

Summary
Aims
Manganese superoxide dismutase (MnSOD), one of the most crucial antioxidant enzymes in the central nervous system, is thought to be one of the major mechanisms by which cells counteract the injuries of reactive oxygen species after cerebral ischemia. In this study, we used a novel synthesized compound (MnTm4PyP) with highly effective superoxide dismutase activity to study the therapeutic potential of MnSOD and the possible underlying mechanisms in cerebral ischemia.
Methods
Primary cultured cortical neurons were used to examine the protective effect of the compounds. Mice with middle cerebral artery occlusion were used as ischemic stroke animal model. Animals were pretreated with MnTm4PyP intravenously 30 min before surgery. At 24 h after surgery, neurological behavior and histological function were observed. Infarcted cortex tissues and cultured neurons were collected for investigation of the oxidative stress signaling pathways.
Results
In vitro studies revealed that MnSOD mimic MnTm4PyP pretreatment significantly increased viability of neurons after injury by H 2 O 2. Intracellular superoxide radical levels were eliminated. In vivo experiments demonstrated MnTm4PyP pretreatment reduced infarct volume and improved neurological function. The MnSOD mimic alleviated oxidative stress and apoptosis.
Conclusion
MnSOD is an effective therapeutic target in ischemic stroke prevention because of its antioxidant effects and oxidative stress regulation.
Keywords: Ischemic stroke, Manganese superoxide dismutase, Middle cerebral artery occlusion, Neuroprotection
Introduction
Ischemic stroke is caused by glucose and oxygen deficiency resulting from the reduction or complete blockage in blood flow to the brain. The three primary natural causes of ischemia are thrombosis, embolism, and systemic decrease in blood perfusion 1. At present, the risk of intracranial hemorrhagic transformation limits the use of thrombolytic therapy 2. Increased levels of reactive oxygen species (ROS), including superoxide radical (), hydrogen peroxide (H2O2), and hydroxyl radical (OH−), are major causes of tissue injury after cerebral ischemia, because they cause destruction of cellular proteins, lipids, and DNA and disruption of normal cellular signaling and gene regulation 3. is of particular importance. Compelling evidence has indicated that cerebral ischemia and traumatic brain injury can cause increased formation, which is considered a component of acute brain injury 4, 5. The interaction of with nitric oxide produces peroxynitrite, a highly toxic molecule that causes further tissue damage and is considered as the trigger molecule for apoptosis after an ischemic stroke 6. As ROS precursors, hydrogen peroxide (H2O2) can induced apoptotic cell death so that H2O2‐induced apoptosis serves as a valuable model for the studies of neuroprotection related to oxidative stress.
Numerous studies have focused on the therapeutic efficacy of various antioxidant compounds in both animal models and humans 7, 8, 9, 10. However, concerns about the bioavailability of these compounds have not been resolved yet 11, 12. Superoxide dismutase (SOD) is an endogenous enzyme that is part of the first line of defense through its elimination of by converting it into H2O2 and O2. SOD enzymes have been shown to reduce ischemic and traumatic brain injuries, thus leading to the recent renewal of interest in upregulating the synthesis of different SODs 13, 14, 15. In addition, manganese superoxide dismutase (MnSOD or SOD2) knockout mice have exhibited neonatal lethality from neurodegeneration and cardiomyopathy 16, 17. Previous studies have demonstrated that MnSOD deficiency exacerbates cerebral infarction after cerebral ischemia and that reperfusion after cerebral ischemia significantly reduces the expression of MnSOD 18, 19. This cause‐and‐effect relationship between MnSOD and cerebral ischemia implies that MnSOD could be an effective therapeutic target for the treatment of stroke. Overexpression of MnSOD has been shown to be neuroprotective 20. However, the properties of the enzyme (e.g., low oral activity, immunogenicity because of nonhuman sources, and short half‐life) largely limit its application as a therapeutic target.
Manganese porphyrins were synthesized as a new class of low molecular weight antioxidants that mimic the high catalytic efficiency of MnSOD in scavenging for . In this study, we tested the capability of MnTm4PyP and MnTPPS (see Figure 1) to mimic the enzymatic function of MnSOD. Their neuroprotective effects and the possible underlying mechanisms were investigated using primary cultured cortical neurons and middle cerebral artery occlusion (MCAO) mice.
Figure 1.
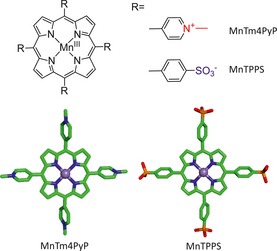
Chemical structure of MnSOD mimics.
Materials and Methods
Materials
Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco BRL (Gaithersburg, MD, USA). Cell Titer 96® AQueous MTS reagent and Cyto Tox 96 Nonradioactive Cytotoxicity Assay Kit were acquired from Promega (Madison, WI, USA). Dihydroethidium (DHE), 2′,7′‐dichlorofluorescein‐diacetate (DCFH‐DA), and Fluo‐3 AM were obtained from Invitrogen (Paisley, UK); antibodies against caspase‐3, β‐actin, and cytochrome c were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against C/EBP homologous protein (CHOP) were obtained from Cell Signaling Technology (Danvers, MA, USA).
