

Summary
Aims
Dysfunction of glutamate uptake, largely mediated by the glutamate–aspartate transporter (GLAST), may lead to retinal cell apoptosis in diabetic retinopathy. The aim of this study is to examine how cell apoptosis and the expression level of GLAST in neural retina of a diabetic rat model are changed and whether the neuroretinal apoptosis could be ameliorated by the administration of glial cell line–derived neurotrophic factor (GDNF).
Methods
Diabetes was induced by intraperitoneal injection of streptozotocin (STZ) in Sprague–Dawley rats. GLAST protein expression levels were determined by Western blotting, whereas apoptosis of retinal neurons was evaluated by TUNEL staining. To assess the role of GDNF in ameliorating the STZ‐induced retinal changes, GDNF/GDNF with siRNA directed against GLAST was injected into the vitreous after STZ injection.
Results
In rat retinas 4 weeks after the onset of STZ‐induced diabetes, TUNEL‐positive cells were significantly increased, whereas GLAST levels were significantly reduced. Intraocular administration of GDNF at the early stage of diabetes remarkably increased the GLAST levels and decreased TUNEL‐positive signals in the retinas. These effects of GDNF were largely abolished by coadministration of GLAST siRNA.
Conclusions
GDNF, administrated at the early stage of diabetes, could rescue retinal cells from neurodegeneration by upregulating the expression of GLAST.
Keywords: Apoptosis, Diabetic retinopathy, Glial cell line–derived neurotrophic factor, Glutamate–aspartate transporter, Streptozotocin
Introduction
Diabetic retinopathy is a leading cause of vision impairment and blindness in the working‐age adults 1, 2, 3. There is much evidence demonstrating that in addition to a disturbance of the retinal vasculature, retinal neuronal and glial elements are also affected in this disease 4, 5, 6, 7. Specifically, degeneration of major retinal neurons has been reported in experimental and human diabetes 6, 8, 9, 10, 11, 12, 13, 14.
The level of glutamate, a major excitatory neurotransmitter, is markedly increased in the vitreous of patients with diabetic retinopathy 15, 16 and in the retinas of experimental diabetes 17, 18, suggesting that diabetic retinopathy may be associated with glutamate‐induced excitotoxicity 19. In the vertebrate retina, the glutamate–aspartate transporter (GLAST), which is exclusively expressed in Müller cells and astrocytes 20, plays a prominent role in the removal of glutamate from the synaptic clefts and thereby terminates glutamate signaling and protects neurons from the excitotoxic action of glutamate 19, 21, 22, 23. The activity of GLAST is greatly reduced in Müller cells, freshly isolated from streptozotocin (STZ)‐induced diabetic rats during the initial 13 weeks 24. Moreover, ganglion cell (GC) death is induced by suppressing GLAST 23.
Glial cell line–derived neurotrophic factor (GDNF) is a growth and survival factor for neuronal cells 25, 26, which is involved in various physiological processes by activating the high‐affinity ligand‐binding receptor GFRα‐1 (GDNFR‐α) 27 and the tyrosine kinase receptor Ret 25. In the retina, GDNF is localized to GCs, outer segments of photoreceptors and pigment epithelial cells 28, whereas GFRα‐1 is predominantly expressed in Müller cells 28, suggesting that Müller cells are a major target of GDNF. GDNF has been reported to stimulate photoreceptor survival in the retinal degeneration 1 (rd1) mouse model, an effect that may be mediated by Müller cells 29, 30, 31, 32, 33, 34. Furthermore, GDNF application decreases GC loss in rats after optic nerve transection by upregulating GLAST protein levels 35. Now that GLAST levels are reduced in diabetic rat retinas 24, it was therefore intriguing whether the increase in neuronal apoptosis under this condition is associated with the reduced GLAST levels and whether GDNF administration could rescue retinal cells from neurodegeneration in STZ‐induced diabetic rats by regulating GLAST expression, just like that observed after optic nerve transection.
Materials and Methods
Animals and Induction of Diabetes
All experimental procedures described here were in accordance with the National Institutes of Health guidelines for the Care and Use of Laboratory Animals and the guidelines of Fudan University on the ethical use of animals.
Adult male Sprague‐Dawley rats (220 ± 20 g) were provided by SLAC Laboratory Animal Co. Ltd. (Shanghai, China) and were housed in standard 12‐h light/dark schedule with food and water ad libitum for at least 7 days before experimentation. Diabetes was induced by a single intraperitoneal (i.p.) injection of STZ (Sigma, St. Louis, MO, USA) (65 mg/kg), freshly dissolved in 0.1 M citrate buffer (pH 4.2), in the morning after an overnight fast. Age‐matched normal rats were injected with an equal volume of citrate buffer as control group. Blood glucose concentrations of the injected animals were first checked 2 and 3 days after the injection and weekly thereafter, using a glucometer (Roche Diagnostics GmbH, Sandhofer, Mannheim, Germany), and animals whose glucose concentrations exceed 16.7 mM were selected for the experiments 10. The final measurements of blood glucose levels were made just before animals were killed.
Intraocular Administration of GDNF
Referring to previous studies 13, 28, 35, multiple intraocular injections of GDNF (0.5 μg/each time) in 3 μL of 0.1 M sterile phosphate‐buffered saline (PBS) or vehicle, for a total of 3 times, were given to one eye of an animal at random every 3 days, beginning 2 weeks after the i.p. injection of STZ. The animals were under anesthesia with sodium pentobarbital (4%, w/v, 0.2 mL/100 g body weight) and topical anesthesia with a drop of 2% lidocaine applied to the eyes. An equal volume of vehicle (0.1 M PBS) was injected into the vitreous space of the other eye as control.
