiple mechanisms, including improvements of cerebrovascular function, neurogenesis, angiogenesis, synaptic plasticity, and neural growth factor secretion 6. Regular physical exercise can reduce or even prevent mild cognitive decline in older people 7 as well as in aged rodents 8 or in AD model mice 9. However, there are contradictory conclusions about the therapeutic efficacy of aerobic exercise against moderate‐to‐severe cognitive impairment 10. It is significant to determine the effects of aerobic exercise on moderate‐ or mid‐stage AD, because clinically a considerable portion of patients with AD already have moderate or even severe cognitive decline when first diagnosed 11.
It is well known that reactive oxygen species (ROS) are involved in the pathogenesis of AD 12. Accumulated ROS can cause oxidative damage to organelles, such as mitochondria, and macromolecules including proteins, lipids, and DNA 13. On the other hand, malfunctional mitochondria overproduce ROS, causing increased β‐and γ‐secretase activity and Aβ production 14. Excessive Aβ, in turn, impairs mitochondrial function and produces excessive ROS, thus forming a vicious cycle, subsequently accelerating AD pathology 15, 16. Despite the widespread view that aerobic exercise can improve the normal body's antioxidant capability and reduce levels of oxidative stress 17, aerobic exercise itself enhances metabolism, which increases ROS production 18. Antioxidant capacity is impaired in moderate stages of AD because of long‐term increases in baseline brain ROS levels 19. Based on this, excessive ROS generated in the course of aerobic exercise may lead to further brain oxidative damages, thus weakening the beneficial role of aerobic exercise in AD treatment. Therefore, in moderate‐ or mid‐stage AD, aerobic exercise and antioxidant combination therapy may be necessary and reasonable.
N‐acetyl‐L‐cysteine (NAC) is a mitochondria‐targeted antioxidant that can be converted into glutathione or combined with ROS directly, playing an antioxidant effect 20. A previous study has shown that NAC can reduce the levels of endogenous ROS and provide a protective effect on Aβ‐induced memory decline in mice 21. It also improves social isolation‐triggered onset of early AD‐related cognitive deficits in a transgenic mouse model 22. In the present study, to explore the combined treatment effects of aerobic exercise and antioxidative stress in moderate‐ or mid‐stage AD, 10‐month‐old APP/PS1 mice that show severe cognitive impairment received aerobic exercise plus NAC treatment for 6 weeks. Spatial learning and memory, oxidative stress, Aβ metabolism, glial inflammatory, and synaptic loss were analyzed and compared with sedentary controls. Our data indicate that aerobic exercise, combined with antioxidant treatment, fails to mitigate pathophysiological changes in moderate‐ or mid‐stage AD.
Materials and Methods
Animals and Experimental Design
Three‐month‐old APP695/PS1‐dE9 transgenic (APP/PS1) mice and their wild‐type (WT) littermates were obtained from the Model Animal Research Center of Nanjing University. Animals were housed at 20–25°C, 60% relative humidity, a 12‐h light/dark cycle (light turned on at 7 am), with food and water available ad libitum. APP/PS1 transgenic and WT mice were randomly separated into a treatment group and a sedentary control group when they were 10 months old. Treatment group mice were housed in cages equipped with a running wheel and introduced to running for 2 h per day (from 9:00 to 11:00 am). All mice were confirmed to voluntarily run, ruling out possible differences due to unwillingness to exercise. The exercising mice were also freely supplied with 1 mg/mL NAC (Sigma‐Aldrich, St. Louis, MO, USA) dissolved in water. Mice were housed two per cage, and their water intakes were measured daily. There was an average of 5 mL per mouse per day; thus, the mean NAC intake was calculated to be 5 mg, which has been used frequently in previous studies 23. The sedentary mice were reared in the standard cages supplied with normal water. After 6‐weeks of treatment, the mice were tested for spatial learning and memory capability. All protocols of animal experiments were conducted in accordance with international standards on animal welfare and the guidelines of the Institute for Laboratory Animal Research of Nanjing Medical University. All efforts were made to minimize animal suffering and to reduce the number of animals used.
Morris Water Maze
The Morris water maze (MWM) test was carried out to evaluate hippocampal related spatial learning and memory ability, as described previously 24. Each mouse was placed in the water, facing the pool wall, at one of four pseudorandomly chosen start positions. If the mouse failed to escape onto the platform within 60 s, it was guided to the platform and allowed to remain there for 10 s. Following each training exercise, the animals were placed in a clean cage and allowed a 30‐min resting interval before the next trial began. Each mouse received four training trials on each of 4 consecutive days. Latency to escape from the water maze was calculated for each trial. The swimming distance, speed and patterns were also analyzed. On the 5th day, the probing test was carried out by removing the platform, and allowing the mouse to swim freely for 60 s. The percentage of time spent in the target quadrant and the number of crossings where the platform had been previously located were calculated.
