

Summary
Aims
Oxidative stress is frequently implicated in the pathology of neurodegenerative diseases. This study aimed to investigate the effects and their underlying mechanism(s) of edaravone upon hydrogen peroxide (H2O2)–induced oxidative stress and apoptosis in HT22 cells, a murine hippocampal neuronal model.
Methods
HT22 cells were treated with H2O2 in the presence of various concentrations of edaravone or in its absence. A CCK‐8 assay, Hoechst 33342 staining, and flow cytometry were used to detect cytotoxicity and apoptosis. In addition, the levels of reactive oxygen species (ROS) and the expression of Bcl‐2, Bax, p‐ERK 1/2, p‐JNK, and p‐P38 proteins in HT22 cells were examined.
Results
Exogenous H2O2 decreased cell viability in a concentration‐dependent manner and was associated with increased apoptosis and ROS production. Moreover, H2O2 significantly activated and upregulated the expression of p‐ERK 1/2, p‐JNK, and p‐P38, while edaravon protected HT22 cells against H2O2‐induced injury by inhibiting the production of ROS and activating the MAPK signaling pathway.
Conclusions
Our results provide the first evidence that edaravone can protect H2O2‐induced cell injury in HT22 neurons via its antioxidant action. These findings suggest that edaravone may be useful in the treatment of neurodegenerative disorders in which oxidative stress has been principally implicated.
Keywords: Edaravone, HT22 cells, Hydrogen peroxide, Neurodegenerative diseases, Neuroprotective effect, Oxidative stress
Introduction
The production of reactive oxygen species (ROS) and their detoxification are normal physiological processes. Nevertheless, an imbalance between ROS production and ROS removal may lead to oxidative stress. ROS are important in neuronal signaling and physiology at low levels. However, they can lead to neuronal dysfunction and cell death at higher levels 1. Several components of ROS can cause damage to cardinal cellular components, such as lipids, proteins, and DNA, initiating subsequent cell death via necrosis or apoptosis 2. Thus, ROS can contribute to neuronal toxicity and have been implicated in both acute injury and chronic neuropathological conditions 3. Growing evidence from experimental models and human brain studies suggests that oxidative stress may play an important role in the common pathway for neurotoxicity in a variety of neurodegenerative diseases, including Alzheimer's disease (AD) 4, Parkinson's disease (PD) 5, Huntington's disease (HD) 6, and amyotrophic lateral sclerosis (ALS) 7. Therefore, the search for neuroprotective drugs that counteract ROS‐induced neuronal death has attracted growing interest.
Hydrogen peroxide (H2O2), one of the major agents generated by oxidative stress, is produced by nearly every stage of the oxidative cycle. Neural cells that are exposed to H2O2 may undergo an apoptotic‐like delayed death and necrosis. Substantial evidence has indicated etiological links between the generation of H2O2 and neurodegenerative diseases 8. Indeed, the H2O2 molecule has been considered as a therapeutic target for the treatment of oxidative stress associated with these diseases. Nevertheless, the effectiveness of drugs that target this component of the disease pathology remains to be determined.
Edaravone (EDA, 3‐methyl‐1‐phenyl‐2‐pyrazolin‐5‐one) has high liposolubility and permeability through the blood–brain barrier and is currently applied as a free radical scavenger in the treatment of acute cerebral vascular diseases 9, 10. Some studies have demonstrated that EDA efficiently scavenges oxygen free radicals by providing a hydrogen atom 11. Oxidative stress has been implicated as a pathogenetic mechanism for neurodegenerative diseases; thus, we suspect that EDA may be a promising therapeutic agent for oxidative stress–induced neurodegenerative disease. However, only a few reports in the literature have previously examined this possibility. To further understand the neuroprotective effects of EDA and develop its pharmaceutical application for the comprehensive management of neurodegenerative disorders, we used immortalized mouse hippocampal neurons (HT22 cells), a line possessing functional cholinergic properties and other characteristics similar to primary hippocampal neurons after differentiation 12, 13, as a cell model to investigate whether EDA attenuates H2O2‐induced neurotoxicity. Our aims were to 1) evaluate the neurocytotoxicity of H2O2 upon HT22 cells and 2) observe the effects of EDA on oxidative stress–induced cell death and elucidate the underlying mechanism(s) of these effects.
