

Summary
Aims
In acute stroke, neurological damage is due to oxidative stress and neuronal apoptotic death. This study investigated whether Nogo‐A 290‐562 residues region (M9), fused to the transduction domain of the HIV trans‐activator (TAT) protein, is neuroprotective against cerebral ischemia and the mechanisms.
Methods
Transient focal cerebral ischemia was induced by middle cerebral artery occlusion in male C57BL/6J mice. TAT‐M9, its mutation or vehicle was applied via intraperitoneal injection at the onset of reperfusion. The neurobehavioral scores, infarction volumes, neuronal apoptosis, and the ratio of Bax/Bcl‐2 were evaluated. Malondialdehyde (MDA), reactive oxygen species (ROS) levels, and NADPH oxidase activation were measured in the presence or absence of the NADPH oxidase inhibitor apocynin or activator tetrabromocinnamic acid (TBCA).
Results
Immunofluorescence results confirmed that TAT‐M9 was transduced into brain parenchyma, and it significantly improved neurological behavior, reduced infarct volumes, protected neuronal cells from apoptosis, inhibited activation of NADPH oxidase, and decreased MDA and ROS contents. Furthermore, apocynin imitated the beneficial effects of TAT‐M9, while TBCA abolished them.
Conclusions
Our results demonstrate that TAT‐M9 administration attenuates cerebral ischemia by inhibiting NADPH oxidase‐mediated oxidative damage and neuronal apoptosis in mice. TAT‐M9 may be a potential treatment for cerebrovascular disease.
Keywords: Apoptosis, Ischemia‐reperfusion injury, NADPH oxidase, Nogo‐A, Oxidative stress
Introduction
Oxidative stress is an important factor underlying neuronal damage caused by ischemic insult 1, 2. Ischemic injury induced by the cerebral artery occlusion is proved to begin with massive generation of reactive oxygen species (ROS) 3, 4. ROS target neurons in the brain, which are particularly vulnerable to ischemia‐reperfusion injury 5. The balance between ROS generation and clearance is destroyed by ischemic injury, resulting in oxidative stress‐induced signaling activation and cell apoptosis. At the same time, neuronal apoptosis is an early event in ischemic injury and severe oxidative stress could lead to exacerbation of neuronal apoptosis, whereas reduction in ROS could contribute to suppression of neuronal apoptosis 6. Thus, therapeutic strategies are needed that target both the oxidative damage and the downstream consequences such as apoptosis.
Nogo‐A, known as myelin‐associated neurite inhibitory factor, inhibits the outgrowth of axonal processes after central nervous system injury. Inhibition of Nogo‐A can enhance axonal sprouting and help neurological recovery in a rodent model of ischemic injury 7, 8. However, the function of neuronal Nogo‐A is still widely uncharacterized. A recent study proved that the neuronal expression of Nogo‐A changed after the set of stroke 9. Another study uncovered that Nogo‐A antibodies promoted neuronal survival after stroke 10. The seemingly conflict results indicate that different interruption with Nogo‐A in different time point could lead to opposite effects. Taken together, Nogo‐A may play a role in promoting survival after ischemic injury. Recently, we have further demonstrated that amino‐Nogo‐A, a long specific region (aa 186‐1004) of Nogo‐A, showed a strong cytoprotective effect against oxidative stress in vitro, and that residues 290‐562 (M9) may be a pivotal domain for this protective effect 11. However, the neuroprotective effect of M9 in vivo is still unclear.
NADPH oxidase is a membrane‐bound enzyme highly localized in the brain that generates ROS in periods of ischemia and reperfusion 12. The activation of NADPH oxidase contributes to ischemic oxidative damage to neurons 13. Several studies have shown elevated expression of NADPH oxidase in the brain following ischemic stroke, with attenuation of cerebral infarction by the NADPH oxidase inhibitor apocynin 14, 15. These results indicate a pivotal role for NADPH oxidase in the pathogenesis of brain ischemic injury and suggest that NADPH oxidase is a promising target in developing treatments for ischemic injury.