Synthesis of Manganese Porphyrins
Metal‐free porphyrins—Tetrakis(1‐methylpyridinium‐4‐yl)porphyrin p‐Toluenesulfonate (Tm4PyP) and Tetraphenylporphyrin Tetrasulfonic Acid Hydrate (TPPSH)—were obtained from TCI Chemicals (Tokyo, Japan). Manganese complexes were prepared by metallation of the porphyrin ligands as described previously with some modifications 21, 22. Briefly, porphyrin was added to manganese acetate in water (1:20) at 80°C. The reaction was monitored by UV‐visible spectroscopy until the completion of metallation. To prepare Mn(III)Tm4PyP, the PF6 salt of the manganese complex was precipitated from the solution with the addition of NH4PF6. The compound was then dissolved in acetone and tetrabutylammonium chloride solution was added, leading to the formation of the chloride salt of porphyrin. The precipitate was washed thoroughly with acetone and dried in vacuo at room temperature. Mn(III)TPPS was prepared by acidification of the solution to pH 3 with 1 N HCl, chromatography on a Dowex 50‐WX8 cation exchange column, and dialysis against water for 2 days. The solution was evaporated to recover the product. Anal. Calcd (Found) for Mn(III)Tm4PyPCl5•2H2O was C 55.92 (55.75), H 4.27 (4.38), N 11.86 (11.72); for Mn(III)TPPSH4Cl•5H2O was C 47.46 (47.62), H 3.44 (3.29), N 5.03 (5.09).
Primary Cortical Neuron Culture and Cell Viability Assay
Primary cortical neuronal cultures were prepared as described previously 23, 24. Eighteen‐day‐old embryonic Sprague Dawley rats were sacrificed, and cortex tissues were obtained quickly. Tissues were incubated in 0.25% trypsin at 37°C for 15 min and then triturated with a Pasteur pipette. Clumps were removed by filtering. Cortical neurons were cultured in poly‐d‐lysine (5 μg/mL)‐coated wells in DMEM (10% FBS, 100 U/mL penicillin, 5 mM 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid, and 500 ng/mL insulin) at 37°C and 5% CO2. Three days after plating, 10 μg/mL of cytarabine was added to the culture medium to reduce the number of glia. The medium was changed every 3 days. Experiments were performed after cells were maintained in culture for 9 days.
Cells were treated with different concentrations of MnSOD mimics for 30 min, washed twice, and then treated with 100 μM of H2O2 for 18 h. Cell viability was measured by a colorimetric assay using Cell Titer 96® AQueous MTS reagent. Half maximal effective concentration (EC50) was analyzed using the Originpro software (Version 8.0; Originlab, Northampton, MA, USA).
Drug Treatment and Animal Ischemic Stroke Model
This study was conducted using the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. All experimental protocols were approved by the Animal Care and Use Committee of the Peking University. Animals were supplied by the Laboratory Animal Center of the Peking University Health Science Center (Beijing, China). Adult male C57BL/six mice (20–25 g) were housed under diurnal lighting conditions and allowed food and water ad libitum. They were randomly divided into three groups: control group (sham, n = 6), MCAO group treated with natural saline (vehicle, n = 6), and MCAO group with MnTm4PyP treatment (MnTm4PyP, n = 6). In the drug‐treated group, 1.5 mg/kg of MnTm4PyP was administered via intravenous (IV) injection 30 min before MCAO. Mice in the vehicle group received 1.5 mg/kg of 0.9% NaCl IV 30 min before MCAO. After drug treatment, stroke was induced in mice through transtemporal coagulation as described previously 25. Briefly, mice were anesthetized with 3.5% chloral hydrate (0.1 mL/10 g, i.p.). A 1‐cm skin incision was made in the midpoint the between left eye and left ear, and a burr hole was drilled through the temporal skull using a high‐speed microdrill. After the removal of the dura mater, bipolar electrocoagulation forceps (ERBOTOM, ERBE, Germany) were used to permanently occlude the exposed left middle cerebral artery. The wound was closed using stitches. Body temperature was kept at 37°C using a heating blanket. The same operation was performed on the sham group, except for the absence of MCAO.
Measurement of Intracellular O 2 − and H 2 O 2 Levels
Dihydroethidium and DCFH‐DA were used to detect intracellular and H2O2 levels, respectively. Primary cultured neurons were treated with 5 μM of MnTm4PyP for 30 min, washed twice, and then treated with 100 μM of H2O2 for 2 h. After three washings with PBS, cells were incubated with DHE or DCFH‐DA at 37°C for 30 min and then washed twice. Cells were observed under a laser‐scanning confocal microscope (Leica, Heidelberg, Germany), with fluorescence intensity corresponding to the levels of intracellular H2O2. Fluorescence intensity data were analyzed using the Leica TCSNT software.
Neurological Behavior and Histological Studies
Mice were sacrificed 24 h post‐MCAO. The brain was collected and sliced into six coronal sections (2 mm thick). They were stained with 0.1% of 2,3,5‐triphenyltetrazolium chloride (TTC) in natural saline at 37°C for 30 min and postfixed with 10% formalin for 2 h. Images of the sections were scanned, and infarcted areas were measured by the Scion Image software (Ver. 4; Scion Corporation, Frederick, MD, USA). Hematoxylin and eosin (HE) staining assays were performed to observe the neuropathology as described previously 26. Terminal deoxynucleotidyl transferase‐mediated dUTP nick end labeling (TUNEL) assays were carried out to detect apoptotic cells in situ according to the method described in 27.