To determine whether GLAST may be involved in effects of GDNF on neurons, intraocular injections of GDNF were made along with small interfering RNAs (siRNAs) directed against GLAST. Sequences of the siRNAs used in this study were as follows: GLAST (123) 5′‐AGAGGAGAUUUCGCUUUCU‐3′ and GLAST (176) 5′‐AAAGCCGUAUCAGCUGAUU‐3′. The rats received intravitreous injections of GDNF of 0.5 μg (dissolved in 1.5 μL of sterile PBS), along with 1.5 μL of an siRNA solution containing 2 μg of GLAST siRNA complexed in Lipofectamine/Opti‐MEM (Invitrogen, Carlsbad, CA, USA) for each time. The injections were given every 3 days, beginning 2 weeks after the injection of STZ in diabetic rats. Control injections contained firefly luciferase siRNA (Luc siRNA) (5′‐CGUACGCGGAAUACUUCGA‐3′) substituted for GLAST siRNA. Retinas were dissected 4 weeks after the i.p. injection of STZ. Any animal with lens damage or visible vitreous hemorrhage was rejected from the study.
Determination of GLAST Levels in Retina
Glutamate–aspartate transporter levels were determined by Western blotting. For procedures of Western blot analysis, refer to the study by 36, with some modifications. Nine animals were assigned to each group. In brief, three rat retinas (as one sample) were freshly isolated and sonicated in lysis buffer containing 1 M Tris–HCl (pH 7.5), 0.5 M EDTA, 10% SDS, NP‐40 (nonidet p‐40), sodium deoxycholate, CHAPS, and Triton X‐100, and a protease inhibitor cocktail (Sigma). Protein concentrations were determined using a standard bicinchoninic acid (BCA) assay kit (Pierce Biotechnology, Rockford, IL, USA). The extracted samples (50 μg, 20 μL in volume) were loaded, subjected to 10% SDS‐PAGE, and then transferred onto PVDF membranes (Millipore Co. Bedford, MA, USA). Non‐specific binding was blocked for 2 h at room temperature in blocking buffer consisting of 20 mM Tris–HCl, pH 7.4, 137 mM NaCl, 0.1% Tween‐20, and 3% bovine serum albumin. The blot was incubated with the anti‐GLAST antibody (1:20000 dilution, Chemicon, Temecula, CA, USA) overnight at 4°C, followed by horseradish peroxidase–conjugated donkey anti‐guinea pig IgG (1:2000 dilution, Jackson Labs, West Grove, PA, USA) for 2 h at room temperature, and finally visualized by enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ, USA). The experiments were performed in triplicates. For the quantification of Western signals, X‐ray films with blotting bands were scanned, and the intensity of the bands was quantified by Image‐Pro analysis system.
To avoid any possible effects of circadian rhythm, all experiments were performed in the morning (09:00–11:00). As GLAST expression may be regulated by dark/light, as shown in bullfrog retina 37, we first examined changes in GLAST expression in rat retina as a function of time in the dark. The animals were first light‐adapted by a constant white light with an intensity of 1600 lux at the cornea level for 2 h and then left in the dark for different periods of time (30, 60, 90, 120 min). Western blotting revealed that the GLAST level reached a peak in retinas dark‐adapted for 90 min, but declined when retinas were dark‐adapted for longer periods (>120 min). As a result, all retinas used in this study were prepared from rats dark‐adapted for 90 min.
Tissue Preparation
After anesthetization, animals were perfused transcardially with 0.9% saline, followed by 4% paraformaldehyde in 0.1 M PB (pH 7.4). The retinas and retinal sections were prepared as previously described with some modifications 38, 39. The isolated eyecups or retinas were immediately postfixed in 4% paraformaldehyde and chilled sequentially in 10% (w/v), 20%, and 30% sucrose in 0.1 M PB at 4°C. The eyecups were then embedded in OCT (Miles Inc., Elkhart, IN, USA), frozen by liquid nitrogen, and sectioned vertically at 14 μm thickness on a freezing microtome (Leica, Nussloch, Germany). The sections were mounted on gelatin–chromium‐coated slides. The isolated retinas and the retinal sections were treated with a modified terminal transferase dUTP nick‐end labeling (TUNEL)‐FITC assay and immunocytochemistry.
In situ Detection of DNA Fragmentation by TUNEL Assay
TUNEL‐FITC was performed in whole isolated retinas or in 14‐μm sections using the DeadEnd™ Fluorometric TUNEL Detection System kit (Promega, Madison, WI, USA), following the manufacturer's instruction. The retinas or sections were counterstained with DAPI (Vector Lab, Burlingame, CA, USA) for labeling nuclei of neurons. The TUNEL‐labeled retinas were whole‐mounted on slides, with the ganglion cell layer (GCL) upward. To distinguish between autofluoresced structures and TUNEL‐positive ones, the tissue was sequentially examined with the rhodamine and FITC filters. Autofluorescent structures were visible under both filters, whereas TUNEL‐positive cells were detectable only with the FITC filter. The tissue was further examined with the UV filter (for DAPI) to confirm the colocalization of TUNEL signals with the cell nuclei. The number of TUNEL‐positive cells was counted in whole‐mounted retinas. Total number of apoptotic cells of each retina, which was scanned at different focal planes, was recorded using a computer‐based Neurolucida system (Microbrightfield, Williston, VT, USA) with a 20× objective on an Olympus microscope (Olympus Corporation, Tokyo, Japan). Five animals were used for each group.
Immunocytochemistry in Combination with TUNEL‐FITC Labeling
To determine the identity of neurons undergoing apoptosis, immunostaining with specific markers for different types of retinal neurons, followed by TUNEL‐FITC labeling, was further performed in diabetic retinas. For the procedures of immunocytochemistry, refer to our previous work 36, 39. Briefly, the whole retinas or sections were preincubated as usual. Rabbit antirecoverin (RC) polyclonal antibody (1:1000 dilution, Chemicon) and mouse antisyntaxin clone HPC‐1 (HPC‐1) monoclonal antibody (1:2000 dilution, Sigma) were, respectively, used for labeling photoreceptors and amacrine cells (ACs). Goat anti‐Brn3a polyclonal antibody (1:400 dilution, Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used for labeling GCs. Immunostaining experiments were carried out by incubating with the corresponding primary antibody for 5 days (for whole retinas) or 3 days (for sections) at 4°C in a medium containing 3% normal donkey serum, 0.2% Triton X‐100, and 0.5% bovine serum albumin in PBS (DT‐PBS). Binding sites of the primary antibody were revealed by incubating with the secondary antibody for 2 h at room temperature in DT‐PBS. Secondary antibodies used in this work were as follows: Alexa Fluor 555‐conjugated donkey anti‐rabbit IgG for labeling RC; Alexa Fluor 555‐conjugated donkey anti‐mouse IgG for HPC‐1; and Alexa Fluor 555‐conjugated donkey anti‐goat IgG for Brn3a (all 1:200 dilution, Invitrogen). After completion of the immunostaining, TUNEL‐FITC labeling was further carried out. Five animals were used for each group.