Brain Sample Preparation
For immunoblot analysis, mice were deeply anesthetized and decapitated. Brains were removed, and the hippocampus was dissected bilaterally and stored at −80°C. For pathological analysis, mice were deeply anesthetized, transcardially perfused with 0.9% saline, followed by 4% paraformaldehyde. Brains were dissected in the mid‐sagittal plane and postfixed overnight at 4°C. One, sagittally sliced, half‐brain was dehydrated in a series of graded ethanol solutions and embedded in paraffin, then serially cut into 5 μm coronal sections using a paraffin slicing machine (Leica RM2135, Nussloch, Germany). The other, half‐brain was dehydrated in a series of graded sucrose solutions, embedded in optimal cutting temperature compound (Fisher Scientific, Pittsburgh, PA, USA), then serially cut into 40 μm sagittal sections using a cryostat (Leica CM1900).
Quantitative Analysis of Aβ Plaque Load
Sagittal brain sections located at 0.48, 0.72, 0.96, 1.20, 1.44, and 1.68 mm lateral to the mid‐sagittal fissure were selected for histofluorescence staining using Thioflavin‐S (Sigma‐Aldrich) as previously described 25. The sections were observed under a digital microscope (Leica Microsystems), and the micrographs of the entire forebrain were photographed at 50× magnification. In each micrograph, boundaries of the following brain regions were demarcated using Adobe Photoshop 6.0 (Adobe Systems Inc., San Jose, CA, USA), according to The Mouse Brain in Stereotaxic Coordinates by Franklin and Paxinos 26: the primary motor cortex, secondary motor cortex, primary somatosensory cortex, visual cortex, hippocampus, and hypothalamus. Percent plaque load in each brain region was determined by standardized region of interest grayscale threshold analysis 27, using Image‐Pro Plus 6.0 Analysis System (Media Cybernetics Inc., San Francisco, CA, USA). Data were expressed as percent plaque load, corresponding to the total amount of area covered with plaques relative to the total brain area.
Immunohistochemistry
Immunohistochemical staining was performed as previously described 28. Briefly, after deparaffinization and rehydration, tissue sections were incubated with mouse monoclonal antibody to synaptophysin (SYP) (1:1000; Millipore, Billerica, MA, USA), mouse monoclonal antibody to glial fibrillary acidic protein (GFAP) (1:1000; Sigma‐Aldrich), or rabbit polyclonal antibody to ionized calcium‐binding adaptor molecule 1(Iba‐1) (1:500; Wako, Osaka, Japan) at 4°C overnight. After PBS washing, sections were incubated with horseradish peroxidase‐conjugated goat anti‐mouse or rabbit IgG (1:200) for 1 h at 37°C and visualized using DAB (Sigma‐Aldrich). The mean integrated optical density (MIOD = IOD/total area) was measured to assess the expression level of SYP, GFAP, and Iba‐1 in the entire hippocampus at 100× magnification using an Image‐Pro Plus 6.0 Analysis System (Media Cybernetics Inc.).
Western Blot
Hippocampal tissues were homogenized and centrifuged at 4°C, and 12000 rpm for 15 min. The samples were resolved on SDS–PAGE, transferred onto PVDF membranes using a Bio‐Rad miniprotein‐III wet transfer unit, then blocked with 5% skim milk dissolved in TBST (pH 7.5, 10 mm Tris–HCl, 150 mm NaCl, and 0.1% Tween 20) at room temperature for 1 h. Immunoblotting was probed with antibodies specific for Aβ1–42 (1:1000; Abcam, Cambridge, United Kingdom),β‐secretase (1:2000; Millipore), γ‐secretase (1:1000, Sigma‐Aldrich), peroxiredoxin‐1 (prdx‐1) (1:200; Santa Cruz BioTech, Santa Cruz, CA, USA), nitrotyrosine (NTS) (1:1000; Millipore), GFAP (1:1500, Sigma‐Aldrich), Iba‐1(1:1000; Wako), and SYP (1:1000; Millipore) at 4°C overnight. Horseradish peroxidase‐conjugated secondary antibodies (Vector Laboratories, Burlingame, CA, USA) were used, and bands were visualized using ECL plus detection system. β‐tubulin was used as an internal control for protein loading and transfer efficiency.
Statistics
Data are presented as means ± SEM. All statistical analyses were performed using SPSS software, version 16.0 (SPSS Inc., Chicago, IL, USA). Group differences in the MWM platform training data were analyzed using repeated‐measures two‐way ANOVA, with day of training as the within‐subject variable, genotype and treatment as the between subjects factors. Student's t‐test was applied for the analyses of Aβ plaque burden. Other experiments were analyzed by two‐way ANOVA with treatment and genotype as between‐subject factors. Significance was accepted at P < 0.05.
Results
Aerobic Exercise Combined with NAC Treatment does not Ameliorate Cognitive Deficits in APP/PS1 Mice
It is well known that APP/PS1 mice develop age‐related cognitive impairments, with occurrence of Aβ‐associated long‐term memory malfunction, which serves as a hallmark of moderate‐stage AD from approximately 6 months of age to about 12 months 29. Consistently, the present Morris water test results showed that 11.5‐month‐old APP/PS1 control mice had spatial learning impairment, as indicated by slower improvements in escape latency across consecutive trials, compared with WT controls (F 3,96 = 23.563, P < 0.001). These mice also displayed spatial memory deficits, exhibited by less time spent in the target quadrant (F 1,24 = 4.855, P = 0.039) and a decreased number of platform crossings in the probe test (F 1,24 = 4.319, P = 0.05). Aerobic exercise, combined with NAC treatment, did not improve learning capability of APP/PS1 mice or WT mice, as there was no significant treatment effect on escape latency during the 4‐day training (F 3,96 = 0.008, P = 0.929) (Figure 1A). Furthermore, aerobic exercise combined with NAC treatment did not affect time spent in the target quadrant (F 1,24 = 0.075, P = 0.787) or platform crossing numbers (F 1,24 = 0.002, P = 0.965) of APP/PS1 mice and WT mice (Figure 1D,E).