To evaluate neurological behavior after stroke, the neurological deficit scoring system with a scale of zero to five was utilized 28. In addition, the body swing test was performed to measure motor asymmetry 29. Briefly, the mouse was held by its tail, and the direction of the swings made by the mouse was recorded. Movement of the mouse head sideways beyond 10° from the body's midline was considered a swing direction. Tests were repeated 20 times for each animal, and the animal was allowed to move freely for 30 second before retesting.
Measurement of Intracellular Ca2+ Levels
Fluo‐3 AM was used to observe the continuous changes in cellular Ca2+ levels as described previously 30. Briefly, primary cultured neurons were treated with 1 or 10 μM MnTm4PyP for 18 h. Cells were incubated with 5 μM Fluo‐3 AM at 37°C and washed with Earle's Balanced Salt Solution. Cellular Ca2+ was observed using a laser‐scanning confocal microscope with an excitation of 488 nm and an emission of 526 nm. Seven visual fields were randomly selected for observation. The basal level was determined after 2 min of observation. Then, 100 μM H2O2 was added to the medium, and changes in intracellular Ca2+ levels, represented by Fluo‐3 fluorescent intensities, were observed for 10 min without interruption. Data from five experiments were analyzed using the Leica TCSNT software.
Western Blot analysis
Western blot was performed as described previously 31. Briefly, tissue samples were derived from the infarct tissue of MCAO mice brain. Cultured cells were pretreated with 1 or 5 μM MnTm4PyP, followed by H2O2 treatment for 18 h. Tissue samples and cultured cells were washed with cold PBS and lysed using NP‐40 lysis buffer. Proteins were determined using the Bradford assay. Equal amounts of total proteins (20 μg) were electrophoresed on an SDS–PAGE gel and transferred to a PVDF immunoblotting membrane. The membrane was blocked with 5% nonfat milk in TBST (TBS with 0.02% of Tween‐20, pH 7.5) for 1 h and then incubated overnight at 4°C with primary antibodies diluted in TBST (1:1000). The membrane was incubated with HRP‐conjugated secondary antibodies (1:6000) at 37°C for 1 h and was visualized using an enhanced chemiluminescent detection kit (Applygen Technologies, Beijing, China). Optical band densities were quantified using the Gel‐Pro Analyzer software.
Statistical Analysis
Data are presented as mean ± SD. Multiple comparisons were performed using the Student's t‐test function of the SPSS software (Version 16.0; SPSS, Chicago, IL, USA). A probability of P < 0.05 was considered statistically significant.
Results
Neuroprotection of MnSOD on Primary Cultured Cortical Neurons
The neuroprotective effects of MnSOD mimics on H2O2‐induced neurotoxicity were examined through MTS assay of primary cultured cortical neuron exposed to H2O2. Results (Figure 2A) showed that 18 h of H2O2 (100 μM) treatment markedly decreased the viability of primary cultured cortical neurons compared with the untreated control group. Pretreatment of cells with MnTm4PyP (3.2–100 μM) increased cell viability in a dose‐dependent manner compared with the H2O2 group. MnTPPS exhibited minimal neuroprotective effects. The dose‐effect curve (Figure 2B) revealed that MnTm4PyP had dose‐dependent neuroprotective behavior at low concentrations (0.78–50 μM). At concentrations above 50 μM, the protective effect declines, and the half‐effective concentration is approximately 5 μM. Thus, we determined that MnTm4PyP is an effective compound that displays high MnSOD activity and used it in subsequent experiments.
Figure 2.
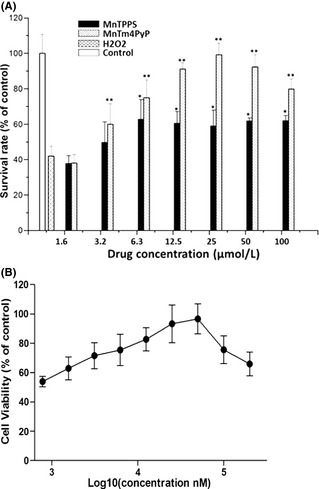
Protective effects of MnSOD mimics on H 2 O 2‐induced toxicity in primary cortical neuron cultures. Cell viability was evaluated using the MTS assay. (A) Neurons were treated with different concentrations of MnTPPS or MnTm4PyP before exposure to H 2 O 2 for 18 h. (B) Protective effects of MnTm4PyP on H 2 O 2‐induced toxicity. Values represent mean ± SD in separate experiments (n = 5). *P < 0.05, *P < 0.01 compared with the H 2 O 2 group.
Levels of Intracellular O 2 − and H 2 O 2 were Reduced by MnSOD mimic
Primary cultured cortical neurons were used to evaluate the ability of the MnSOD mimic to scavenge for intracellular . Representative laser‐scanning confocal microscope images of neurons stained with DHE or DCFH‐DA are shown in Figure 3. Neurons were loaded with DHE after treatment with 100 μM H2O2 for 2 h (Figure 3A). Levels of intracellular markedly increased compared with the control group. In contrast, the levels of intracellular in neurons treated with 5 μM of MnSOD mimic before H2O2 treatment were significantly reduced compared with the H2O2 group. The bright field images showed a morphologically superior integration of synapses of the neurons in the MnTm4PyP group, suggesting that MnSOD mimic treatment attenuated the apoptosis and necrosis induced by H2O2. MnSOD mimic treatment slightly reduced intracellular H2O2 levels (Figure 3B).