Images were taken with a digital camera (Zeiss AxioCam HRc, Carl Zeiss Inc., Oberkochen, Germany) mounted on a Zeiss Axioskop 40 microscope using a 20× objective.
Statistical Analysis
Statistically significant differences between the data obtained in different groups were detected using Student's t‐test, with values of P < 0.05 being taken as significant. All data were presented as mean ± SEM.
Results
Characterization of Diabetic Rat
Table 1 shows the blood glucose levels and body weights for age‐matched control and diabetic rats treated with different experimental manipulations in this study. The average blood glucose levels in all groups of diabetic rats were significantly higher than the control levels (P < 0.001). No significant difference in average blood glucose levels was observed between the diabetic groups treated with different experimental manipulations. The rats with a blood glucose level lower than 16.7 mM were excluded from this work. All diabetic rats gained significantly less weights than their respective control rats (P < 0.001).
Table 1.
Average blood glucose levels and weights of control and diabetic rats treated with different experimental manipulations
| Duration of diabetes | Treatment group | n | Blood glucose (mM) | Weight (g) |
|---|---|---|---|---|
| 4 week | Control | 33 | 5.52 ± 0.37 | 419.53 ± 4.59 |
| 4 week | STZ | 35 | 31.41 ± 1.02*** | 326.72 ± 3.67*** |
| 4 week | STZ + Vehicle | 16 | 30.14 ± 2.04*** | 319.41 ± 2.98*** |
| 4 week | STZ + GDNF | 16 | 29.83 ± 1.64*** | 333.50 ± 1.99*** |
| 4 week | STZ + GLAST siRNA(123) | 11 | 32.33 ± 1.16*** | 299.86 ± 3.66*** |
| 4 week | STZ + GLAST siRNA(176) | 11 | 30.06 ± 1.57*** | 304.71 ± 3.01*** |
| 4 week | STZ + Con Luc siRNA | 11 | 31.30 ± 0.89*** | 310.49 ± 2.63*** |
| 4 week | STZ + GDNF + GLAST siRNA (123) | 9 | 32.25 ± 1.83*** | 328.58 ± 2.07*** |
| 4 week | STZ + GDNF + GLAST siRNA (176) | 14 | 28.86 ± 2.11*** | 336.93 ± 1.62*** |
| 4 week | STZ + GDNF + Con Luc siRNA | 14 | 29.53 ± 0.97*** | 349.03 ± 4.35*** |
Data are expressed as the mean ± SEM. n, number of animals; ***P < 0.001 versus age‐matched control rats.
TUNEL Analysis of Diabetic Retinas
We examined changes in number of apoptotic cells in whole‐mounted retinas in diabetic animals by TUNEL staining analysis. The spatial distribution of TUNEL‐positive nuclei was mapped using Neurolucida system with an Olympus microscope, and the data were expressed as the number of TUNEL‐positive cells per 0.5 cm2 of retina. As shown in Figure 1A, the control rat had few TUNEL‐positive cells in the whole mount. In contrast, the diabetic rat exhibited much more TUNEL‐positive cells (Figure 1B), and these cells appeared randomly in all parts of the retina, mostly as individual cells or as loosely associated groups at different focal depths in the whole mount, noted by focusing through the tissue. Figure 1E shows the microphotographs of a whole‐mounted retina of diabetic rat counterstained with DAPI after TUNEL staining. As evident from the Figure 1E3, a merged image of E1 and E2, the TUNEL‐positive cell (E1) is also labeled by DAPI (E2). Statistical analysis revealed that the average number of TUNEL‐positive cells in diabetic rats was 73.4 ± 5.91 (Figure 1F), significantly higher than that (11.1 ± 0.81) in control ones (P < 0.001, n = 5). We also counted the number of TUNEL‐positive cells in the outer and inner retina separately in control and diabetic retinas. The outer retina, including the outer nuclear layer (ONL), the outer plexiform layer (OPL), and horizontal cells, accounts for ~37.5% of the thickness of the entire neural retina, whereas the inner retina, including the inner nuclear layer (INL) (except horizontal cells), the inner plexiform layer (IPL), and the GCL, accounts for the remaining ~62.5%. The average number of TUNEL‐positive cells in diabetic rats was 47.8 ± 6.56 in the outer retina (vs. 6.84 ± 1.37 in control rats, P < 0.01) and 25.5 ± 5.28 in the inner retina (vs. 4.30 ± 0.88 in control rats, P < 0.05).
Figure 1.