Figure 1.
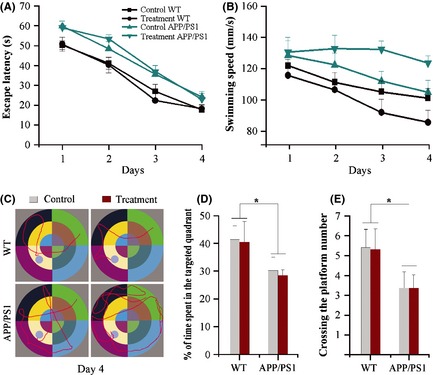
Spatial learning and memory analysis of 11.5‐month‐old wild‐type (WT) and APP/PS1 mice with or without aerobic exercise and N‐acetyl‐L‐cysteine (NAC) combination treatment from 10 months of age. (A and B) APP/PS1 mice showed longer escape latency and higher swimming speed than WT mice during 4 days of Morris water maze training. Aerobic exercise and NAC combination treatment did not affect escape latency of both WT mice and APP/PS1 mice. Swimming speeds decreased in the aerobic exercise and NAC‐treated WT mice, but increased in the aerobic exercise and NAC‐treated APP/PS1 mice. (C) Tracings of the typical swim patterns on the 4th day. WT mice reached the hidden platform with small loops. In contrast, APP/PS1 mice, especially those with aerobic exercise and NAC combination treatment, swam randomly with most time spent in the outer portion of the pool before finding the hidden platform. (D and E) WT mice displayed a higher percentage of time spent in each quadrant and number of crossing the platform than APP/PS1 mice in the probe test on the 5th day. *P < 0.05, between genotype comparisons. Data represent means ± SEM from 7 mice per group.
In addition, the treatment had no effect on swimming speed (F 3,96 = 1.742, P = 0.199). However, APP/PS1 mice exhibited increased swimming speed when compared with WT mice (F 3,96 = 5.373, P = 0.029). This result was attributed to a progressive decrease in swimming speed over the 4‐day training period of WT mice, but not APP/PS1 mice (F 3,96 = 7.790, P = 0.01) (Figure 1B). Swim tracing analysis showed that all WT mice used weaving or looping search patterns, with slow swimming speeds, to reach the hidden platform following 1–2 days of training. In contrast, even on day 4, a considerable proportion of APP/PS1 mice swam randomly within the entire pool, indicating that these mice did not remember the location of the hidden platform and found it due to chance (Figure 1C). Taken together, these behavioral data demonstrate that 6 weeks of aerobic exercise and antioxidant combination does not attenuate cognitive deficits in 10‐month‐old APP/PS1 mice.
Aerobic Exercise Combined with NAC Treatment does not Attenuate Aβ Deposition and Production in APP/PS1 Brain
Aβ accumulation plays a critical role in AD pathology processes 1. Previous studies have shown that specific neural activity affects Aβ production and deposition 30. Thus, it is necessary to identify whether exercise associated neural activity has specific effects on Aβ accumulation in motor‐related brain regions. Thioflavin‐S staining results show that aerobic exercise combined with NAC treatment did not attenuate Aβ plaque burden in motor‐related brain regions, such as the primary motor cortex, secondary motor cortex, and primary somatosensory cortex or other brain regions such as the visual cortex, hippocampus, and hypothalamus (Figure 2A,B). No Thioflavin‐S‐labeled Aβ plaques were found in brain tissues of WT mice.
Figure 2.
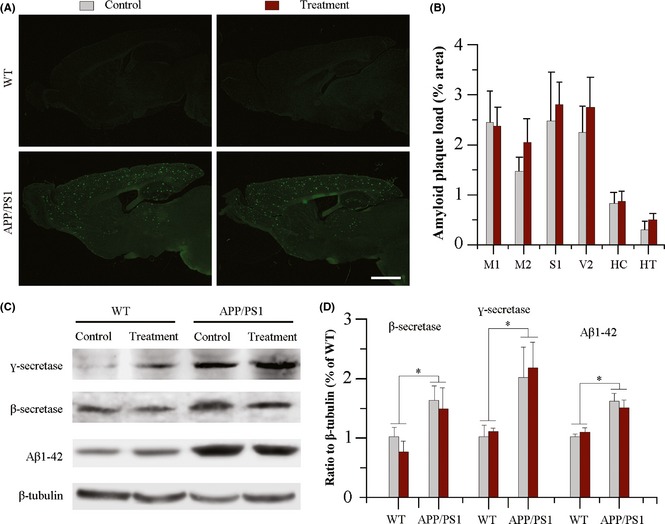
Analysis of Aβ deposition and production in the brain of 11.5‐month‐old wild‐type and APP/PS1 mice with or without aerobic exercise and N‐acetyl‐L‐cysteine (NAC) combination treatment from 10 months of age. (A) Both APP/PS1 mice, with or without aerobic exercise and NAC combination treatment, showed Aβ plaque accumulation in the forebrain, revealed by Thioflavin‐S florescence staining. Scale bar = 1.5 mm. (B) Aerobic exercise and NAC combination therapy did not affect Aβ plaque load in the primary motor cortex (M1), secondary motor cortex (M2), primary somatosensory cortex (S1), visual cortex (V2), hippocampus (HC), and hypothalamus (HT) of APP/PS1 mice. Data represent mean ± SEM from 4 mice per group. (C and D) Western blotting and densitometry quantification revealed that β‐secretase, ү‐secretase, and Aβ1–42 expression increased in the hippocampus of APP/PS1 mice, but was not significantly changed following aerobic exercise and NAC combination therapy. Data represent mean ± SEM from 3 mice per group performed in triplicate. *P < 0.05, between genotype comparisons.