Figure 3.
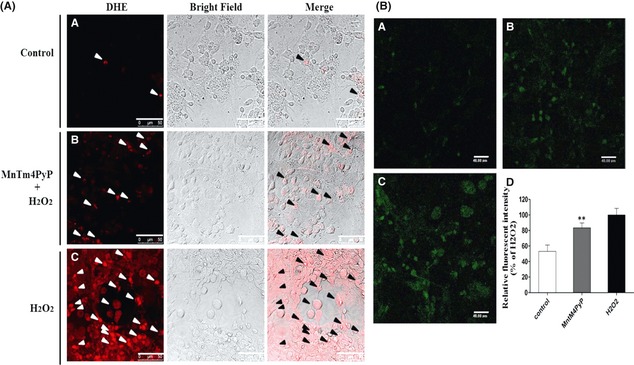
MnSOD mimic eliminates intracellular superoxide radical and H2O2 levels. (A) Representative images of laser scanning confocal microscopy. Arrows indicate red fluorescence representing the presence of intracellular superoxide radical. (B) Representative images of laser scanning confocal microscopy. Green fluorescence represents intracellular H2O2 levels. (B‐A) Cells treated with PBS; (B‐B) cells treated with 5 μM MnTm4PyP before H2O2 (100 μM) exposure for 2 h; (B‐C) cells treated with 100 μM H2O2 for 2 h; (B‐D) 0 Quantification of fluorescence intensity from mean ± SD, (n = 6). **P < 0.01 compared with the H2O2 group.
MnSOD Mimic Ameliorated Neuropathology and Improved Neurological Function
2,3,5‐Triphenyltetrazolium chloride staining of mice in the sham group who underwent a similar operating procedure 24 h postsurgery except for the absence of left MCAO did not reveal any infarct area (Figure 4A). An infarct area was observed in the left cortex (28.70 ± 4.94% of the hemisphere) of mice in the vehicle group treated with 0.9% NaCl. In the MnTm4PyP treatment group, infarct volume was significantly decreased (13.06 ± 2.57% of the hemisphere) compared with the vehicle group. Infarct volume was reduced from 28.70 ± 4.94% to 13.06 ± 2.57% after 1.5 mg/kg of MnTm4PyP treatment 30 min before MCAO. The ischemic cerebral cortex of mice in the vehicle group exhibited marked tissue damage, as indicated by condensed, irregularly shaped nuclei (Figure 4B). Tissue damage was ameliorated in MnTm4PyP group mice. The amount of TUNEL‐positive apoptotic cells was remarkably decreased in the MnTm4PyP group compared with the vehicle group (Figure 4C).
Figure 4.
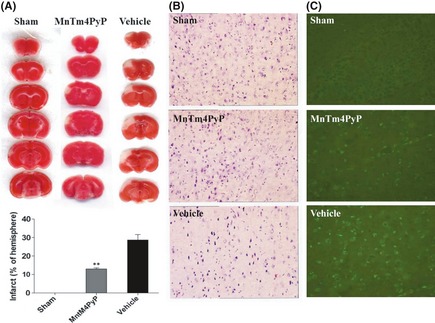
Neuroprotective effects of the MnSOD mimic in ischemic stroke. (A) The infarct area of each group was evaluated by 2,3,5‐triphenyltetrazolium chloride staining. (B) Hematoxylin and eosin staining of infarcted cortex. (C) Terminal deoxynucleotidyl transferase‐mediated dUTP nick end labeling‐positive cells in infarcted cortex. Quantitative analyses of infarct volume by Scion Image software. Values represent mean ± SD of separate experiments (n = 6). **P < 0.01 compared with the vehicle group.
We utilized the neurological scoring system and swing test to evaluate neurological function (Figure 5). Mice from the sham group did not display any neurological deficit, and their neurological score is zero. In contrast, mice in the vehicle group had the highest neurological deficit score, which concurs with the results of our infarct volume measurements and histological studies. MnTm4PyP treatment could rescue the neurological deficit score. Similarly, mice from the sham group did not show any motor asymmetry. The swing rate (53.03 ± 5.74%) was close to the theoretical value of 50%. The vehicle group had a rate of 93.38 ± 4.58%, suggesting severe motor asymmetry. Treatment with the MnSOD mimic reduced the neurological deficit to 60.27 ± 6.83% (Figure 5B). These findings demonstrate that the MnSOD mimic has a neuroprotective effect on MCAO mice in terms of neuropathology and neurological function.
Figure 5.
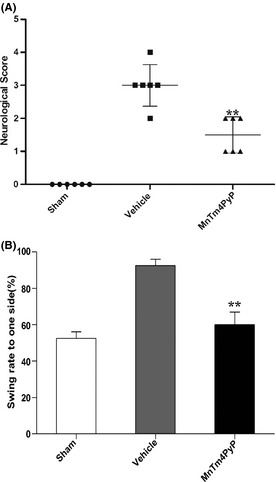
Neuroprotective effects of MnTm4PyP on neurological function in ischemic stroke. (A) Neurological deficit score. (B) Motor asymmetry evaluation. Values represent mean ± SD of separate experiments (n = 6). **P < 0.01 compared with the vehicle group.