Materials and Methods
Materials
H2O2 (30%) and EDA were purchased from Merck (Darmstadt, Hessen, Germany). Fetal bovine serum (FBS) was obtained from Atlanta Biologicals (Norcross, GA, USA). A cell counting kit‐8 (CCK‐8) was acquired from Dojin Kagaku (Kumamoto, Kyushu, Japan). A ROS detection kit was purchased from the Beyotime Institute of Biotechnology (Guangzhou, China). Hoechst 33342 was procured from Invitrogen/Life Technologies (Carlsbad, CA, USA). The following primary antibodies were used for Western blotting: antiphospho‐Erk1/2 (Thr202/Thr204), antiphospho‐P38 (Thr180/Thr182), antiphospho‐JNK (Thr183/Thr185), anti‐Erk1/2, anti‐JNK, anti‐P38, and anti‐Bcl‐2, anti‐Bax; all antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). All other routine cell culture supplies and reagents were purchased from Sigma, Invitrogen, and Fisher.
HT22 Cell Culture, Differentiation, and Treatment
HT22 cells were cultured in DMEM media supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin as previously described 12. The method used for differentiation of HT22 cells has been previously described in detail 13. When the cell density reached 70–80%, the cells were treated with the indicated concentration of H2O2 for 24 h in the absence or presence of EDA (final concentrations ranged from 10 to 100 μM).
Cytotoxicity Assays
Cytotoxicity/cell viability in response to the different treatments was evaluated using a CCK‐8 assay. Briefly, after each treatment, 10 μL/well of WST‐8 reagent was added and the HT22 cells were incubated for 2 h at 37°C and 5% CO2. The absorbance of the samples was measured at 450 nm using a microplate reader with a background control as the blank. The cell survival ratio was expressed as the percentage of the control. The morphological changes were also monitored under an inverted phase contrast microscope with a digital camera.
Flow Cytometric Analysis Using annexin V and Propidium Iodide
The percentage of apoptotic cells in culture was evaluated by flow cytometric analysis using annexin V–FITC and propidium iodide (PI) fluorescence as previously described 14. Briefly, HT22 cells were seeded in six‐well plates at 2 × 105 cells/well and treated as described above. Cells were then washed twice with phosphate‐buffered saline (PBS) and stained with annexin V–FITC and PI in binding buffer (10 mM Hepes, 140 mM NaCl, and 2.5 mM CaCl2). Ten thousand events were collected for each sample. The stained cells were analyzed using Cell‐Quent software in the FL1‐H and FL2‐H channels.
In parallel with the flow cytometric analysis, Hoechst 33342 staining in living cells was performed to discriminate apoptotic cell death induced by the treatments. Highly condensed, marginalized, or fragmented chromatin in apoptotic cells is stained bright blue with Hoechst 33342 13.
Detection of Intracellular ROS Generation
The intracellular ROS were detected by an oxidation‐sensitive fluorescent probe (DCFH‐DA). After treatment, the cells were washed twice in PBS, collected and adjusted to 1 × 107/ml, and incubated with DCFH‐DA at 37°C for 20 min. The DCFH‐DA was intracellularly deacetylated by a nonspecific esterase, which was further oxidized by ROS to produce the fluorescent compound 2,7‐dichlorofluorescein (DCF). The fluorescent signal intensity of DCF was detected by FACScan flow cytometry at an excitation wavelength of 488 nm and an emission wavelength of 535 nm.
Western Blot Analysis
After treatment, the HT22 cells were lysed with an appropriate amount of boiling, denaturing lysate buffer (1% SDS, 1 mM sodium orthovanadate, 10 mM Tris‐Cl, pH 7.4) supplemented with a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). The total protein quantification and Western blot procedures were performed routinely as previously described 15. The following dilution rates for the different primary antibodies, all obtained from Cell Signaling Technology, were used: Bcl‐2, 1:1000; Bax, 1:1000; p‐JNK, 1:1000; p‐P38, 1:1000; p‐ERK1/2, 1:1000; JNK, 1:1000; P38, 1:500; ERK1/2, 1:2000 and GAPDH, 1:500.
Statistical Analysis
All of the statistical analyses were performed using the SPSS 11.0 software (SPSS Inc., Chicago, IL, USA). All of the data are expressed as the mean ± SE Significant differences were analyzed with Student's t‐test and one‐way analysis of variance (ANOVA) with the significance level set at P < 0.05.