We further examined whether aerobic exercise, combined with NAC treatment, affects Aβ generation‐related enzymes and soluble Aβ levels in the hippocampus using western blot. Compared with WT controls, APP/PS1 control mice had high levels of β‐secretase enzyme (F 1,8 = 19.032, P = 0.002), ү‐secretase enzyme (F 1,8 = 8.265, P = 0.021), and Aβ1–42 (F 1,8 = 26.386, P = 0.001), neither of which was rescued by the combination therapy (F β‐secretase 1,8 = 2.567, P = 0.148; F γ‐secretase 1,8 = 0.090, P = 0.772; F Aβ1–42 1,8 = 0.002, P = 0.968, respectively) (Figure 2C,D). Collectively, these results show that 6 weeks of aerobic exercise and antioxidant combination does not decrease Aβ deposition and production in 10‐month‐old APP/PS1 mice.
Aerobic Exercise Combined with NAC Treatment does not Significantly Decrease Oxidative Stress in APP/PS1 Brain
Aβ neurotoxicity and oxidative stress have synergical effects in promoting neurodegeneration 12, 16. To evaluate the influence of aerobic exercise combined with NAC treatment on brain redox status of APP/PS1 mice, the specific oxidative stress marker NTS 31 and the antioxidant enzyme Prdx‐1 32 were measured in the hippocampus of APP/PS1 and WT mice using western blot. APP/PS1 mice displayed a prominent increase in NTS protein level compared with WT controls (F 1,8 = 22.091, P = 0.002). The treatment decreased NTS expression in APP/PS1 mice, but did not reach a significant level (F 1,8 = 0.086, P = 0.777) (Figure 3A,B). In agreement with a recent study revealing that a compensatory overexpression of Prdx‐1 plays a protective role in cultured neurons treated with Aβ 33, APP/PS1 mice also expressed high level of Prdx‐1 protein (F 1,8 = 9.887, P = 0.016) (Figure 3A,B). However, aerobic exercise plus NAC treatment did not significantly affect Prdx‐1 expression (F 1,8 = 0.108, P = 0.752).
Figure 3.

Analysis of oxidative stress in the hippocampus of 11.5‐month‐old wild‐type and APP/PS1 mice with or without aerobic exercise and N‐acetyl‐L‐cysteine (NAC) combination treatment from 10 months of age. (A and B) Western blotting and densitometry quantification revealed that the oxidative stress marker nitrotyrosine and the antioxidative marker Prdx‐1 increased in the hippocampus of APP/PS1 mice. Levels were not significantly changed after aerobic exercise and NAC combination therapy. Data represent mean ± SEM from 3 mice per group performed in triplicate. *P < 0.05, between genotype comparisons.
Aerobic Exercise Combined with NAC Treatment does not Prevent Synaptic Loss in APP/PS1 Brain
Synaptophysin, a representative presynaptic membrane protein, is primarily present within vesicles, and its expression level can be used to evaluate synapse loss 34. Consistent with the previous literature 35, 36, SYP expression dramatically decreases in the hippocampus of APP/PS1 mice, as revealed by immunohistochemistry (F 1,12 = 5.058, P = 0.042) and western blot quantitation (F 1,8 = 6.308, P = 0.031). Aerobic exercise combined with NAC treatment only resulted in a nonsignificant increase in SYP in both APP/PS1 mice and WT littermates (F IHC 1,12 = 0.201, P = 0.661; F Western 1,8 = 2.862, P = 0.122) (Figure 4).
Figure 4.
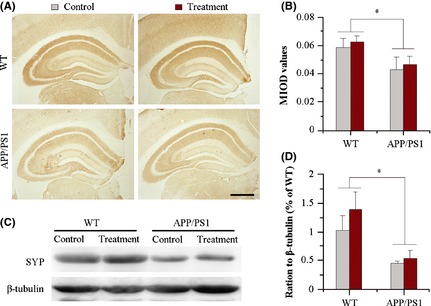
Analysis of synaptophysin (SYP) expression in the hippocampus of 11.5‐month‐old wild‐type (WT) and APP/PS1 mice with or without aerobic exercise and N‐acetyl‐L‐cysteine (NAC) combination treatment from 10 months of age. (A and B) Immunohistochemistry and mean integrated optical density (MIOD) analysis demonstrated that SYP immunoreactivity was decreased in APP/PS1 mice, and not affected by aerobic exercise and NAC combination. Scale bar = 500 μm. Data represent mean ± SEM from 4 mice per group. (C and D) Consistently, western blotting and densitometry quantification showed that the combination therapy did not change decreased SYP expression in the hippocampus of APP/PS1 mice, compared with WT mice. Data represent mean ± SEM from 3 mice per group performed in triplicate. *P < 0.05, between genotype comparisons.