MnTm4PyP Ameliorated Oxidative Stress In vitro and In vivo
MnTm4PyP can reduce intracellular levels and is potentially neuroprotective. Western blot analysis was performed to study the mechanisms underlying this neuroprotective effect. Cortical tissues for molecular analysis were collected from brains with MCAO‐induced injury in the ipsilateral cortex (Figure 4A). Expression levels of cleaved‐caspase‐3, cytochrome c, and CHOP in the MnTm4PyP group were markedly reduced compared with the vehicle group (Figure 6A). Furthermore, in vitro analysis revealed that MnTm4PyP can attenuate the H2O2‐induced increase in these oxidative markers in a dose‐dependent manner (Figure 6B). We investigated the expression of ER stress hallmark GRP78 because we previously found that ER stress is usually involved in oxidative stress in neurons.
Figure 6.
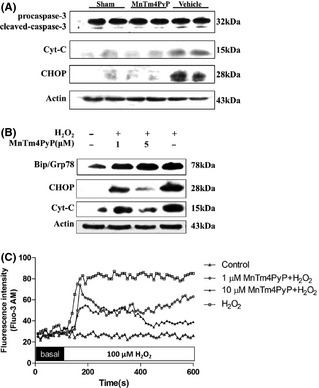
Mechanism of neuroprotection of the MnSOD mimic. (A) Expression of caspase‐3, cytochrome c and C/EBP homologous protein (CHOP) derived from infarct tissue of middle cerebral artery occlusion mice brain. (B) Expression of Bip/Grp78, cytochrome c, and CHOP derived from primary cultured neurons pretreated with 1 or 5 μM of MnTm4PyP before H2O2 treatment for 18 h. (C) Changes in intracellular Ca2+ levels of neurons within 10 min of H2O2 treatment with (1 or 10 μM) or without MnTm4PyP.
Because disturbed intracellular Ca2+ homeostasis is closely related to oxidative stress and ER stress, changes in the cytosolic Ca2+ levels of neurons were tested (Figure 6C). Cytosolic Ca2+ levels remained at base line in the group treated with MnTm4PyP for 18 h compared with the control group. After the addition of H2O2, cellular Ca2+ levels remarkably increased. For cells treated with MnTm4PyP, after the immediate increase upon treatment with H2O2, cellular Ca2+ decreased in a dose‐dependent manner. Within 10 min of observation, neurons treated with 10 μM MnTm4PyP had comparatively normal Ca2+ homeostasis compared with the H2O2 injury group. This finding suggests that normal Ca2+ homeostasis would not be interrupted when neurons are protected by the MnSOD mimic. Results indicate that ER stress is not markedly influenced by MnTm4PyP protection based on the markers of ER stress and intracellular Ca2+ level.
Discussion
The core brain tissue in ischemic stroke that is exposed to the most dramatic reduction in blood flow is severely injured and subsequently undergoes necrotic cell death. The necrotic core is surrounded by a zone where less severely affected tissue is rendered functionally silent by reduced blood flow but remains metabolically active. This region represents an opportunity for therapy and is referred to as the “ischemic penumbra” 3, 32. Mechanisms such as increased production, elevated Ca2+ by excitotoxicity, and inflammatory responses are vital in ischemia 33. Increased levels of ROS are major causes of tissue injury after cerebral ischemia, particularly levels. These findings suggest that MnSOD, the first‐line‐of‐defense enzyme for scavenging , can be an effective therapeutic target.
Hence, we used low molecular weight compounds that mimic MnSOD activity to observe the potential of MnSOD as a treatment target for stroke. MnTm4PyP eliminated in a concentration‐dependent behavior, whereas MnTPPS possessed a relatively lower MnSOD activity. These findings are consistent with previous reports 12. We cultured cortical neurons for in vitro assays to examine the protective effect of MnSOD mimics against H2O2‐induced neuronal injury. Results showed that MnTm4PyP has better protective effects than MnTPPS in the H2O2‐induced neuronal injury model (Figure 2A). These findings indicate that MnTPPS has lower neuroprotective activity than MnTm4PyP, probably due to its relatively low MnSOD activity and its inability to penetrate the membrane because of its high negative charges. Further studies of MnTm4PyP using this model suggested that it has dose‐dependent neuroprotective effects (Figure 2B). Therefore, we selected MnTm4PyP as the more suitable MnSOD mimic for the evaluation of the therapeutic potential of MnSOD in subsequent experiments.
A specific probe for testing intracellular was used to determine whether MnTm4PyP can function as an elimination agent. Results suggest that MnTm4PyP can scavenge with high efficacy and efficiency (Figure 3A). Interestingly, we also observed slightly reduced intracellular H2O2 levels (Figure 3B), which is consistent with the previous reports that MnTm4PyP is a mimic of both SOD and catalase 34, 35. The efficiency of MnTm4PyP elimination was higher than that of H2O2 reduction. Our findings can be used to design new analogs that possess high efficiency in eliminating both and H2O2 levels.