Results
Morphological Changes after H2O2 or/and EDA Treatment
Under an inverted microscope, control HT22 cells appeared to have complete packing membranes, a normal shape and round nuclei (Figure 1A). In contrast, incomplete cellular membranes, cellular swelling, vacuole degeneration as well as pyknosis of the chromatin were observed in the H2O2 treatment groups. The extent of cell damage was exacerbated by the increasing level of H2O2. Cells treated with 1000 μM H2O2 were shrunken, and no cells with normal morphology could be seen (Figure 1B–D). The number of viable cells in the EDA‐treated groups was increased compared to the H2O2 group. Cellular swelling, pyknosis of the chromatin, and vacuole degeneration in the EDA cotreatment groups were observed at a lower frequency than in the H2O2 group. These changes were concentration dependent within the range of 10–60 μM EDA (Figure 1E,F).
Figure 1.
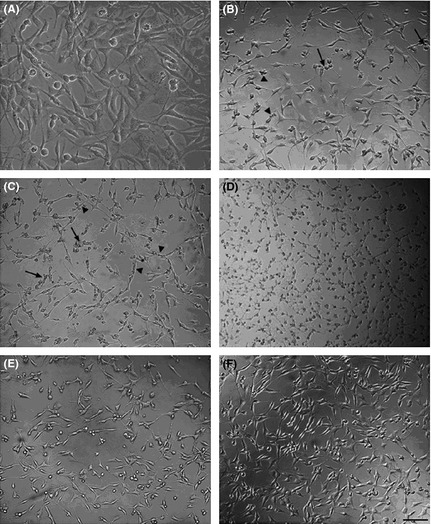
The effect of H2O2 in the absence and presence of EDA on the morphology of HT22 cells. (A) Normal morphologic features of HT22 cells were present in the control group (B–D). Cell morphology changed after treating with different concentrations of H2O2 (250 μM, 500 μM, 1000 μM). The characteristic features of an incomplete cellular membrane, cellular swelling (arrows), and vacuole degeneration were present (black triangle) (E, F). Cotreatment with EDA (30 μM, 60 μM) ameliorated H2O2‐induced HT22 cell damage. Scale bar = 50 μm.
Cell Viability after H2O2 or/and EDA Treatment
When the HT22 cells were treated with increasing doses of H2O2 for 24 h, as shown in Figure 2A, survival decreased with increasing concentrations of H2O2: from 84.6 ± 4.1% at 125 μM H2O2 to 2.87 ± 0.5% at 1000 μM H2O2. At 500 μM H2O2, approximately 40 ± 6.7% of the HT22 cells remained viable, and we selected 500 μM as the optimal dose of H2O2 for the subsequent experiments.
Figure 2.
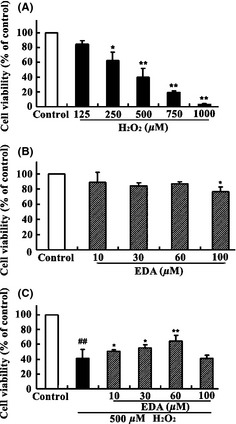
The protective effect of EDA on the H2O2‐induced decrease in cell viability measured by the CCK‐8 assay. (A) HT22 cells were incubated with different concentrations of H2O2 for 24 h. (B) HT22 cells were incubated with different concentrations of EDA for 24 h. (C) HT22 cells were treated with H2O2 (500 μM) for 24 h in the presence of various concentrations of EDA. The values represent the percentage relative to the control (no treatment) and are the mean ± SE (n ≥ 6). The data were analyzed using a one‐way ANOVA followed by post hoc Tukey's multiple comparison tests. ## P < 0.01 versus control, *P < 0.05 and **P < 0.01 versus H2O2.
To investigate whether EDA confers protection against H2O2‐induced cell damage in HT22 cells, we detected the cell viability of the HT22 cells after 500 μM H2O2 incubation for 24 h with EDA co‐incubation at various concentrations from 10 to 100 μM. Our results show that cotreatment with EDA prevented the loss of cell viability induced by treatment with 500 μM H2O2 in a dose‐dependent manner (P < 0.05) (Figure 2C). The application of EDA from 10 to 100 μM alone showed no cytotoxicity toward the HT22 cells (Figure 2B).