Aerobic Exercise Combined with NAC Treatment does not Ameliorate Reactive Gliosis in APP/PS1 Brain
Reactive astrocytes and microglia are hallmarks of AD pathology 37. Aerobic exercise has been shown to attenuate glial inflammatory response in APP/PS1 mice 38. We examined whether this change also occurred in adult APP/PS1 mice treated with a combination of aerobic exercise and NAC. As shown in Figure 5A, GFAP‐positive astrocytes and Iba‐1‐positive microglia were dramatically activated in the hippocampus of treated and control APP/PS1 mice compared with their WT littermates. Aerobic exercise combined with NAC treatment caused only a negligible decrease in GFAP and Ibal‐1 expression in APP/PS1 mice in SYP in both APP/PS1 mice and WT littermates, as revealed by quantitation of immunohistochemistry (F GFAP 1,12 = 1.943, P = 0.187; F Iba‐1 1,12 = 0.245, P = 0.630) and western blotting data (F GFAP 1,8 = 0.405, P = 0.542; F Iba‐1 1,8 = 0.390, P = 0.550) (Figure 5B–D).
Figure 5.

Analysis of glial inflammation in the hippocampus of 11.5‐month‐old wild‐type (WT) and APP/PS1 mice with or without aerobic exercise and N‐acetyl‐L‐cysteine (NAC) combination treatment from 10 months of age. (A) The immunohistochemistry revealed that glial fibrillary acidic protein (GFAP)‐positive astrocytes (upper two panels) and Iba‐1‐positive microglia (lower two panels) were prominently activated in the hippocampus of APP/PS1 mice with or without the combination therapy, compared with WT littermates. Scale bar = 500 μm. (B) The quantification analysis revealed higher values of mean integrated optical density (MIOD) of GFAP and Iba‐1 in APP/PS1 mice, which was not affected by aerobic exercise and NAC combination. Data represent mean ± SEM from 4 mice per group. (C and D) Consistently, western blotting and densitometry quantification showed that the combination treatment did not affect upregulated expression of GFAP and Ibal‐1 in the hippocampus of APP/PS1 mice, compared with WT mice. Data represent mean ± SEM from 4 mice per group performed in triplicate. *P < 0.05, between genotype comparisons.
Discussion
Alzheimer's disease is a devastating neurodegenerative disease, but no promising treatment strategies are currently available 1, 2. There are increasing evidences that exercise has beneficial effects on cognition and brain function; however, its therapeutic efficacy on AD needs to be defined 10. The objective of the current study was to investigate whether aerobic exercise, combined with antioxidative treatment, ameliorates or stabilizes moderate‐ or mid‐stage Alzheimer‐like pathophysiology in APP/PS1 mice. The results show that spatial cognitive dysfunction, Aβ deposit, oxidative stress, glial inflammatory, and synaptic loss are not reduced in 10‐month‐old APP/PS1 mice receiving aerobic exercise and NAC combination therapy for 6 weeks. These outcomes are in agreement with epidemiological studies demonstrating that cognitive stimulation, physical exercise, and various other nonpharmacological interventions, in combination with each other, do not protect against permanent, irreversible end‐point phenotypic AD 11.
In contrast to the present results, previous studies have shown that aerobic exercise before the onset of AD‐like neuropathology can slow AD progression in several animal models, including TgCRND8 mice 9, APP mice 39, APP/PS1 mice 9, 40, and 3xTg‐AD mice 41. Interestingly, Herring et al. 42 have recently reported that short‐term voluntary exercise during pregnancy is able to mitigate AD‐related pathology in TgCRND8 offspring. Consistent with basic researches, several epidemiological studies have suggested that physical exercise may slow the onset of age‐related cognitive decline and improve cognition in older adults with mild cognitive impairment 43, 44. A recent clinical study has revealed that physical activities can reduce the rate of cognitive decline in early stage patients with AD over a 1‐year period 45. Taken together, the results from experimental, epidemiological, and clinical studies highlight that aerobic exercise has beneficial effects on the prevention of early stage AD onset and progression, but does not counteract moderate‐ or late‐ stage AD pathology.
These findings support the view that the therapeutic effect of aerobic exercise on AD is critically dependent on the timing of the treatment 46. Thus, aerobic exercise, if initiated at a time when brain function is healthy or relatively healthy, can be beneficial in preventing or delaying the onset of AD. In contrast, if neurological health is compromised, aerobic exercise does not protect against neurodegeneration. Previous research has suggested that aerobic exercise improves cognitive functions via multiple mechanisms, including improving neurogenesis 47, blood flow 48, neurotrophic factor production 49, and Aβ clearance 9. These neuroprotective mechanisms seem to mainly rely on the integrity of neurovascular coupling, in that energy substrate and oxygen supply meets neuronal metabolic demand in response to various stimuli or physical activities 50. Altered neurovascular coupling 51 and an impaired blood‐brain barrier 52 are evident even before the onset of AD. Based on this, dysfunction of neurogliovascular units may be primarily responsible for hindering the neuroprotective effects of aerobic exercise in moderate‐ or late‐stage AD 53. However, this presumption warrants further experimental evidence.