We then examined the protective potential by which the MnSOD mimic affects MCAO mice. As shown in Figures 4 and 5, a left MCAO causes infarction in the cortical after TTC staining. Histological studies showed obvious tissue damage, while the TUNEL assay suggested the marked presence of apoptotic cells in the infarcted cortex. Mice with MCAO suffered severe neurological deficit and lacked motor symmetry. Treatment with the MnSOD mimic at 1.5 mg/kg 30 min before MCAO significantly ameliorated infarct volume, tissue damage, and apoptosis. The resulting neurological function impairment was alleviated compared with the vehicle group, indicating that the MnSOD mimic possesses protective properties in cerebral ischemic stroke.
To understand the molecular mechanism through which MnSOD protects neurons, Western blot analysis was performed to verify the regulation of oxidative stress. Cerebral ischemia resulted in the opening of mitochondrial transition pores, which resulted in the release of normally sequestered proapoptotic proteins, such as cytochrome c, from the intermembrane space into the cytosol 36. Findings suggest that cytochrome c is significantly increased in the infarct cortex of MCAO mouse and that MnSOD mimic treatment alleviates this increase in cytochrome c (Figures 6). Caspase‐3 has been identified as a key mediator of apoptosis in animal ischemic stroke models 3. In this study, we observed a significant upregulation of the active form of caspase‐3 in the infarct cortex of MCAO mice. Consistently, the MnSOD mimic attenuated the increase in active caspase‐3 levels after MCAO, indicating that MnSOD may be involved in mitochondrial ROS production and its protective role after permanent ischemia. MnSOD can inhibit mitochondrial cytochrome c release to cytosol, thereby preventing apoptosis. Thus, it functions as an endogenous mitochondrial antioxidant after permanent cerebral ischemia. CHOP, also known as Gadd153, is a downstream proapoptotic gene that can be induced by severe ER stress via the ATF6, IRE1, and PERK‐eIF2α pathways 37. Although the exact role of CHOP in mediating apoptosis is not fully understood, this molecule may act as a proapoptotic transcription factor that can be induced by severe ER and oxidative stresses 38. Findings of this study suggest that treatment with the MnSOD mimic can ameliorate the upregulation of CHOP in the infarcted cortex.
Our previous study on oxidative stress‐induced ER stress indicated that the upregulation of CHOP may be a sign of ER stress involvement. Thus, we performed an in vitro assay to examine whether ER stress is involved in MnSOD neuroprotection in this study. Similar to in vivo results, treatment with H2O2 of primary cultured neurons resulted in significant upregulation of cytochrome c and CHOP. The MnSOD mimic also attenuated the increase in their expression levels. Glucose‐regulated protein 78 (GRP78, also known as binding immunoglobulin protein Bip) is the hallmark of ER stress 30, 31. Results of this study suggest that GRP78 expression after MnSOD treatment is not significantly different compared with the H2O2‐treated group, indicating that MnSOD does not disturb the homeostasis of the ER.
Sustained elevations and excessive calcium influx of cytosolic Ca2+ is considered to be a major cause of cell death 39, 40. ER stress, including upexpression of XBP1 and ER‐resident chaperone GRP78, would be triggered following cell exposure to calcium ionophores 41, 42. By releasing Ca2+ from ER reservoirs and taking up Ca2+ from the cytoplasm, the ER is responsible for cellular Ca2+ homeostasis 39. A steady Ca2+ level in the ER lumen is crucial not only for Ca2+ related signal transduction but also for protein synthesis 43. An interruption in Ca2+ level affects many ER functions, including protein folding, quality control and protein degradation 44. Excessive modification of cellular Ca2+ has been reported to result in ER stress and cell death 45, 46. Results of this study showed a remarkable increase in cytoplasm Ca2+ levels after H2O2 treatment because of oxidant‐induced (e.g. superoxide and hydroxyl radicals) calcium influx through voltage‐gated calcium channels 47. The MnSOD mimic effectively scavenges for cellular superoxide and hydroxyl radicals, thus contributing to the inhibition of early calcium overload state induced by oxidants. Long‐term Ca2+ levels normalized in the presence of the MnSOD mimic in the culture medium probably due to the inactivation of the influx of extracellular Ca2+ and possible ER stress, as evidenced by the levels of the ER stress hallmark GRP78. Our findings indicate that MnSOD may be a good treatment target because of its noninvolvement in Ca2+ homeostasis and severe ER stress.
This study demonstrates the neuroprotective properties of MnSOD in vitro and in vivo. (1) MnSOD, as a direct antioxidant, can significantly scavenge for intracellular and maintain ROS levels within the proper oxidative environment. (2) MnSOD is involved in mitochondrial ROS production and the mitochondrial protective role after permanent ischemia by inhibiting the release of mitochondrial cytochrome c to the cytosol, thereby preventing apoptosis. (3) MnSOD can reduce the expression of CHOP, which is a proapoptotic transcription factor induced by ER stress that mediates DNA damage. However, the specific mechanism through which MnSOD regulates mitochondrial functions, oxidative stress, and apoptosis, as well as MnSOD regulation, requires further investigation. Findings of this study indicate that MnSOD is an effective therapeutic target for cerebral ischemic stroke treatment.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (20901005), Beijing Nova Program (No. 2010010), and the National Basic Research Program of China (No. 2010CB12300).