H2O2‐induced Apoptosis in HT22 Cells and Protective Effects of EDA
Apoptotic levels were quantified to examine the extent of H2O2‐induced apoptosis and evaluate whether EDA protects against H2O2‐induced apoptosis. The HT22 cells were incubated with different concentrations of H2O2 alone or in combination with EDA (10–100 μM) and H2O2 (500 μM) for 24 h. A flow cytometric analysis was used to quantify the rate of cell apoptosis. As shown in Figure 3, the percentage of apoptotic cells increased as the concentration of H2O2 increased. The apoptotic percentage was 7.61 ± 0.75% in the control group, which was significantly lower than in the H2O2 groups, which exhibited apoptotic rates of 37.79 ± 2.62% when treated with 250 μM H2O2, 45.67 ± 4.32% when treated with 500 μM H2O2, and 95.59 ± 0.89% when treated with 1000 μM (P < 0.01 or P < 0.05) (Figure 3A). However, EDA cotreatment clearly decreased H2O2‐induced apoptosis. Our data show that EDA cotreatment significantly (P < 0.05) and dose dependently decreased the apoptotic rate induced by H2O2, with a saturation at 60 μM EDA for the protective benefit (100 μM EDA showed no more protective effects compared to the 60 μM group, P > 0.05) (Figure 3B).
Figure 3.
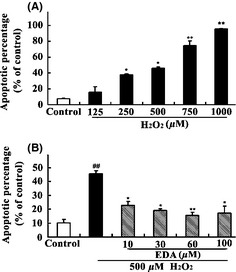
The protective effect of EDA against H2O2‐induced apoptosis measured by flow cytometry. (A) HT22 cells were incubated with different concentrations of H2O2 for 24 h. (B) HT22 cells were treated with H2O2 (500 μM) for 24 h in the presence of various concentrations of EDA (10, 30, 60, 1000 μM). Cell apoptosis was assessed by flow cytometry. The values represent the percentage relative to the control and are the mean ± SE (n ≥ 6). The data were analyzed using a one‐way ANOVA followed by post hoc Tukey's multiple comparison tests. ## P < 0.01 versus control, * P < 0.05 and ** P < 0.01 versus H2O2.
Hoechst 33342 staining revealed that the HT22 cells displayed apoptotic morphology, characterized by chromatin condensation, nuclear shrinkage, and the formation of apoptotic bodies after treatment with H2O2. With increasing concentrations of H2O2, the proportion of cells displaying apoptotic morphology gradually increased. In cells treated with 1000 μM H2O2, normal nuclear morphology was rarely observed. However, cotreatment with EDA reduced the apoptotic fraction of HT22 cells. This observation was consistent with the results obtained from the CCK‐8 assay and flow cytometry. Taken together, these findings suggest that EDA is capable of attenuating H2O2‐induced cell apoptosis.
EDA Inhibited the Generation of ROS Induced by H2O2 in HT22 Cells
To elucidate the effects of EDA on H2O2‐induced oxidative stress, the levels of ROS production in cells were measured using the fluorescence probe DCF. In H2DCF‐DA‐loaded HT22 cells, H2O2 application increased the fluorescence intensity, indicating the generation of ROS. After exposing the cells to H2O2 for 24 h, the M1 peak gradually shifted to the right (Figure 4A), indicating that the ROS production gradually increased with increasing concentrations of H2O2. Moreover, HT22 cells treated with 500 μM H2O2 for 24 h exhibited an approximately 2‐fold increase in fluorescence intensity compared to the control group (Figure 4B). Furthermore, our data show that cotreatment with EDA significantly inhibited the elevated intracellular levels of ROS induced by H2O2 in a significant (P < 0.05), dose‐dependent manner.
Figure 4.