Evidences indicate that oxidative stress plays a critical role in the onset and development of AD 54. A vicious cycle among accumulated ROS, mitochondrial oxidative damage, and Aβ production has been seen as a central mechanism in AD progression 12. Moreover, impaired antioxidant capacity occurs in APP/PS1 mice 55, which may lead to excessive ROS production that cannot be adequately cleared during high metabolic exercise, exacerbating mitochondrial dysfunction, and Aβ overproduction. Based on this, exercising APP/PS1 mice were given NAC supplementation. The results show a slight improvement in the oxidative situation in exercising APP/PS1 mice receiving NAC, as revealed by insignificant decreases in NTS and Prdx‐1 expression in the hippocampus, compared with sedentary APP/PS1 controls. These data suggest that NAC supplementation encounters excessive ROS generated during aerobic exercise. However, the levels of oxidative stress are still higher in these mice than their WT littermates, indicating that NAC alone is not enough to decrease oxidative stress caused by the long‐term Aβ aggregation, glial cell activation, and mitochondrial dysfunction.
The present results are contrary to previous studies showing that NAC can ameliorate cognitive dysfunction in adult mice receiving Aβ injection, and young APP/PS1 mice suffering from social isolation 21, 22. However, the results are consistent with others stating that vitamin E supplementation in young, rather than aged, Tg2576 mice brings about a significant reduction in Aβ levels and Aβ deposition 56. These results provide insight into the hypothesis that oxidative stress is an early event in AD pathogenesis, and may be suppressed by antioxidants during the early stages of the disease.
Besides the timing of the treatment, the dose of antioxidants may also affect the effective treatment for AD. For example, Quinn et al. 57 reported that administration of a relatively low melatonin dose (0.08 mg/day) to 14‐month‐old Tg2576 mice for 4 months results in no effects on Aβ burden or oxidative damage in the brain. In contrast, Dragicevic et al. 58 showed that a 1‐month treatment of 18–20‐month‐old mice with high dose of melatonin (0.5 mg/day) reverses mitochondrial dysfunction caused by Aβ peptides. Based on this, it cannot exclude that a higher intensity or a longer time of aerobic exercise with a higher dose of NAC intake or other antioxidants such as melatonin 58 and coenzyme Q10 59 may have therapeutic potential in the treatment of mid‐stage or even late‐stage AD. The possibility remains to be determined in future studies.
In summary, the results suggest that aerobic exercise combined with antioxidant therapy has no therapeutic benefit on the mid‐stage Alzheimer‐like pathophysiology of APP/PS1 mice, although its positive effect has been found at the onset or in the early stage of this familial AD model. These results suggest that the beneficial effects of aerobic exercise on familial AD are gradually reduced and eventually lost, as the disease progresses. In this regard, preventive or early intervention of aerobic exercise is necessary for decreasing the risk or slowing the progression of AD.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 30971020 and No. 81271210), Jiangsu Province Xinwei Project Key Discipline of Rehabilitation Medicine, PAPD Foundation of Jiangsu Higher Education Institutions, and Qing Lan Project.
The first two authors contributed equally to this work.
References
- 1. Cummings JL. Alzheimer's disease. N Engl J Med 2004;351:56–67. [DOI] [PubMed] [Google Scholar]
- 2. Ferrer I. Defining Alzheimer as a common age‐related neurodegenerative process not inevitably leading to dementia. Prog Neurobiol 2012;97:38–51. [DOI] [PubMed] [Google Scholar]
- 3. Castellani RJ, Rolston RK, Smith MA. Alzheimer disease. Dis Mon 2010;56:484–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mullane K, Williams M. Alzheimer's therapeutics: Continued clinical failures question the validity of the amyloid hypothesis‐but what lies beyond? Biochem Pharmacol 2013;85:289–305. [DOI] [PubMed] [Google Scholar]
- 5. Bassuk SS, Manson JE. Epidemiological evidence for the role of physical activity in reducing risk of type 2 diabetes and cardiovascular disease. J Appl Physiol 2005;99:1193–1204. [DOI] [PubMed] [Google Scholar]
- 6. van Praag H. Exercise and the brain: Something to chew on. Trends Neurosci 2009;32:283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liang KY, Mintun MA, Fagan AM, et al. Exercise and Alzheimer's disease biomarkers in cognitively normal older adults. Ann Neurol 2010;68:311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Albeck DS, Sano K, Prewitt GE, Dalton L. Mild forced treadmill exercise enhances spatial learning in the aged rat. Behav Brain Res 2006;168:345–348. [DOI] [PubMed] [Google Scholar]