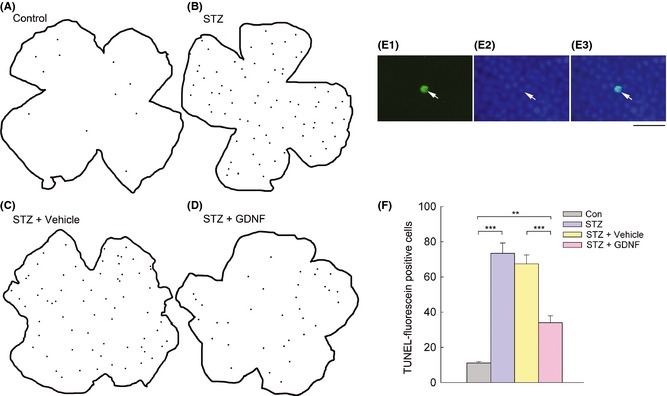
TUNEL analysis revealing the changes in apoptotic cells in rat retinas under different conditions. (A–D) Neurolucida drawings showing TUNEL‐FITC signal distribution in four representative whole‐mounted retinas, respectively, from age‐matched control (A), diabetic (for 4 weeks) (B), vehicle‐treated diabetic (C), and GDNF‐treated diabetic rats (D). Black spots within the retinal contours indicate all individual TUNEL‐positive cells that were seen at different focal planes. (E) Microphotographs of the whole‐mounted diabetic retina, labeled by TUNEL (E1) and DAPI (E2). (E3) is the merged image of (E1) and (E2). The TUNEL signal was well overlapped with the DAPI‐labeled nucleus. Scale bar, 20 μm. (F) Histograms showing quantitative analysis of the average number of TUNEL‐positive cells from different groups. Note that more TUNEL‐positive cells in diabetic retinas (STZ) were observed (***P < 0.001 vs. control (Con), n = 5). GDNF administration (STZ + GDNF) resulted in a significant decrease in the number of TUNEL‐positive cells in diabetic retinas (***P < 0.001 vs. vehicle‐treated diabetic retinas [STZ + Vehicle]), even though the number was still higher than that in control retinas (**P < 0.01).
To determine the identity of neurons undergoing apoptosis, TUNEL‐FITC staining was performed, in combination of immunocytochemical labeling with specific markers for photoreceptors, ACs and GCs in diabetic retinas. Figure 2A1 and A2 shows micrographs of a whole‐mounted retina double‐stained by the antibody against RC, a specific photoreceptor marker 40, 41, and TUNEL. At the focal depth of the ONL, at which photoreceptors are located, RC immunoreactivity was mainly localized to the membranes of photoreceptors (Figure 2A1). Figure 2A2 shows the image of the same retina stained by TUNEL. As shown in the merged image (Figure 2A4) of Figure 2A1, A2, and A3, the corresponding DAPI image of A1, it was clear that some RC‐positive photoreceptors showed TUNEL‐positive signals, and these signals were well overlapped with the DAPI‐labeled nuclei (arrows). TUNEL was also carried out after immunostaining against HPC‐1, a specific marker for ACs 42, 43, in diabetic retinas. HPC‐1 labeled somata and processes of ACs, with stronger immunostaining in the membranes and in the processes located in the IPL (Figure 2B1, D3). TUNEL signals were observed in some HPC‐1 labeled ACs (arrows in Figure 2B2, B4), which were overlapped with the DAPI‐labeled nuclei (arrow in Figure 2B3). The specific ganglion cell marker Brn3a 44, 45 was used for double staining with TUNEL. As shown in Figure 2C1–C4, TUNEL staining was seen in some of Brn3a‐labeled GCs (arrows). The vertical sections were double‐stained by TUNEL (green), along with RC, HPC‐1, and Brn3a (red), in which retinal layers could be seen clearly. TUNEL‐positive signals were present in some photoreceptors (arrows in Figure 2D1, D2), ACs (arrows in Figure 2D3, D4), and GCs (arrows in Figure 2D5, D6). All these were consistent with the results obtained in the whole‐mounted retinas, which demonstrated that TUNEL‐positive cells were seen at different focal depths.
Figure 2.
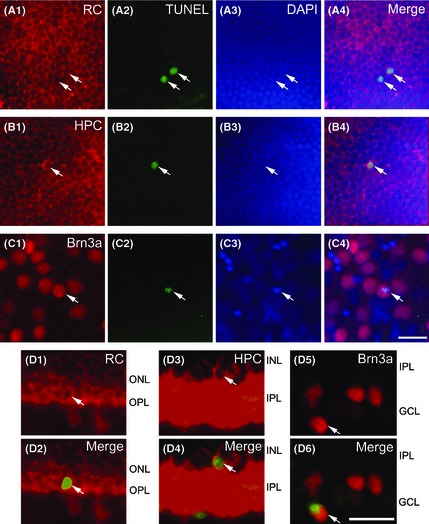
TUNEL staining of several types of retinal neurons in diabetic rat retinas. (A1–C1) Microphotographs of whole‐mounted retinas, showing the immunostaining (red) with RC (A1), HPC (B1), and Brn3a (C1). (A2–C2) Corresponding images of TUNEL signals (green) of A1–C1. (A3–C3) Counterstained images with DAPI (blue). (A4–C4) Merged images of the three showing staining by RC‐, HPC‐, and Brn3a, and TUNEL and DAPI for corresponding retinas. Arrowheads indicate some of triple‐labeled cells. Note that TUNEL‐positive signals were observed in some RC‐positive photoreceptors (A1, A2, A4), HPC‐labeled ACs (B1, B2, B4), and Brn3a‐positive GCs (C1, C2, C4). TUNEL signals were well overlapped with the DAPI images (A3–C3). (D1, D3, D5) Micrographs of retinal vertical sections immunostained with RC, HPC or Brn3a. Double‐labeled elements (red for RC, HPC, and Brn3a, and green for TUNEL‐positive signals) appear yellowish. (D2, D4, D6) TUNEL signals were seen in some photoreceptors (D2), ACs (D4), and GCs (D6). ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer; scale bar: 20 μm.
Reduced GLAST Protein Levels in Diabetic Retinas
Changes in GLAST protein levels in diabetic retinas were measured by Western blot analysis, and some representative results are shown in Figure 3A. For each experiment, three retinas were used for analysis. The anti‐GLAST antibody detected two bands at ~66 kDa, as described previously 35, 46, 47. These two bands represent different glycosylated forms of GLAST 35, 46, 47, which have no functional significance for transporter activity 47. The GLAST protein level in the diabetic rat retina was evidently lower than that in the control retina. For statistical analysis, GLAST protein levels were standardized to β‐actin levels in the same lanes, with the mean values for the GLAST/β‐actin ratios in control retinas being set as 1. As shown in Figure 3B, the mean value of GLAST protein levels in diabetic retinas was reduced to 65.3% of control (P < 0.01, n = 3).
Figure 3.