References
- 1. Zemke D, Smith JL, Reeves MJ, Majid A. Ischemia and ischemic tolerance in the brain: an overview. Neurotoxicology 2004;25:895–904. [DOI] [PubMed] [Google Scholar]
- 2. Loh KP, Qi J, Tan BK, Liu XH, Wei BG, Zhu YZ. Leonurine protects middle cerebral artery occluded rats through antioxidant effect and regulation of mitochondrial function. Stroke 2010;41:2661–2668. [DOI] [PubMed] [Google Scholar]
- 3. Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke 2009;40:331–339. [DOI] [PubMed] [Google Scholar]
- 4. Fabian RH, DeWitt DS, Kent TA. In vivo detection of superoxideanion production by the brain using a cytochrome c electrode. J Cereb Blood Flow Metab 1995;15:242–247. [DOI] [PubMed] [Google Scholar]
- 5. Peters O, Back T, Lindauer U, et al. Increased formation of reactive oxygen species after permanent and reversible middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab 1998;18:196–205. [DOI] [PubMed] [Google Scholar]
- 6. Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 1999;22:391–397. [DOI] [PubMed] [Google Scholar]
- 7. Uyama O, Matsuyama T, Michishita H, Nakamura H, Sugita M. Protective effects of human recombinant superoxide dismutase on transient ischemic injury of CA1 neurons in gerbils. Stroke 1992;23:75–81. [DOI] [PubMed] [Google Scholar]
- 8. Young AR, Ali C, Duretête A, Vivien D. Neuroprotection and stroke: time for a compromise. J Neurochem 2007;103:1302–1309. [DOI] [PubMed] [Google Scholar]
- 9. Dunn DE, He DN, Yang P, Johansen M, Newman RA, Lo DC. In vitro and in vivo neuroprotective activity of the cardiac glycoside oleandrin from Nerium oleander in brain slice‐based stroke models. J Neurochem 2011;119:805–814. [DOI] [PubMed] [Google Scholar]
- 10. Lapchak PA, Schubert DR, Maher PA. Delayed treatment with a novel neurotrophic compound reduces behavioral deficits in rabbit ischemic stroke. J Neurochem 2011;116:122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haun SE, Kirsch JR, Helfaer MA, Kubos KL, Traystman RJ. Polyethylene glycol‐conjugated superoxide dismutase fails to augment brain superoxide dismutase activity in piglets. Stroke 1991;22:655–659. [DOI] [PubMed] [Google Scholar]
- 12. Batinić‐Haberle I, Rebouças JS, Spasojević I. Superoxide dismutase mimics: chemistry, pharmacology, and therapeutic potential. Antioxid Redox Signal 2010;13:877–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sheng H, Kudo M, Mackensen GB, Pearlstein RD, Crapo JD, Warner DS. Mice overexpressing extracellular superoxide dismutase have increased resistance to global cerebral ischemia. Exp Neurol 2000;163:392–398. [DOI] [PubMed] [Google Scholar]
- 14. Pineda JA, Aono M, Sheng H, et al. Extracellular superoxide dismutase overexpression improves behavioral outcome from closed head injury in the mouse. J Neurotrauma 2001;18:625–634. [DOI] [PubMed] [Google Scholar]
- 15. Didion SP, Kinzenbaw DA, Fegan PE, Didion LA, Faraci FM. Overexpression of CuZn‐SOD prevents lipopolysaccharide‐induced endothelial dysfunction. Stroke 2004;35:1963–1967. [DOI] [PubMed] [Google Scholar]
- 16. Li Y, Huang TT, Carlson EJ, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 1995;11:376–381. [DOI] [PubMed] [Google Scholar]
- 17. Lebovitz RM, Zhang H, Vogel H, et al. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase‐deficient mice. Proc Natl Acad Sci U S A 1996;93:9782–9787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim GW, Kondo T, Noshita N, Chan PH. Manganese superoxide dismutase deficiency exacerbates cerebral infarction after focal cerebral ischemia/reperfusion in mice: implications for the production and role of superoxide anions. Stroke 2002;33:809–815. [DOI] [PubMed] [Google Scholar]
- 19. Jung JE, Kim GS, Narasimhan P, Song YS, Chan PH. Regulation of Mn‐superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J Neurosci 2009;29:7003–7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maier CM, Hsieh L, Crandall T, Narasimhan P, Chan PH. Evaluating therapeutic targets for reperfusion‐related brain hemorrhage. Ann Neurol 2006;59:929–938. [DOI] [PubMed] [Google Scholar]
- 21. Faulkner KM, Liochev SI, Fridovich I. Stable Mn(III) porphyrins mimic superoxide dismutase in vitro and substitute for it in vivo . J Biol Chem 1994;269:23471–23476. [PubMed] [Google Scholar]
- 22. Serpa C, Schabauer J, Piedade AP, et al. Photoacoustic measurement of electron injection efficiencies and energies from excited sensitizer dyes into nanocrystalline TiO2 films. J Am Chem Soc 2008;130:8876–8877. [DOI] [PubMed] [Google Scholar]
- 23. Xia Q, Feng X, Yuan L, Wang K, Yang XD. Brain‐derived neurotrophic factor protects neurons from GdCl3‐induced impairment in neuron‐astrocyte co‐cultures. Sci China Chem 2010;53:2193–2199. [Google Scholar]