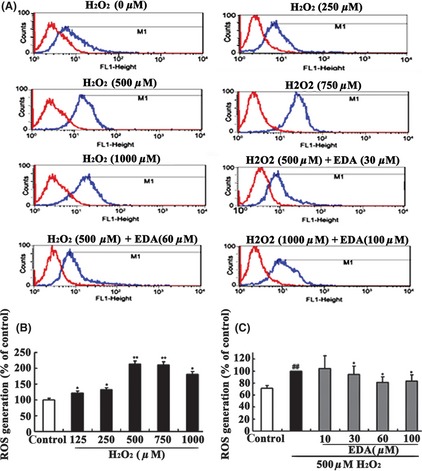
The protective effect of EDA on H2O2‐induced ROS generation measured by flow cytometry. HT22 cells treated with different concentrations of H2O2 in the absence and presence of EDA were loaded with H2 DCF‐DA to detect the generation of intracellular ROS. (A) The M1 peak gradually shifted to the right compared to the control group. However, the M1 peak shifted to the left compared to the H2O2 group when the cells were co‐incubated with EDA. (B) H2O2 significantly increased the intracellular level of ROS compared to the control group (**P < 0.01 or *P < 0.05). (C) Cotreatment with EDA reduced the levels of ROS (P < 0.05 compared to the H2O2 group). The values represent the percentage relative to the control (no treatment) and are the mean ± SE (n ≥ 6). The data were analyzed by a one‐way ANOVA followed by post hoc Tukey's multiple comparison tests. ## P < 0.01 versus control,*P < 0.05 and **P < 0.01 versus H2O2.
EDA Inhibited MAPK Family Activation Stimulated by H2O2
To further explore the mechanism of the protective effects of EDA, we next examined the effect of EDA on potential pathways that are activated during apoptosis. We first examined the protein expression levels of Bcl‐2 and Bax. As shown in Figure 5, H2O2 treatment resulted in decreased Bcl‐2 expression and increased Bax expression compared to the control groups, a tendency that was reversed by EDA cotreatment. Moreover, previous studies have demonstrated that H2O2‐induced oxidative stress may trigger apoptosis by activation of mitogen‐activated protein kinase (MAPK) pathways 16, 17. To clarify the mechanism of action underlying EDA protection, MAPK activities were investigated using Western blot analysis. Compared to the control group, we found that treating the HT22 cells with H2O2 for 24 h increased the phosphorylation of ERK1/2, JNK1/2, and P38 in a concentration‐dependent manner. However, cotreatment with EDA decreased the expression levels of p‐JNK, p‐P38, and p‐ERK1/2 (Figure 6). These results suggest that EDA protects HT22 cells against H2O2‐induced oxidative injury by inhibiting the phosphorylation of MAPK pathway members.
Figure 5.
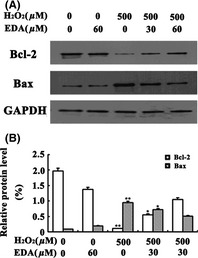
The effects upon Bcl‐2 and Bax expression following EDA treatment. HT22 cells were treated with 500 μM H2O2 and cotreated with different concentrations of EDA for 24 h. Then, the cells were harvested and assayed for the expression of Bcl‐2 and Bax. The results are presented as the mean ± SE from at least three independent experiments. The expression of GAPDH confirms equal protein loading. Panel A shows the protein expression in the HT22 cells. Panel B depicts the statistical results (**P < 0.01, *P < 0.05).
Figure 6.
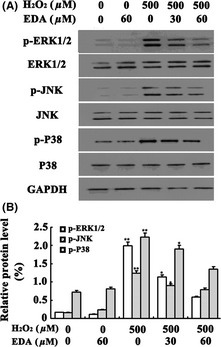
The effects upon ERK, JNK, and P38 expression following EDA treatment. HT22 cells were treated with 500 μM H2O2 and cotreated with different concentrations of EDA for 24 h. Then, the cells were harvested and assayed for the expression of p‐ERK1/2, p‐JNK1/2, and p‐P38. The results are presented as the mean ± SE from at least three independent experiments. The expression of GAPDH confirms equal protein loading. Panel A shows the protein expression in the HT22 cells. Panel B depicts the statistical results (**P < 0.01, *P < 0.05).
Discussion
Oxidative stress refers to the cytologic consequences of a mismatch between the production of free radicals and the ability of the cell to defend against them. Substantial data have been assembled to investigate the essential role of oxidative stress in the regulation of diverse cellular events such as proliferation, differentiation, adhesion, oxidative damage, and cell death 18, 19. Several components of ROS, such as H2O2, may lead to various forms of reversible and irreversible oxidative modification of proteins, lipids, and DNA, initiating subsequent cell death via necrosis or apoptosis and leading to cellular damage 2, 20.