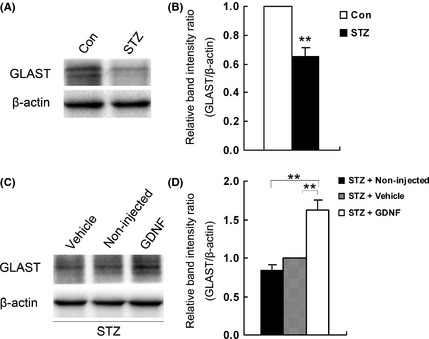
Western blot analysis of GLAST protein levels under different conditions. (A) Representative immunoblots comparing GLAST levels in age‐matched control and STZ‐induced diabetic (for 4 weeks) retinas. Anti‐GLAST antibody revealed two bands at ~66 kDa. (B) Densitometric analysis revealing a significant decrease in GLAST protein levels in diabetic animals (**P < 0.01, n = 3, 9 rats). β‐actin served as loading control, and the mean value for the GLAST/β‐actin ratio in normal retinas was set as 1. (C) Representative immunoblots comparing GLAST levels in GDNF‐treated, vehicle‐treated, and non‐injected diabetic rat retinas. (D) Densitometric analysis revealing that GDNF administration significantly increased GLAST levels in diabetic retinas (**P < 0.01 vs. vehicle‐treated diabetic group, n = 3, 9 rats). The mean value for the GLAST/β‐actin ratio in the vehicle‐treated group was set as 1. Note that the GLAST levels in non‐ and vehicle‐injected diabetic retinas were similar.
Effects of GDNF on GLAST Protein Levels and Apoptotic Status in Diabetic Retinas
Effects of GDNF administration on GLAST protein levels in diabetic retinas were first evaluated by Western blotting. As shown in Figure 3C, the GLAST level in the vehicle‐treated diabetic retinas was not significantly different from that in the non‐injected diabetic one, but multiple intraocular injections of GDNF greatly increased the GLAST level in the diabetic retina. On average, the GLAST levels in diabetic retinas, as expressed in GLAST/β‐actin, were considerably increased to 1.62 ± 0.13 (vs. 1.0 for the vehicle‐injected group) (Figure 3D, P < 0.01).
The possible neuroprotective role of GDNF was assessed by examining whether GDNF administration could reverse the apoptotic status in diabetic retinas. A representative result is illustrated in Figure 1D. Compared with the vehicle‐treated diabetic retina (Figure 1C), TUNEL‐positive cells in the diabetic retina were largely reduced in number following GDNF administration. Statistical analysis revealed that the average number of TUNEL‐positive cells was reduced from 67.4 ± 5.04 in vehicle‐treated rats to 34.0 ± 3.89 in GDNF‐treated rats (P < 0.001, n = 5, Figure 1F). It should be indicated that the number of apoptotic cells in diabetic retinas with GDNF administration was close to, but still higher than that in control retinas (11.1 ± 0.81, P < 0.01) (Figure 1F, also compare Figure 1D with Figure 1A). The average numbers of TUNEL‐positive cells observed in the outer and inner retina were both reduced in the GDNF‐treated group (20.8 ± 4.69 in the outer retina, vs. 44.8 ± 6.76 for the vehicle‐treated group; 13.2 ± 3.85 in the inner retina, vs. 22.7 ± 5.84 for the vehicle‐treated group).
GLAST siRNA Administration Diminishes the Effect of GDNF
We further examined how the GDNF effect could be changed by RNA interference which is supposed to downregulate the GDNF‐increased GLAST levels. We first determined the knockdown efficiency of GLAST siRNAs by measuring GLAST levels 7 days after final siRNA injection. Injections of Luc siRNA did not change the GLAST mRNA (Figure 4A) and protein levels (Figure 4B) in diabetic retinas. However, both GLAST mRNA (Figure 4A) and protein levels (Figure 4B) became obviously lower following GLAST siRNA injections, with GLAST siRNA (176) exhibiting a higher knockdown efficiency than that of GLAST siRNA (123). Effects of GLAST siRNAs on GLAST levels were further tested in GDNF‐treated diabetic retinas, and a representative result is shown in Figure 4C. The enhanced GLAST level by GDNF was dramatically reduced following GLAST siRNA (176) coadministration. Figure 4D shows the statistical results, in which the ratio of GLAST/β‐actin of the vehicle‐injected diabetic group was set as 1. The relative GLAST level in the GDNF + GLAST siRNAs (176) of the diabetic group was reduced to 0.23 ± 0.02 (P < 0.01, n = 3). Administration of GLAST siRNA (123) yielded similar results, but with a lower knockdown efficiency (data not shown for presentation clarity). These results suggest that intraocular injection of GLAST siRNA (176) could effectively suppress the upregulated GLAST induced by GDNF.
Figure 4.
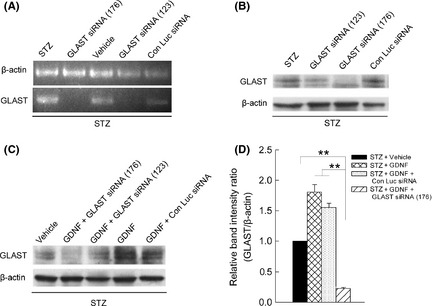
Suppression of GLAST expression by siRNA administration. (A–B) Representative RT‐PCR data and immunoblots showing the changes in GLAST mRNA (A) and protein levels (B) in retinas, treated with STZ alone and along with GLAST siRNA (123)/(176) and control luciferase siRNA (Con Luc siRNA). (C) Representative immunoblots showing the changes in GLAST levels in STZ‐induced diabetic retinas, treated by GDNF alone and along with siRNA (176)/(123) or Con Luc siRNA. (D) Densitometric analysis showing that the GDNF‐induced increase in GLAST levels in diabetic retinas was significantly inhibited by coadministration of GLAST siRNAs (176) (**P < 0.01 vs. GDNF‐treated diabetic or GDNF + Con Luc siRNA‐treated diabetic groups, n = 3, 9 rats). GLAST/β‐actin ratio in vehicle‐treated diabetic retinas was set as 1. Note that coadministration with control Luc siRNA had no effect.