To test the M9 domain as putative neuroprotectant in stroke, we fused the most widely utilized protein transduction domains, HIV TAT (trans‐activator gene product) with M9 protein (TAT‐M9), and made the agent more stable and cross the blood–brain barrier (BBB) into the mouse brain. We then investigated its neuroprotective effects against stroke damage induced by middle cerebral artery occlusion (MCAO) in mice. Furthermore, we tested the hypothesis that TAT‐M9 treatment protects against cerebral ischemic injury via inhibition of NADPH oxidase‐mediated oxidative stress.
Materials and Methods
Construction, Purification, and Transduction of TAT‐M9 and TAT‐M9CA Fusion Proteins
Trans‐activator‐M9 was constructed and purified as described in the article by Mi YJ, et al. 11. For the delivery of TAT fusion proteins into the mouse brain, 10 mg/kg TAT proteins were intraperitoneally injected into mice, and the transducible effect of TAT fusion proteins was analyzed by immunofluorescence and Western blotting using anti‐6 × His antibody. TAT‐M9CA was constructed as the domain of 424, 464, and 559 cysteine mutating into alanine and then injected through the same procedure used for TAT‐M9.
Experimental Animals and Drug Injection
The C57BL/6J mice in this study were obtained from the Experimental Animal Center at the Fourth Military Medical University. The animals were housed at 21 ± 2°C, with 60–70% humidity for at least 1 week before surgery. They were also under a fixed 12‐h light/dark cycle and had free access to food and water. Procedures involving animals and their care conformed to the Guidelines for Animal Experimentation of the Fourth Military Medical University (Xi'an, China), which are in compliance with Ethics Committee for Animal Experimentation of the Fourth Military Medical University.
Tetrabromocinnamic acid (TBCA; EMD Chemicals, Gibbstown, NJ, USA) has a high specificity inhibition for CK2 and activates NADPH oxidase. In this study, TBCA (20 nmol in 2 μL of 50% dimethyl sulfoxide [DMSO] in PBS) was injected intracerebroventricularly (i.c.v) 30 min before induction of ischemia (bregma: 1.0 mm lateral, 0.2 mm posterior, 3.1 mm deep) 16. Fifty percent DMSO in PBS was used as a vehicle 16. Apocynin (Sigma‐Aldrich, St Louis, MO, USA), which was dissolved in DMSO and phosphate‐buffered saline, and 2.5 mg/kg body weight were administered intravenously (2.5 mg/kg) 15 min before onset of MCAO 16.
In the first part of the study, for the dose‐dependent study, C57BL/6J mice were randomly assigned to 0 mg/kg TAT‐M9 + MCAO, 1 mg/kg TAT‐M9 + MCAO, 3 mg/kg TAT‐M9 + MCAO, and 10 mg/kg TAT‐M9 + MCAO. Then male 10‐week‐old C57BL/6J mice weighing 20–25 g were randomly assigned to Sham, MCAO, Vehicle + MCAO, TAT‐M9 + MCAO, and TAT‐M9CA + MCAO groups. In the second part, the animals were randomly assigned to Sham, MCAO, TAT‐M9 + MCAO, apocynin + MCAO, Vehicle + MCAO, TBCA + TAT‐M9 + MCAO, and Vehicle + TAT‐M9 + MCAO groups. In the third part, C57BL/6J mice were randomly assigned to MCAO, TAT‐M9 + MCAO, apocynin + TAT‐M9 + MCAO, and Vehicle + TAT‐M9 + MCAO groups.
Induction of Transient Focal Cerebral Ischemia
Focal cerebral ischemia was induced by MCAO in mice using an intraluminal filament technique as described previously 17, 18. The animals were anesthetized with 2% isoflurane delivered by a facemask at a flow rate of 2 L/min oxygen. After 1 h of MCAO, the filament was withdrawn and regional cerebral blood flow was restored to normal. PBS, TAT‐M9 or TAT‐M9CA fusion protein (10 mg/kg) was immediately injected intraperitoneally after retraction of the thread occlusion. Regional cerebral blood flow (rCBF) was monitored through a disposable microtip fiber optic probe (diameter, 0.5 mm) connected through a master probe to a laser Doppler computerized main unit (Periflux 5000; Perimed AB, Jarfalla‐Stockholm, Sweden). MCAO was considered adequate if rCBF sharply decreased to 30% of the baseline level; reperfusion was accompanied with rCBF recovered up to 80% of baseline; otherwise, animals were excluded from analysis.