- 9. Adlard PA, Perreau VM, Pop V, Cotman CW. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer's disease. J Neurosci 2005;25:4217–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sofi F, Valecchi D, Bacci D, et al. Physical activity and risk of cognitive decline: A meta‐analysis of prospective studies. J Intern Med 2011;269:107–117. [DOI] [PubMed] [Google Scholar]
- 11. Korczyn AD. AD: Are we intervening too late? J Neural Transm 2011;118:1359. [DOI] [PubMed] [Google Scholar]
- 12. Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA. Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol 2006;65:631–641. [DOI] [PubMed] [Google Scholar]
- 13. Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008;60:748–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shen C, Chen Y, Liu H, et al. Hydrogen peroxide promotes Abeta production through JNK‐dependent activation of gamma‐secretase. J Biol Chem 2008;283:17721–17730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reddy PH. Amyloid precursor protein‐mediated free radicals and oxidative damage: Implications for the development and progression of Alzheimer's disease. J Neurochem 2006;96:1–13. [DOI] [PubMed] [Google Scholar]
- 16. Tamagno E, Guglielmotto M, Aragno M, et al. Oxidative stress activates a positive feedback between the gamma‐ and beta‐secretase cleavages of the beta‐amyloid precursor protein. J Neurochem 2008;104:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Corbi G, Conti V, Russomanno G, et al. Is physical activity able to modify oxidative damage in cardiovascular aging? Oxid Med Cell Longev 2012;2012:728547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Powers SK, Nelson WB, Hudson MB. Exercise‐induced oxidative stress in humans: Cause and consequences. Free Radic Biol Med 2011;51:942–950. [DOI] [PubMed] [Google Scholar]
- 19. Grundman M, Grundman M, Delaney P. Antioxidant strategies for Alzheimer's disease. Proc Nutr Soc 2002;61:191–202. [DOI] [PubMed] [Google Scholar]
- 20. Pocernich CB, Butterfield DA. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim Biophys Acta 2012;1822:625–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fu AL, Dong ZH, Sun MJ. Protective effect of N‐acetyl‐L‐cysteine on amyloid beta‐peptide‐induced learning and memory deficits in mice. Brain Res 2006;1109:201–206. [DOI] [PubMed] [Google Scholar]
- 22. Hsiao YH, Kuo JR, Chen SH, Gean PW. Amelioration of social isolation‐triggered onset of early Alzheimer's disease‐related cognitive deficit by N‐acetylcysteine in a transgenic mouse model. Neurobiol Dis 2012;45:1111–1120. [DOI] [PubMed] [Google Scholar]
- 23. Liu J, Cao L, Chen J, et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 2009;459:387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li L, Ding J, Marshall C, Gao J, Hu G, Xiao M. Pretraining affects Morris water maze performance with different patterns between control and ovariectomized plus D‐galactose‐injected mice. Behav Brain Res 2011;217:244–247. [DOI] [PubMed] [Google Scholar]
- 25. Styren SD, Hamilton RL, Styren GC, Klunk WE. X‐34, a fluorescent derivative of Congo red: A novel histochemical stain for Alzheimer's disease pathology. J Histochem Cytochem 2000;48:1223–1232. [DOI] [PubMed] [Google Scholar]
- 26. Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates, 2nd edn Amsterdam: Elsevier, 2008. [Google Scholar]
- 27. Fishman CE, Cummins DJ, Bales KR, et al. Statistical aspects of quantitative image analysis of beta‐amyloid in the APP(V717F) transgenic mouse model of Alzheimer's disease. J Neurosci Methods 2001;108:145–152. [DOI] [PubMed] [Google Scholar]
- 28. Hua X, Lei M, Zhang Y, et al. Long‐term D‐galactose injection combined with ovariectomy serves as a new rodent model for Alzheimer's disease. Life Sci 2007;80:1897–1905. [DOI] [PubMed] [Google Scholar]
- 29. Trinchese F, Liu S, Battaglia F, Walter S, Mathews PM, Arancio O. Progressive age‐related development of Alzheimer‐like pathology in APP/PS1 mice. Ann Neurol 2004;55:801–814. [DOI] [PubMed] [Google Scholar]
- 30. Bero AW, Yan P, Roh JH, et al. Neuronal activity regulates the regional vulnerability to amyloid‐β deposition. Nat Neurosci 2011;14:750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Butterfield DA, Reed T, Sultana R. Roles of 3‐nitrotyrosine‐ and 4‐hydroxynonenal‐modified brain proteins in the progression and pathogenesis of Alzheimer's disease. Free Radic Res 2011;45:59–72. [DOI] [PubMed] [Google Scholar]
- 32. Bell KF, Hardingham GE. CNS peroxiredoxins and their regulation in health and disease. Antioxid Redox Signal 2011;14:1467–1477. [DOI] [PubMed] [Google Scholar]
- 33. Cimini A, Gentile R, Angelucci F, et al. Neuroprotective effects of PrxI over‐expression in an in vitro human Alzheimer's disease model. J Cell Biochem 2013;114:708–715. [DOI] [PubMed] [Google Scholar]