Administration of GLAST siRNA (176) also modified the GDNF‐induced apoptosis changes in diabetic retinas. By coadministrating GLAST siRNA (176) with GDNF, the number of TUNEL‐positive neurons in diabetic retinas (62.5 ± 4.67) was much higher than that in diabetic rats treated only with GDNF (30.0 ± 4.25) and that for GDNF + Con Luc siRNA‐treated diabetic ones (26.5 ± 3.07) (P < 0.001) (Figure 5).
Figure 5.
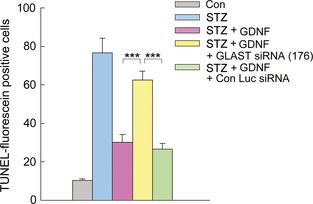
Quantitative analysis of apoptotic cell numbers in whole‐mounted retinas under different conditions. Bar chart showing that the GDNF‐induced decrease in number of TUNEL‐positive neurons was antagonized by coadministration of GLAST siRNA (176). Note that coinjection of GLAST siRNA (176) and GDNF resulted in a significant increase in the number of TUNEL‐positive cells in diabetic retinas (***P < 0.001 vs. GDNF‐treated diabetic or GDNF + Con Luc siRNA‐treated diabetic retinas,n = 5).
Taken together, intraocular administration of GDNF effectively rescues retinal cells from neurodegeneration in STZ‐induced diabetic rats, which could be largely due to GDNF‐induced upregulation of GLAST.
Discussion
In the present work, we demonstrated that the number of apoptotic cells was significantly increased in whole‐mounted rat retinas 4 weeks after the onset of STZ‐induced diabetes, with more apoptotic cells (65.2%) in the outer retina and relatively lower cells (34.8%) in the inner retina. This result is consistent with the previous results obtained in whole‐mounted retinas 8, 11 and in retinal sections of STZ‐induced diabetic rats and mice 12, 48, 49. It has been emphasized that whole‐mounted retina preparations could be more appropriate for quantifying retinal neuron apoptosis shown by TUNEL staining at the early stage of diabetes when only a small number of apoptotic cells exists 8.
Neuroexcitotoxicity due to excessive synaptic glutamate, among others, may be a cause of degeneration of retinal neurons in diabetes. Elevated vitreoretinal glutamate levels have been reported in both patients with diabetes and experimental diabetes 15, 16, 17, 18. This could be due to the inhibition and/or deficiency of GLAST, which is mainly localized to retinal Müller cells 20 and removes glutamate from extracellular space 19, 21, 22, 23. In this work, we showed that GLAST protein levels, determined in retinas from rats dark‐adapted for 90 min when GLAST expression reached a peak, showed a big reduction (Figure 3A, B) after 4 weeks of STZ diabetes. This is consistent with a recent work showing that GLAST expression was reduced in retinas of rats 4, 8, and 12 weeks after STZ‐induced diabetes 50. Functionally, it was also shown 24 that a reduced activity of GLAST in rat Müller cell was detected after 4 weeks of diabetes.
Now that downregulation and/or dysfunction of GLAST may lead to degeneration of retinal neurons in STZ‐induced diabetic retinas, administration of GDNF that has been shown to increase GLAST levels in rMC Müller cell line and in vivo 35 is expected to ameliorate cell apoptosis during diabetes. This supposition is supported by the major finding of this work that GDNF administration, which induced a significant increase in GLAST levels (Figure 3C, D), reduced the total number of TUNEL‐positive cells in STZ‐induced diabetic rat retinas (Figure 1D, F). We further demonstrated that the neuroprotective effects of GDNF were largely blocked (Figure 5) when the expression of GLAST in diabetic rats was suppressed by GLAST siRNA (176). As shown in Figure 5, in the presence of GLAST siRNA (176), GDNF‐induced reduction in apoptotic cell number in diabetic retinas became much less significant. These results suggest that the GDNF effects reported in this work were largely mediated by upregulation of GLAST expression, which could remove glutamate from extracellular space more effectively, thereby reducing neuroexcitotoxicity due to excessive glutamate. It is noteworthy that, although much compromised, the apoptotic cell number obtained in STZ‐induced diabetic retinas was still higher than the normal level after GDNF treatment. This suggests that factors other than neuroexcitotoxicity, such as insulin deprivation, inflammation, oxidative stress, and exposure to advanced glycation end products 5, 51, could be also involved in the degeneration of retinal neurons during diabetes.
How GLAST expression was upregulated by GDNF in the retina is poorly understood. It is known that GDNF exerts its action mainly through the high‐affinity ligand‐binding receptor GFRα‐1 27 and tyrosine kinase receptor Ret 25. Müller cells may be a major target for GDNF, as evidenced by GLAST immunoreactivity for GFRα‐1 being mostly localized to these cells 28. The intracellular signal transduction pathways for regulation of GLAST by GDNF, as shown by a study conducted in rMC‐1 cells and in vivo, involve activation of phosphoinositide‐3 kinase (PI3K) and modulation by Src kinase 35. While the precise mechanism for GDNF‐induced regulation of GLAST in STZ‐induced diabetic retinas remains to be further explored, the data presented in the present work provide evidence, suggesting that GFRα‐1 may be a potential target for ameliorating retinal damages during diabetes.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by grants from the National Program of Basic Research sponsored by the Ministry of Science and Technology of China (2011CB504602), the National Natural Science Foundation of China (31171055, 31070967, 30930034, 31121061), and the 211 Project sponsored by the Ministry of Education of China.