Neurological Deficit Evaluation and Infarct Assessment
Neurological deficits were monitored 72 h after vascular occlusion using the 6‐point scoring system 19 by a blinded observer. Animals were decapitated and brains were dissected into 6 equidistant coronal slices and immediately stained with 2% 2,3,5‐triphenyltetrazolium chloride (TTC). The infarct volumes were evaluated as described previously 18.
Terminal Transferase Biotinylated‐dUTP Nick End Labeling
Samples from different groups were used for experiments (n = 5 for each group). Twenty‐four hours after reperfusion, neuronal apoptosis was assessed in situ by terminal deoxynucleotide transferase‐mediated dUTP nick end labeling (TUNEL) (Roche) and was quantitatively evaluated with the method described previously 17. Briefly, 32 pixels of 0.10 mm2 were placed by light microscope with 100 × magnification and then the total number of positively stained cells in these pixels was counted and expressed as cells per millimeter squared.
Western Blot Analysis
The ischemic penumbra was identified and harvested as described previously 20, weighed and homogenized (10%) in ice‐cold RIPA buffer (Beyotime Institute of Biotechnology, Nantong, China) with 1 × Roche complete protease inhibitor cocktail and 1 mmol/L phenylmethylsulfonyl fluoride on ice. The Western blot was performed as described in our previous study 17. The following primary antibodies were used: anti‐6 × His, 1:500; anti‐Bcl‐2, 1:200; anti‐Bax, 1:1000; anti‐Activation caspase‐3, 1:200; anti‐NADPH oxidase p47phox, 1:500; and anti‐NADPH oxidase gp91phox, 1:1000 (Abcam, Cambridge, MA, USA) For each primary antibody, the appropriate secondary horseradish peroxidase‐conjugated anti‐rabbit immunoglobulin G (IgG) antibody at a 1:10,000 dilution (KangWei Inc., Beijing, China.) was used. Immunoreactive proteins were then visualized using enhanced chemiluminescence (ECL).
Measurement of Cerebral ROS and MDA Levels
The ischemic penumbra was harvested, and lipid peroxidation was measured according to the method described by Ohkawa H, et al. 21 by a detection kit purchased from Beyotime Institution of China. The absorbance of the supernatant was measured by spectrophotometry at 532 nm. ROS levels were determined using detection kits from Western Tang Bio Institution, Shanghai, China, and the absorbance of the supernatant was measured by spectrophotometry at 490 nm.
Immunofluorescence
For analysis of TAT protein transfusion, the mice in the experiment were anesthetized and transcardially perfused with PBS and 4% paraformaldehyde. The brain sections were prepared and stained as described in our previous study 22. Different brain areas in each section were observed and photographed using a microscope with a 40 × objective (Olympus, BX51, Japan) from the same part of the brain in each group.
Statistical Analysis
The software SPSS 11.0 for Windows (SPSS Inc, Chicago, IL, USA) was used to conduct statistical analyses. All values, except for neurological scores, are presented as mean ± SEM and were analyzed by one‐way analysis of variance, and between‐group differences were detected with the post hoc Student‐Newman–Keuls test. The neurological deficit scores were expressed as median (range) and were analyzed with the Kruskal–Wallis test followed by the Mann–Whitney U‐test with the Bonferroni correction. Values of P ≤ 0.05 were considered statistically significant.