A number of oxidants are produced as by‐products of normal aerobic cell metabolism, and ROS levels are particularly high in neurodegenerative disorders. The central nervous system is especially vulnerable to oxidative stress due to the relatively low levels of cellular antioxidants, high oxygen consumption, and high concentrations of lipids and metal cations available for the generation of free radicals 21. Thus, oxidative stress has been considered to be one of the major risk factors to exacerbate neuronal damage, via different molecular pathways, in many neurodegenerative disorders. Currently, there is growing evidence showing that oxidative stress induced by ROS or free radicals plays a key role in the pathogenesis of neurodegenerative disorders, including AD, PD, HD, and amyotrophic lateral sclerosis 4, 5, 6, 7, 22. Therefore, the removal of excess ROS or the suppression of their generation by antioxidants may be effective in preventing degeneration of neural cells.
H2O2, formed as a natural by‐product of enzymatic oxidase action, is an endogenous source of hydroxyl free radicals normally produced in cells, including neurons. Exogenous H2O2 is able to cross membranes and thus directly alter the intracellular concentration. Furthermore, H2O2 is a major ROS that can induce oxidative stress by increasing the production of other ROS, subsequently leading to cell damage and death. Moreover, H2O2 is involved in the regulation of a variety of cellular events, such as cell proliferation, signal transduction, DNA damage, and cell apoptosis 20, 21. Therefore, H2O2 has been extensively used as an inducer of neuronal injury to both explore the mechanisms of oxidative stress and evaluate the neuroprotective potential of new pharmacotherapies 23. In this study, we also used H2O2 to generate an in vitro model of oxidative stress.
Edaravone, a lipophilic molecule with rapid access to the intracellular space, is a powerful free radical scavenger that has been clinically used to reduce neuronal damage following cerebral ischemic stroke 9, 10. It protects cells from injury by directing the scavenging activities of the hydroxy radical (OH.) and inhibiting lipoxygenase activity and 15‐HPETE. Furthermore, it can also directly neutralize peroxy radicals (LOO.) but does not scavenge for O2 . 24. Consequently, EDA has been evaluated as a neuroprotective compound 25. Recently, EDA has been offered as a viable candidate for the treatment of oxidative stress–induced neurodegenerative disease 26, 27, but until now, research on its effect and probable mechanisms in the treatment of such diseases was lacking. Therefore, in this study, we attempted to verify the antioxidative role of EDA in a hippocampal cell model of oxidative injury.
Our results provide direct evidence of EDA protection against H2O2‐induced HT22 cell injury based on the CCK‐8 method. Exogenous H2O2 decreased cell viability in a concentration‐dependent manner, and 500 μM H2O2 caused a significant decrease 40 ± 6.7% in HT22 cell viability. However, cotreatment with EDA for 24 h increased cell viability in a concentration‐dependent manner. As an extension of the cell viability results, we evaluated cell apoptosis and the generation of ROS. When HT22 cells were exposed to H2O2, cell apoptosis and ROS levels increased. In contrast, cotreatment with EDA decreased both the apoptotic rate of HT22 cells and the production of ROS generation. These protective effects may contribute to the antioxidative activity of EDA.
After discovering that EDA exerted a protective effect against H2O2‐induced cytotoxicity in HT22 cells, we studied the potential pathway responsible for these effects. The MAPKs, a family of serine/threonine protein kinases, are involved in many cellular processes, including cell growth, differentiation, inflammation, and cell death 28. MAPKs are key kinases in signal transduction pathways, and members of each major MAPK subfamily (JNK, P38, and ERK1/2) have been implicated in neuronal injury and disease 29. The unphosphorylated forms of the MAPKs are virtually inactive, and phosphorylation stimulates their activity. The oxidative stress–induced ROS activation of JNK, ERK1/2, and/or P38 has been described in various types of cells, and ROS activation of the MAPK cascade might be a common mechanism by which oxidative stress induces neuronal cell death or neurodegenerative disorders 30, 31. In this study, we examined the expression of the three signaling proteins in the MAPK pathway. We found that activation of JNK, P38, and ERK1/2 in HT22 cells was significantly increased following exposure to H2O2, a result which is consistent with studies on other cell models 32, 33. However, when the cells were cotreated with EDA, activation of the MAPKs was reduced, suggesting that EDA protects cells from oxidative stress–induced apoptosis by inhibiting the MAPK signaling pathway.