References
- 1. Merimee TJ. Mechanisms of disease‐diabetic retinopathy‐a synthesis of perspectives. N Engl J Med 1990;322:978–983. [DOI] [PubMed] [Google Scholar]
- 2. Klein R, Klein BEK. Vision Disorders in Diabetes. National Diabetes Data Group. Diabetes in America. Bethesda, MD: National Institutes of Health; NIH publication No. 95‐1468;1995:293–337. [Google Scholar]
- 3. Sato S, Yamamoto T, Yamashita H. Diabetic retinopathy‐state of arts of diagnosis and treatment. Nihon Rinsho 2006;3:161–168. [PubMed] [Google Scholar]
- 4. Barber AJ. A new view of diabetic retinopathy: a neurodegenerative disease of the eye. Prog Neuropsychopharmacol Biol Psychiatry 2003;27:283–290. [DOI] [PubMed] [Google Scholar]
- 5. Bringmann A, Pannicke T, Grosche J, et al. Müller cells in the healthy and diseased retina. Prog Retin Eye Res 2006;25:397–424. [DOI] [PubMed] [Google Scholar]
- 6. Kern TS, Barber AJ. Retinal ganglion cells in diabetes. J Physiol 2008;586:4401–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fletcher EL, Downie LE, Ly A, et al. A review of the role of glial cells in understanding retinal disease. Clin Exp Optom 2008;91:67–77. [DOI] [PubMed] [Google Scholar]
- 8. Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Penn State Retina Res G. Neural apoptosis in the retina during experimental and human diabetes ‐ Early onset and effect of insulin. J Clin Invest 1998;102:783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lieth E, Gardner TW, Barber AJ, Antonetti DA. Penn State Retina Res G. Retinal neurodegeneration: early pathology in diabetes. Clin Experiment Ophthalmol 2000;28:3–8. [DOI] [PubMed] [Google Scholar]
- 10. Aizu Y, Oyanagi K, Hu JG, Nakagawa H. Degeneration of retinal neuronal processes and pigment epithelium in the early stage of the streptozotocin‐diabetic rats. Neuropathology 2002;22:161–170. [DOI] [PubMed] [Google Scholar]
- 11. Park SH, Park JW, Park SJ, et al. Apoptotic death of photoreceptors in the streptozotocin‐induced diabetic rat retina. Diabetologia 2003;46:1260–1268. [DOI] [PubMed] [Google Scholar]
- 12. Martin PM, Roon P, Van Ells TK, Ganapathy V, Smith SB. Death of retinal neurons in streptozotocin‐induced diabetic mice. Invest Ophthalmol Vis Sci 2004;45:3330–3336. [DOI] [PubMed] [Google Scholar]
- 13. Seki M, Tanaka T, Nawa H, et al. Involvement of brain‐derived neurotrophic factor in early retinal neuropathy of streptozotocin‐induced diabetes in rats ‐ Therapeutic potential of brain‐derived neurotrophic factor for dopaminergic amacrine cells. Diabetes 2004;53:2412–2419. [DOI] [PubMed] [Google Scholar]
- 14. Fletcher EL, Phipps JA, Ward MM, Puthussery T, Wilkinson‐Berka JL. Neuronal and glial cell abnormality as predictors of progression of diabetic retinopathy. Curr Pharm Des 2007;13:2699–2712. [DOI] [PubMed] [Google Scholar]
- 15. Ambati J, Chalam KV, Chawla DK, et al. Elevated gamma‐aminobutyric acid, glutamate, and vascular endothelial growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch Ophthalmol 1997;115:1161–1166. [DOI] [PubMed] [Google Scholar]
- 16. Deng J, Wu DZ, Gao R. Detection of glutamate and gamma‐aminobutyric acid in vitreous of patients with proliferative diabetic retinopathy. Yan ke xue bao 2000;16:199–202. [PubMed] [Google Scholar]
- 17. Lieth E, Barber AJ, Xu BY, et al. Penn State Retina Res G. Glial reactivity and impaired glutamate metabolism in short‐term experimental diabetic retinopathy. Diabetes 1998;47:815–820. [DOI] [PubMed] [Google Scholar]
- 18. Kowluru RA, Engerman RL, Case GL, Kern TS. Retinal glutamate in diabetes and effect of antioxidants. Neurochem Int 2001;38:385–390. [DOI] [PubMed] [Google Scholar]
- 19. Pulido JE, Erie JC, Arroyo J, Bertram K, Lu M‐J, Shippy SA. A role for excitatory amino acids in diabetic eye disease. Exp Diabetes Res 2007;2007:36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rauen T, Rothstein JD, Wassle H. Differential expression of three glutamate transporter subtypes in the rat retina. Cell Tissue Res 1996;286:325–336. [DOI] [PubMed] [Google Scholar]
- 21. Rauen T, Taylor WR, Kuhlbrodt K, Wiessner M. High‐affinity glutamate transporters in the rat retina: a major role of the glial glutamate transporter GLAST‐1 in transmitter clearance. Cell Tissue Res 1998;291:19–31. [DOI] [PubMed] [Google Scholar]
- 22. Pow DV, Barnett NL. Changing patterns of spatial buffering of glutamate in developing rat retinae are mediated by the Muller cell glutamate transporter GLAST. Cell Tissue Res 1999;297:57–66. [DOI] [PubMed] [Google Scholar]
- 23. Vorwerk CK, Naskar R, Schuettauf F, et al. Depression of retinal glutamate transporter function leads to elevated intravitreal glutamate levels and ganglion cell death. Invest Ophthalmol Vis Sci 2000;41:3615–3621. [PubMed] [Google Scholar]
- 24. Li Q, Puro DG. Diabetes‐induced dysfunction of the glutamate transporter in retinal Müller cells. Invest Ophthalmol Vis Sci 2002;43:3109–3116. [PubMed] [Google Scholar]
- 25. Trupp M, Arenas E, Fainzilber M, et al. Functional receptor for GDNF encoded by the c‐ret proto‐oncogene. Nature 1996;381:785–789. [DOI] [PubMed] [Google Scholar]
- 26. Lin LFH, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF ‐ a glial‐cell line derived neurotrophic factor for midbrain dopaminergic‐neurons. Science 1993;260:1130–1132. [DOI] [PubMed] [Google Scholar]