Results
Assessment of in Vivo Transduction of TAT‐M9 into Mouse Brains
According to our previous studies, M9 is the region of Nogo‐A protein from residues 290‐562 (Figure 1A). The M9 fusion protein, attached to TAT, was constructed and purified (Figure 1B). A high level of TAT‐M9 in brain tissue was detected with Western blotting 4 h after intraperitoneal injection; this reached a peak at 12 h (Figure 1C). At 6 h after intraperitoneal injection, anti‐6 × His immunofluorescence revealed a robust transduction in the ischemic penumbra in TAT‐M9‐treated animals, as well as in TAT‐M9CA‐treated animals. No protein transduction was detectable in vehicle‐treated control animals (Figure 1D).
Figure 1.
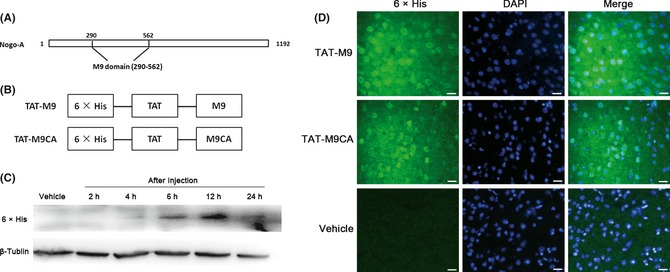
Construction and transduction of trans‐activator (TAT)‐M9 fusion proteins. (A) The structure of the M9 domain. (B) The construction of TAT‐M9CA and TAT‐M9. (C) Western blot showing TAT‐M9 uptake in the brain after intraperitoneal injection. (D) TAT‐M9 was double‐stained with the nuclear marker DAPI and rapidly taken up into the brain. TAT‐M9 showed widespread uptake in the brain using anti‐6 × His antibody to test the signal 6 h after intraperitoneal injection compared with vehicle. TAT‐M9CA showed similar results. Scale Bar = 20 μm.
Protective Effect of TAT‐M9 was Dose‐Dependent
First, we used the middle cerebral artery occlusion model for exploring the dose‐dependent effect of TAT‐M9. The animals were treated with vehicle (PBS), 2, 6 or 10 mg/kg doses of TAT‐M9 at the onset of reperfusion injury. With the dosage of 10 mg/kg, the infarct volumes were significantly reduced compared with vehicle group, together with a ameliorated neurological behavior 72 h after reperfusion injury (Figure 2A,B).
Figure 2.
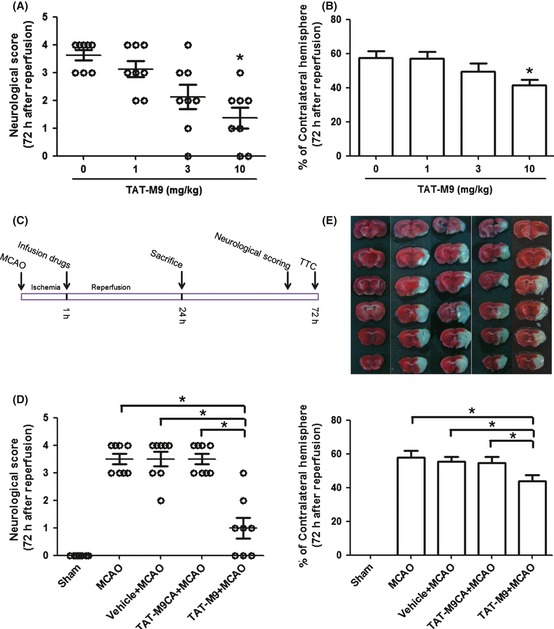
Trans‐activator (TAT)‐M9 improved neurological scores and reduced infarct volumes 72 h after reperfusion injury. 10 mg/kg TAT‐M9 significantly improved neurological scores (A) and reduced infarct volumes (B) compared with the control group, whereas 1 or 3 mg/kg showed no significant difference compared with the mice without TAT‐M9 injection (n = 8 per group). *P < 0.05 versus 0 mg/kg TAT‐M9 + middle cerebral artery occlusion (MCAO) group. (C) A diagram of the experimental design for drug treatment and assessment of the ischemic injury. (D) TAT‐M9 significantly improved neurological scores compared with TAT‐M9CA or vehicle (n = 8 per group). (E) Representative 2,3,5‐triphenyltetrazolium chloride sections outlining the infarcted areas. Infarct volumes of mice were significantly reduced in TAT‐M9‐treated group, whereas TAT‐M9CA or vehicle showed no such effects (n = 8 per group). *P < 0.05 versus TAT‐M9 group.