- 34. Clare R, King VG, Wirenfeldt M, Vinters HV. Synapse loss in dementias. J Neurosci Res 2010;88:2083–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fonseca MI, Zhou J, Botto M, Tenner AJ. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer's disease. J Neurosci 2004;24:6457–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Malthankar‐Phatak G, Poplawski S, Toraskar N, Siman R. Combination therapy prevents amyloid‐dependent and ‐independent structural changes. Neurobiol Aging 2012;33:1273–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schwab C, McGeer PL. Inflammatory aspects of Alzheimer disease and other neurodegenerative disorders. J Alzheimers Dis 2008;13:359–369. [DOI] [PubMed] [Google Scholar]
- 38. Ke HC, Huang HJ, Liang KC, Hsieh‐Li HM. Selective improvement of cognitive function in adult and aged APP/PS1 transgenic mice by continuous non‐shock treadmill exercise. Brain Res 2011;1403:1–11. [DOI] [PubMed] [Google Scholar]
- 39. Maesako M, Uemura K, Kubota M, et al. Exercise is more effective than diet control in preventing high fat diet‐induced β‐amyloid deposition and memory deficit in amyloid precursor protein transgenic mice. J Biol Chem 2012;287:23024–23033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu HL, Zhao G, Cai K, Zhao HH, Shi LD. Treadmill exercise prevents decline in spatial learning and memory in APP/PS1 transgenic mice through improvement of hippocampal long‐term potentiation. Behav Brain Res 2011;218:308–314. [DOI] [PubMed] [Google Scholar]
- 41. García‐Mesa Y, López‐Ramos JC, Giménez‐Llort L, et al. Physical exercise protects against Alzheimer's disease in 3xTg‐AD mice. J Alzheimers Dis 2011;24:421–454. [DOI] [PubMed] [Google Scholar]
- 42. Herring A, Donath A, Yarmolenko M, et al. Exercise during pregnancy mitigates Alzheimer‐like pathology in mouse offspring. FASEB J 2012;26:117–128. [DOI] [PubMed] [Google Scholar]
- 43. Lautenschlager NT, Cox KL, Flicker L, et al. Effect of physical activity on cognitive function in older adults at risk for Alzheimer disease: A randomized trial. JAMA 2008;300:1027–1037. [DOI] [PubMed] [Google Scholar]
- 44. Baker LD, Bayer‐Carter JL, Skinner J, et al. High‐intensity physical activity modulates diet effects on cerebrospinal amyloid‐β levels in normal aging and mild cognitive impairment. J Alzheimers Dis 2012;28:137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Winchester J, Dick MB, Gillen D, et al. Walking stabilizes cognitive functioning in Alzheimer's disease (AD) across one year. Arch Gerontol Geriatr 2013;56:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Richter H, Ambrée O, Lewejohann L, et al. Wheel‐running in a transgenic mouse model of Alzheimer's disease: Protection or symptom? Behav Brain Res 2008;190:74–84. [DOI] [PubMed] [Google Scholar]
- 47. van Praag H, Kempermann G, Gage FH. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci 1999;2:266–270. [DOI] [PubMed] [Google Scholar]
- 48. Querido JS, Sheel AW. Regulation of cerebral blood flow during exercise. Sports Med 2007;37:765–782. [DOI] [PubMed] [Google Scholar]
- 49. Cotman CW, Berchtold NC, Christie LA. Exercise builds brain health: Key roles of growth factor cascades and inflammation. Trends Neurosci 2007;30:464–472. [DOI] [PubMed] [Google Scholar]
- 50. Willie CK, Cowan EC, Ainslie PN, et al. Neurovascular coupling and distribution of cerebral blood flow during exercise. J Neurosci Methods 2011;198:270–273. [DOI] [PubMed] [Google Scholar]
- 51. Stanimirovic DB, Friedman A. Pathophysiology of the neurovascular unit: Disease cause or consequence? J Cereb Blood Flow Metab 2012;32:1207–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jeynes B, Provias J. The case for blood‐brain barrier dysfunction in the pathogenesis of Alzheimer's disease. J Neurosci Res 2011;89:22–28. [DOI] [PubMed] [Google Scholar]
- 53. Nicolakakis N, Hamel E. Neurovascular function in Alzheimer's disease patients and experimental models. J Cereb Blood Flow Metab 2011;31:1354–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nunomura A, Perry G, Aliev G, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 2001;60:759–767. [DOI] [PubMed] [Google Scholar]
- 55. Lovell MA, Xiong S, Lyubartseva G, Markesbery WR. Organoselenium (Sel‐Plex diet) decreases amyloid burden and RNA and DNA oxidative damage in APP/PS1 mice. Free Radic Biol Med 2009;46:1527–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sung S, Yao Y, Uryu K, et al. Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer's disease. FASEB J 2004;18:323–325. [DOI] [PubMed] [Google Scholar]
- 57. Quinn J, Kulhanek D, Nowlin J, et al. Chronic melatonin therapy fails to alter amyloid burden or oxidative damage in old Tg2576 mice: Implications for clinical trials. Brain Res 2005;1037:209–213. [DOI] [PubMed] [Google Scholar]
- 58. Dragicevic N, Copes N, O'Neal‐Moffitt G, et al. Melatonin treatment restores mitochondrial function in Alzheimer's mice: A mitochondrial protective role of melatonin membrane receptor signaling. J Pineal Res 2011;51:75–86. [DOI] [PubMed] [Google Scholar]
- 59. Yang X, Dai G, Li G, Yang ES. Coenzyme Q10 reduces beta‐amyloid plaque in an APP/PS1 transgenic mouse model of Alzheimer's disease. J Mol Neurosci 2010;41:110–113. [DOI] [PubMed] [Google Scholar]