- 27. Jing SQ, Wen DZ, Yu YB, et al. GDNF‐induced activation of the Ret protein tyrosine kinase is mediated by GDNFR‐alpha, a novel receptor for GDNF. Cell 1996;85:1113–1124. [DOI] [PubMed] [Google Scholar]
- 28. Koeberle PD, Ball AK. Neurturin enhances the survival of axotomized retinal ganglion cells in vivo: combined effects with glial cell line‐derived neurotrophic factor and brain‐derived neurotrophic factor. Neuroscience 2002;110:555–567. [DOI] [PubMed] [Google Scholar]
- 29. Frasson M, Picaud S, Leveillard T, et al. Glial cell line‐derived neurotrophic factor induces histologic and functional protection of rod photoreceptors in the rd/rd mouse. Invest Ophthalmol Vis Sci 1999;40:2724–2734. [PubMed] [Google Scholar]
- 30. Harada T, Harada C, Nakayama N, et al. Modification of glial‐neuronal cell interactions prevents photoreceptor apoptosis during light‐induced retinal degeneration. Neuron 2000;26:533–541. [DOI] [PubMed] [Google Scholar]
- 31. Wahlin KJ, Campochiaro PA, Zack DJ, Adler R. Neurotrophic factors cause activation of intracellular signaling pathways in Müller cells and other cells of the inner retina, but not photoreceptors. Invest Ophthalmol Vis Sci 2000;41:927–936. [PubMed] [Google Scholar]
- 32. Bringmann A, Reichenbach A. Role of Müller cells in retinal degeneration. Front Biosci 2001;6:E72–E92. [DOI] [PubMed] [Google Scholar]
- 33. Harada C, Harada T, Quah HMA, et al. Potential role of glial cell line‐derived neurotrophic factor receptors in Müller glial cells during light‐induced retinal degeneration. Neuroscience 2003;122:229–235. [DOI] [PubMed] [Google Scholar]
- 34. Hauck SM, Kinkl N, Deeq CA, Swiatek‐de Lange M, Schöffmann S, Ueffing M. GDNF family ligands trigger indirect neuroprotective signaling in retinal glial cells. Mol Cell Biol 2006;26(7):2746–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Koeberle PD, Bahr M. The upregulation of GLAST‐1 is an indirect antiapoptotic mechanism of GDNF and neurturin in the adult CNS. Cell Death Differ 2008;15:471–483. [DOI] [PubMed] [Google Scholar]
- 36. Ke JB, Zhong YM. Expression of somatostatin receptor subtype 5 in rat retinal amacrine cells. Neuroscience 2007;144:1025–1032. [DOI] [PubMed] [Google Scholar]
- 37. Xu LY, Zhao JW, Yang XL. GLAST expression on bullfrog Muller cells is regulated by dark/light. NeuroReport 2004;15:2451–2454. [DOI] [PubMed] [Google Scholar]
- 38. Liu LL, Wang L, Zhong YM, Yang XL. Expression of sigma receptor 1 mRNA and protein in rat retina. Neuroscience 2010;167:1151–1159. [DOI] [PubMed] [Google Scholar]
- 39. Xu GZ, Tian J, Zhong YM, Yang XL. Natriuretic peptide receptors are expressed in rat retinal ganglion cells. Brain Res Bull 2010;82:188–192. [DOI] [PubMed] [Google Scholar]
- 40. Haverkamp S, Wässle H. Immunocytochemical analysis of the mouse retina. J Comp Neurol 2000;424:1–23. [PubMed] [Google Scholar]
- 41. Cuenca N, Deng P, Linberg KA, et al. The neurons of the ground squirrel retina as revealed by immunostains for calcium binding proteins and neurotransmitters. J Neurocytol 2002;31:649–666. [DOI] [PubMed] [Google Scholar]
- 42. Koizumi A, Watanabe SI, Kaneko A. Persistent Na+ current and Ca2+ current boost graded depolarization of rat retinal amacrine cells in culture. J Neurophysiol 2001;86:1006–1016. [DOI] [PubMed] [Google Scholar]
- 43. Raymond ID, Vila A, Huynh UC, Brecha NC. Cyan fluorescent protein expression in ganglion and amacrine cells in a thy1‐CFP transgenic mouse retina. Mol Vis 2008;14:1559–1574. [PMC free article] [PubMed] [Google Scholar]
- 44. Nadal‐Nicolás FM, Jiménez‐López M, Sobrado‐Calvo P, et al. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naïve and optic nerve‐injured retinas. Invest Ophthalmol Vis Sci 2009;50:3860–3868. [DOI] [PubMed] [Google Scholar]
- 45. Zhang PP, Yang XL, Zhong YM. Cellular localization of P2Y₆ receptor in rat retina. Neuroscience 2012;220:62–69. [DOI] [PubMed] [Google Scholar]
- 46. Furuta A, Rothstein JD, Martin LJ. Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci 1997;17:8363–8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Conradt M, Storck T, Stoffel W. Localization of N‐glycosylation sites and functional role of the carbohydrate units of GLAST‐1, a cloned rat brain L‐glutamate/L‐aspartate transporter. Eur J Biochem 1995;229:682–687. [DOI] [PubMed] [Google Scholar]
- 48. Feit‐Leichman RA, Kinouchi R, Takeda M, et al. Vascular damage in a mouse model of diabetic retinopathy: relation to neuronal and glial changes. Invest Ophthalmol Vis Sci 2005;46:4281–4287. [DOI] [PubMed] [Google Scholar]
- 49. Layton CJ, Chidlow G, Casson RJ, et al. Monocarboxylate transporter expression remains unchanged during the development of diabetic retinal neuropathy in the rat. Invest Ophthalmol Vis Sci 2005;46:2878–2885. [DOI] [PubMed] [Google Scholar]
- 50. Yu X, Xu Z, Mi M, et al. Dietary taurine supplementation ameliorates diabetic retinopathy via anti‐excitotoxicity of glutamate in streptozotocin‐induced Sprague–Dawley rats. Neurochem Res 2008;33:500–507. [DOI] [PubMed] [Google Scholar]
- 51. Antonetti DA, Barber AJ, Bronson SK, et al. Diabetic retinopathy ‐ Seeing beyond glucose‐induced microvascular disease. Diabetes 2006;55:2401–2411. [DOI] [PubMed] [Google Scholar]