TAT‐M9 Attenuated Cerebral Ischemic Injury
The diagram of experimental design for drug treatment and assessment of the ischemic injury is shown in Figure 2C. 72 h after ischemia and reperfusion, intraperitoneal injection of TAT‐M9 significantly improved the neurological scores compared with the TAT‐M9CA, vehicle‐treated or MCAO groups (Figure 2D). In MCAO, vehicle‐treated and TAT‐M9CA‐treated groups, reproducible brain infarcts were obtained. The infarct was significantly smaller in TAT‐M9‐treated animals and the trend reached statistical significance (Figure 2E).
TAT‐M9 Inhibited Cellular Apoptosis in the Ischemic Penumbra
Apoptotic cells were identified through TUNEL staining. The number of TUNEL‐positive cells was significantly reduced in the region of ischemic damage in the cerebral cortex in the TAT‐M9‐treated group compared with the vehicle‐treated group (Figure 3B,C,D). In ischemic animals treated with the vehicle and in untreated animals, a considerable amount of activated caspase 3 was detected in the penumbra 24 h after thread occlusion. The significantly lower amount of caspase 3 was found in animals treated with TAT‐M9 (Figure 4A).
Figure 3.
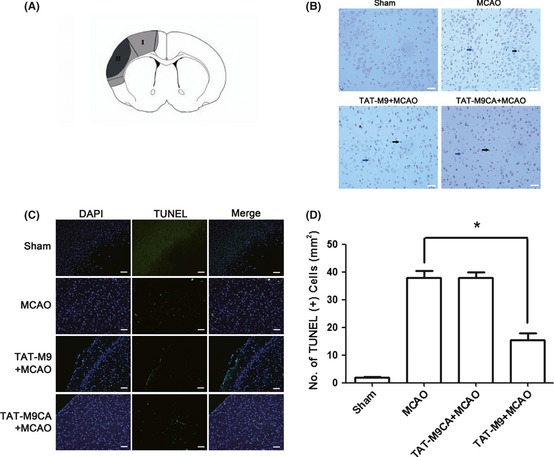
Trans‐activator (TAT)‐M9 inhibited apoptotic cell death. (A) The gray region (I) is defined as the ischemic penumbra, and the black area (region II) is defined as the ischemic core. (B) Representative photomicrographs of TUNEL in the ischemic penumbra by immunohistology. The blue arrow indicates a viable cell, and the black arrow indicates a TUNEL‐positive cell. Scale Bar = 20 μm. (C) Representative photomicrographs of TUNEL in the ischemic penumbra by immunofluorescence. Scale Bar = 20 μm. (D) Quantitative analysis of the number of TUNEL‐positive cells in the ischemic penumbra. TAT‐M9 significantly decreased the number of TUNEL‐positive cells, whereas the TAT‐M9CA showed no such effect. Values are presented as mean ± SEM (n = 5 per group).
Figure 4.
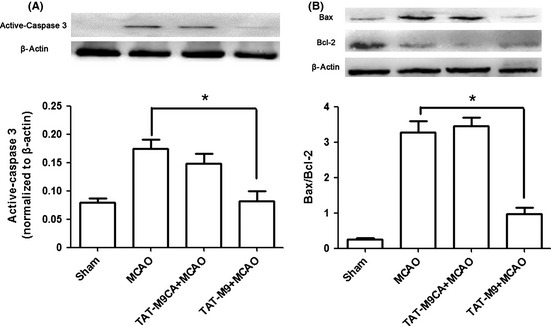
Effect of trans‐activator (TAT)‐M9 on the expression of Bcl‐2, Bax, and caspase‐3 proteins. (A) Apoptosis in the brain was analyzed by Western blot of caspase‐3 activation of the ischemic penumbra. The level of activated caspase‐3 was significantly lower in animals treated with TAT‐M9. Results were normalized with β‐actin. Results are presented as mean ± SEM (n = 3 per group). (B) The TAT‐M9‐treated group showed a lower ratio of Bax/Bcl‐2 compared with TAT‐M9CA or middle cerebral artery occlusion (MCAO) group. Data are mean ± SEM of three mice per group. *P < 0.05 versus MCAO group.