Taken together, our results demonstrate that H2O2 can induce cell apoptosis through the production of ROS and activation of the MAPK signaling pathway. To the best of our knowledge, we show for the first time that EDA, a radical scavenger, can inhibit the production of ROS and the expression of the MAPK signaling pathway and thus confer protection for HT22 cells 34. Currently, many studies suggest that oxidative stress–induced damage is involved in many neurodegenerative diseases, including AD. Thus, antioxidant treatment may be an effective method for the treatment of these diseases. As a radical scavenger, EDA has been used to treat brain stroke/ischemia, and as shown in our study, it can also protect HT22 cells from H2O2‐induced cell apoptosis, indicating its potential as an antioxidant candidate in the treatment of neurodegenerative diseases.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
We are grateful for support from the National Nature Science Foundation of China (No. 30970966), the Fundamental Research Funds for the Central Universities (No. 11ykpy23), and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry.
The first two authors contributed equally to this work.
References
- 1. Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem 1997;272:20313–20316. [DOI] [PubMed] [Google Scholar]
- 2. Gorman AM, McGowan A, O'Neill C, Cotter T. Oxidative stress and apoptosis in neurodegeneration. J Neurol Sci 1996;139(Suppl):45–52. [DOI] [PubMed] [Google Scholar]
- 3. Pettmann B, Henderson CE. Neuronal cell death. Neuron 1998;20:633–647. [DOI] [PubMed] [Google Scholar]
- 4. Choi J, Sullards MC, Olzmann JA, et al. Oxidative damage of DJ‐1 is linked to sporadic Parkinson and Alzheimer diseases. J Biol Chem 2006;281:10816–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Caviness JN, Lue L, Adler CH, Walker DG. Parkinson's disease dementia and potential therapeutic strategies. CNS Neurosci Ther 2011;17:32–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stoy N, Mackay GM, Forrest CM, et al. Tryptophan metabolism and oxidative stress in patients with Huntington's disease. J Neurochem 2005;93:611–623. [DOI] [PubMed] [Google Scholar]
- 7. Bellingham MC. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: What have we learned in the last decade? CNS Neurosci Ther 2011;17:4–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ishikawa Y, Satoh T, Enokido Y, Nishio C, Ikeuchi T, Hatanaka H. Generation of reactive oxygen species, release of L‐glutamate and activation of caspases are required for oxygen‐induced apoptosis of embryonic hippocampal neurons in culture. Brain Res 1999;824:71–80. [DOI] [PubMed] [Google Scholar]
- 9. Edaravone Acute Infarction Study Group . Effect of a novel free radical scavenger, edaravone (MCI‐186), on acute brain infarction. Randomized, placebo‐controlled, double‐blind study at multicenters. Cerebrovasc Dis 2003;15:222–229. [DOI] [PubMed] [Google Scholar]
- 10. Yang J, Liu M, Zhou J, Zhang S, Lin S, Zhao H. Edaravone for acute intracerebral haemorrhage. Cochrane Database Syst Rev 2011:CD007755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kawasaki T, Kitao T, Nakagawa K, et al. Nitric oxide‐induced apoptosis in cultured rat astrocytes: Protection by edaravone, a radical scavenger. Glia 2007;55:1325–1333. [DOI] [PubMed] [Google Scholar]
- 12. Liu J, Li L, Suo WZ. HT22 hippocampal neuronal cell line possesses functional cholinergic properties. Life Sci 2009;84:267–271. [DOI] [PubMed] [Google Scholar]
- 13. Zhao Z, Lu R, Zhang B, et al. Differentiation of HT22 neurons induces expression of NMDA receptor that mediates homocysteine cytotoxicity. Neurol Res 2012;34:38–43. [DOI] [PubMed] [Google Scholar]