- 24. Feng XD, Xia Q, Yuan L, Yang XD, Wang K. Impaired mitochondrial function and oxidative stress in rat cortical neurons: implications for gadolinium‐induced neurotoxicity. Neurotoxicology 2010;31:391–398. [DOI] [PubMed] [Google Scholar]
- 25. Liesz A, Suri‐Payer E, Veltkamp C, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med 2009;15:192–199. [DOI] [PubMed] [Google Scholar]
- 26. Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 2002;298:1781–1785. [DOI] [PubMed] [Google Scholar]
- 27. Kabra D, Thiyagarajan M, Kaul CL, Sharma SS. Neuroprotective effect of 4‐amino 1, 8‐napthalimide, a poly (ADP ribose) polymerase inhibitor in middle cerebral artery occlusion induced focal cerebral ischemia in rat. Brain Res Bull 2004;62:425–433. [DOI] [PubMed] [Google Scholar]
- 28. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989;20:84–91. [DOI] [PubMed] [Google Scholar]
- 29. Borlongan CV, Sanberg PR. Elevated body swing test: a new behavioral parameter for rats with 6‐hydroxydopamine‐induced hemiparkinsonism. J Neurosci 1995;15:5372–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Feng XD, Xia Q, Yuan L, Huang HF, Yang XD, Wang K. Gadolinium triggers unfolded protein responses (UPRs) in primary cultured rat cortical astrocytes via promotion of an influx of extracellular Ca2+ . Cell Biol Toxicol 2011;27:1–12. [DOI] [PubMed] [Google Scholar]
- 31. Xia Q, Feng X, Huang H, Du L, Yang X, Wang K. Gadolinium‐induced oxidative stress triggers endoplasmic reticulum stress in rat cortical neurons. J Neurochem 2011;117:38–47. [DOI] [PubMed] [Google Scholar]
- 32. Ginsberg MD. The new language of cerebral ischemia. Am J Neuroradiol 1997;18:1435–1445. [PMC free article] [PubMed] [Google Scholar]
- 33. Loh KP, Huang SH, De Silva R, Tan BK, Zhu YZ. Oxidative stress: apoptosis in neuronal injury. Curr Alzheimer Res 2006;3:327–337. [DOI] [PubMed] [Google Scholar]
- 34. Sharma SS, Gupta S. Neuroprotective effect of MnTMPyP, a superoxide dismutase/catalase mimetic in global cerebral ischemia is mediated through reduction of oxidative stress and DNA fragmentation. Eur J Pharmacol 2007;561:72–79. [DOI] [PubMed] [Google Scholar]
- 35. Day BJ, Fridovich I, Crapo JD. Manganic porphyrins possess catalase activity and protect endothelial cells against hydrogen peroxide‐mediated injury. Arch Biochem Biophys 1997;347:256–262. [DOI] [PubMed] [Google Scholar]
- 36. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007;35:495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Benavides A, Pastor D, Santos P, Tranque P, Calvo S. CHOP plays a pivotal role in the astrocyte death induced by oxygen and glucose deprivation. Glia 2005;52:261–275. [DOI] [PubMed] [Google Scholar]
- 38. Katsoulieris E, Mabley JG, Samai M, Sharpe MA, Green IC, Chatterjee PK. Lipotoxicity in renal proximal tubular cells: relationship between endoplasmic reticulum stress and oxidative stress pathways. Free Radic Biol Med 2010;48:1654–1662. [DOI] [PubMed] [Google Scholar]
- 39. Abramov AY, Canevari L, Duchen MR. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci 2003;23:5088–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Verkhratsky A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev 2005;85:201–279. [DOI] [PubMed] [Google Scholar]
- 41. Li XA, Lee AS. Competitive inhibition of a set of endoplasmic reticulum protein genes (GRP78, GRP94, and ERp72) retards cell growth and lowers viability after ionophore treatment. Mol Cell Biol 1991;11:3446–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kudo T, Kanemoto S, Hara H, et al. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ 2008;15:364–375. [DOI] [PubMed] [Google Scholar]
- 43. Burdakov D, Petersen OH, Verkhratsky A. Intraluminal calcium as a primary regulator of endoplasmic reticulum function. Cell Calcium 2005;38:303–310. [DOI] [PubMed] [Google Scholar]
- 44. Michalak M, Robert Parker JM, Opas M. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium 2002;32:269–278. [DOI] [PubMed] [Google Scholar]
- 45. Hayashi T, Saito A, Okuno S, Ferrand‐Drake M, Dodd RL, Chan PH. Damage to the endoplasmic reticulum and activation of apoptotic machinery by oxidative stress in ischemic neurons. J Cereb Blood Flow Metab 2005;25:41–53. [DOI] [PubMed] [Google Scholar]
- 46. Shen C, Li Z, Yang X, Wang K. La3+ binds to BiP/GRP78 and induces unfolded protein response in HepG2 cells. Chem Biol Interact 2008;176:196–203. [DOI] [PubMed] [Google Scholar]
- 47. Josephson RA, Silverman HS, Lakatta EG, Stern MD, Zweier JL. Study of the mechanisms of hydrogen peroxide and hydroxyl free radical‐induced cellular injury and calcium overload in cardiac myocytes. J Biol Chem 1991;266:2354–2361. [PubMed] [Google Scholar]