Effect of TAT‐M9 on the Expression of Bcl‐2 and Bax Proteins
Neuronal cellular apoptosis in the ischemic penumbra was accompanied by a decrease in Bcl‐2 expression and an increase in Bax expression. TAT‐M9 injection prevented the decrease in the expression of Bcl‐2 protein and suppressed the increase in Bax expression. The changes in Bax/Bcl‐2 ratios were statistically significant between the TAT‐M9 + MCAO group and the MCAO group (Figure 4B).
NADPH Oxidase was Involved in Neuroprotection of TAT‐M9
For reflecting the activation of NADPH oxidase 23, 24, the NADPH oxidase subunits gp91phox and p47phox were tested by Western blot. As showed in Figure 5A, the activation of NADPH oxidase was increased after MCAO. The TAT‐M9‐treated group showed a lower level of NADPH oxidase compared with the MCAO group. Pretreatment with TBCA significantly reversed the protective effect of TAT‐M9, while apocynin pretreatment significantly reduced the activation of NADPH oxidase. The infarct volume was significantly larger in the TBCA + TAT‐M9 + MCAO group compared with TAT‐M9‐treated animals; this was accompanied with lower neurological scores. In contrast, apocynin pretreatment inhibited the activation of NADPH oxidase, improved the neurological scores, and reduced the infarction volumes compared with the MCAO group (Figure 5B,C).
Figure 5.
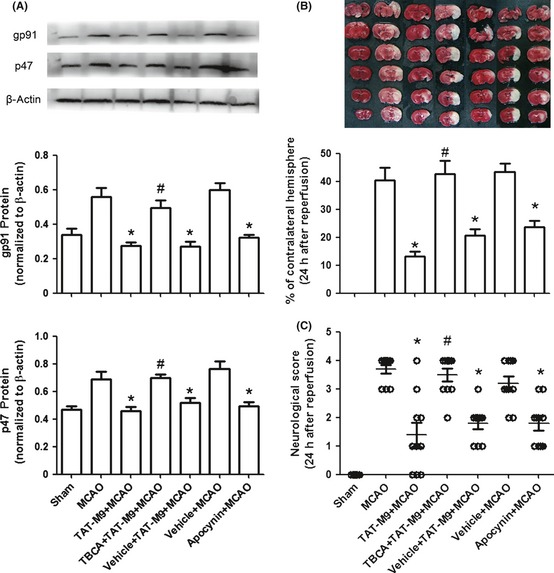
NADPH oxidase‐mediated anti‐oxidation of TAT‐M9. (A) Western blot of NADPH oxidase. Data are mean ± SEM of three mice per group. (B) The reduction in infarct size by TAT‐M9 was reversed by pretreatment with tetrabromocinnamic acid (TBCA), a novel agonist of NADPH oxidase. The NADPH oxdiase inhibitor apocynin could reduce infarction volumes. Infarct volume values given are mean ± SEM (n = 6 per group). (C) Neurological deficits were significantly improved in those animals treated with TAT‐M9 and apocynin but reversed in TBCA pretreatment group (n = 10 per group). *P < 0.05 versus middle cerebral artery occlusion (MCAO) group; #P < 0.05 versus TAT‐M9 + MCAO group.