- 14. Luan P, Zhou H, Zhang B, et al. Basic Fibroblast Growth Factor Protects C17.2 Cells from Radiation‐induced Injury through ERK1/2. CNS Neurosci Ther 2012;18:767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu J, Rasul I, Sun Y, et al. GRK5 deficiency leads to reduced hippocampal acetylcholine level via impaired presynaptic M2/M4 autoreceptor desensitization. J Biol Chem 2009;284:19564–19571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Valencia A, Moran J. Reactive oxygen species induce different cell death mechanisms in cultured neurons. Free Radic Biol Med 2004;36:1112–1125. [DOI] [PubMed] [Google Scholar]
- 17. Ruffels J, Griffin M, Dickenson JM. Activation of ERK1/2, JNK and PKB by hydrogen peroxide in human SH‐SY5Y neuroblastoma cells: Role of ERK1/2 in H2O2‐induced cell death. Eur J Pharmacol 2004;483:163–173. [DOI] [PubMed] [Google Scholar]
- 18. Bergamini CM, Gambetti S, Dondi A, Cervellati C. Oxygen, reactive oxygen species and tissue damage. Curr Pharm Des 2004;10:1611–1626. [DOI] [PubMed] [Google Scholar]
- 19. Martindale JL, Holbrook NJ. Cellular response to oxidative stress: Signaling for suicide and survival. J Cell Physiol 2002;192:1–15. [DOI] [PubMed] [Google Scholar]
- 20. Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: A metabolic by‐product or a common mediator of ageing signals? Nat Rev Mol Cell Biol 2007;8:722–728. [DOI] [PubMed] [Google Scholar]
- 21. Brown DR, Schmidt B, Kretzschmar HA. Effects of oxidative stress on prion protein expression in PC12 cells. Int J Dev Neurosci 1997;15:961–972. [DOI] [PubMed] [Google Scholar]
- 22. He F, Luan P, He R, et al. Effect of edaravone on Aβ1‐40 induced enhancement of voltage‐gated calcium channel current. CNS Neurosci Ther 2012;18:89–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saitoh M, Nishitoh H, Fujii M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal‐regulating kinase (ASK) 1. EMBO J 1998;17:2596–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Watanabe T, Yuki S, Egawa M, Nishi H. Protective effects of MCI‐186 on cerebral ischemia: Possible involvement of free radical scavenging and antioxidant actions. J Pharmacol Exp Ther 1994;268:1597–1604. [PubMed] [Google Scholar]
- 25. Kaur C, Ling EA. Antioxidants and neuroprotection in the adult and developing central nervous system. Curr Med Chem 2008;15:3068–3080. [DOI] [PubMed] [Google Scholar]
- 26. Pan YH, Wang YC, Zhang LM, Duan SR. Protective effect of edaravone against PrP106‐126‐induced PC12 cell death. J Biochem Mol Toxicol 2010;24:235–241. [DOI] [PubMed] [Google Scholar]
- 27. Xiong N, Xiong J, Khare G, et al. Edaravone guards dopamine neurons in a rotenone model for Parkinson's disease. PLoS ONE 2011;6:e20677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pearson G, Robinson F, Beers GT, et al. Mitogen‐activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr Rev 2001;22:153–183. [DOI] [PubMed] [Google Scholar]
- 29. Harper SJ, Wilkie N. MAPKs: New targets for neurodegeneration. Expert Opin Ther Targets 2003;7:187–200. [DOI] [PubMed] [Google Scholar]
- 30. Ouyang M, Shen X. Critical role of ASK1 in the 6‐hydroxydopamine‐induced apoptosis in human neuroblastoma SH‐SY5Y cells. J Neurochem 2006;97:234–244. [DOI] [PubMed] [Google Scholar]
- 31. Kim SD, Moon CK, Eun SY, Ryu PD, Jo SA. Identification of ASK1, MKK4, JNK, c‐Jun, and caspase‐3 as a signaling cascade involved in cadmium‐induced neuronal cell apoptosis. Biochem Biophys Res Commun 2005;328:326–334. [DOI] [PubMed] [Google Scholar]
- 32. Kim SK, Woodcroft KJ, Oh SJ, Abdelmegeed MA, Novak RF. Role of mechanical and redox stress in activation of mitogen‐activated protein kinases in primary cultured rat hepatocytes. Biochem Pharmacol 2005;70:1785–1795. [DOI] [PubMed] [Google Scholar]
- 33. Yeo JE, Kim JH, Kang SK. Selenium attenuates ROS‐mediated apoptotic cell death of injured spinal cord through prevention of mitochondria dysfunction; in vitro and in vivo study. Cell Physiol Biochem 2008;21:225–238. [DOI] [PubMed] [Google Scholar]
- 34. Inokuchi Y, Imai S, Nakajima Y, et al. Edaravone, a free radical scavenger, protects against retinal damage in vitro and in vivo. J Pharmacol Exp Ther 2009;329:687–698. [DOI] [PubMed] [Google Scholar]