NADPH Oxidase‐Mediated Anti‐Oxidation of TAT‐M9
Compared with the Sham group, the MCAO group showed a strong increase in ROS production in penumbra tissue. Treatment with TAT‐M9 significantly reduced the increase in ROS generation. TBCA pretreatment could reverse this protective effect of TAT‐M9, while apocynin could simulate the decrease in ROS production (Figure 6A). In this study, malondialdehyde (MDA) was used as a marker of lipid peroxidation, which is one of the hallmarks of ROS‐induced injury in the brain. After 24 h of reperfusion, MDA content was significantly elevated in MCAO mice. The administration of TAT‐M9 or apocynin prevented the formation of MDA compared with the MCAO group and TBCA pretreatment could reverse the reduction in MDA (Figure 6B).
Figure 6.
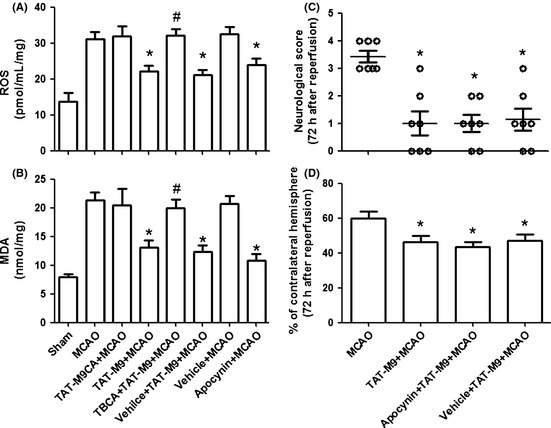
Effect of trans‐activator (TAT)‐M9 on malondialdehyde (MDA), reactive oxygen species (ROS) production, and the effect of apocynin on TAT‐M9 neuroprotection. TAT‐M9 and apocynin administration reduced ROS production (A) and MDA level (B), whereas tetrabromocinnamic acid (TBCA) reversed TAT‐M9 the protective effects. Values are expressed as mean ± SEM (n = 6 per group). *P < 0.05 versus middle cerebral artery occlusion (MCAO) group, #P < 0.05 versus TAT‐M9 + MCAO group. Improved neurological behaviors (C) and reduced infarct volumes (D) were detected in apocynin+TAT‐M9 + MCAO group and vehicle+TAT‐M9 + MCAO group, but both groups showed no significant differences compared with TAT‐M9 group. *P < 0.05 versus MCAO group.
In addition, compared with MCAO group, both apocynin + TAT‐M9 + MCAO group and vehicle + TAT‐M9 + MCAO group improved neurological behaviors (Figure 6C) and reduced infarct volumes (Figure 6D) at 72 h after reperfusion injury. However, the two groups showed no differences in comparison with TAT‐M9 group.
Conclusion
Stroke is one leading cause of death and disability worldwide. Data from the World Health Organization indicate that 15 million people worldwide suffer from stroke annually, placing a heavy burden on family and society 25. Currently, thrombolysis with tissue plasminogen activator (t‐PA) is the basic treatment for ischemic stroke, but the neuroprotective effect is still limited 26, 27.
Nogo‐A has received much attention as a principal inhibitor of neurite outgrowth 22, 28. Numerous studies have demonstrated that blockage of Nogo‐A enhances axonal regeneration, thus reducing long‐term ischemia‐induced neuronal damage. However, in a recent study, Cheatwood et al. 9 proved that Nogo‐A was expressed in cortical neurons, and the expression of Nogo‐A varied over time after MCAO in rats. They emphasized that Nogo‐A expression in neurons reached high peak at 28 days after the set of MCAO. As the cortical area they tested was similar to the ischemic penumbra areas, they believed neuronal Nogo‐A to be a potential target for stroke therapy. But on the other hand, we could clearly see from their results that Nogo‐A expression decreased sharply in the early phase after stroke. This result indicated the different role that Nogo‐A may play in different time points. Kilic et al. also demonstrated that Nogo‐A −/− mice suffered more severe neuronal damage when compared with wild‐type animals and deactivation of Nogo‐A went along with decreased neuronal survival, as well as protracted neurological recovery 10. Although these results seem paradoxical compared with previous findings, together they indicate that in the ischemic model, the blockage of Nogo‐A at different time points may result in various effects.
Our recent study showed that amino‐Nogo‐A upregulated the anti‐apoptotic Bcl‐2 level, countered the reactive oxygen radicals and induced cytoprotection in vitro against exogenous hydrogen peroxide injury, and suggested that residues 290‐562 (M9) may be a pivotal domain for these protective effects 11. This new protein seems to be a promising agent for stroke treatment. However, it is recognized that presently even drugs that were apparently effective in improving cell viability in vitro can easily fail in animal models or even human. Whether M9 is neuroprotective against cerebral ischemic injury in vivo is still unknown.
For this reason, we examined the effect of M9 on brain ischemia‐reperfusion injury in mice. As M9 could not pass the blood–brain barrier, we used a biologically active M9 protein attached to TAT‐His by recombinant technology. In the present study, we proved that TAT‐M9 was able to cross the blood–brain barrier after intraperitoneal infusion and enter cells. We also found that the TAT‐M9 concentration reached the highest levels 12 h after intraperitoneal injection, as demonstrated by Western blotting.
Furthermore, we demonstrated that treatment with TAT‐M9 immediately after reperfusion of MCAO significantly decreased the brain infarct volume and improved neurological outcomes with a concentration of 10 mg/kg. The neuroprotection occurred without altering physiological parameters during ischemia. We also compared the differences in infarct volumes and neurological deficits between TAT‐M9‐treated mice and TAT‐M9CA‐treated mice. Our results suggested that TAT‐M9 treatment could reduce neuronal damage but not TAT by itself or other parts of the protein.
In ischemic stroke, the neurons in the penumbra undergo apoptosis after ischemic insult. Apoptotic events were revealed by activation of caspase as well as TUNEL staining of brain areas. From the current study, the number of TUNEL‐positive cells and the caspase 3 activation in the penumbra area was significantly reduced in the TAT‐M9‐treated group compared with TAT‐M9CA‐treated group. These results provide the first evidence that TAT‐M9 can protect the brain against apoptotic injury induced by ischemia.
The balance between pro‐ and anti‐apoptotic proteins contributes to the process of apoptosis. The Bcl‐2 family is involved in ischemia‐induced neuronal cell apoptosis and regulating mitochondrial functions. Bcl‐2 is classified as the anti‐apoptotic protein and Bax the pro‐apoptotic protein. Our results support that with TAT‐M9 treatment, the activation of Bax after ischemia injury was inhibited, while Bcl‐2 was activated. This finding was consistent with the caspase and TUNEL experiment results above.
Brain ischemia‐reperfusion triggers the activation of oxidative stress, which is an imbalance between the generation and elimination of ROS in the biological system. Severe oxidative stress can cause neuronal apoptosis via multiple pathways 29. NADPH oxidase 2 is a multiunit enzyme composed of membrane‐bound subunits (p22phox and gp91phox), cytoplasmic subunits (p40phox, p47phox, and p67phox), and the small G protein Rac‐1, while it is widely expressed in the central nervous system including cortex and hippocampus 30, 31. For stroke‐induced brain injury, the activation of NADPH oxidase 2 represents a major pathological mechanism among the free radical producing‐enzyme systems. Activated NADPH oxidase was shown in ischemic stroke, while inhibition of NADPH oxidase resulted in reduced cerebral infarct volumes in the ischemic brain 32, 33. These results indicate that NADPH oxidase plays an important role in ischemic‐induced neuronal damage. We proved in our preliminary experiments that TAT‐M9 could increase extracellular signal‐regulated protein kinases (ERK1/2) activation, and ERK1/2 activation was proved to be related to the subunits of NADPH oxidase 10, 34.
Thus, we hypothesized that TAT‐M9 might suppress the activity of NADPH oxidase and alter the ROS generation induced by ischemic injury. The activation of NOX2 is dependent upon forming an active complex with several phox subunits including p47phox and gp91phox. We therefore tested the hypothesis by analyzing gp91phox and p47phox expression in the present study. To better understand the role of NADPH oxidase in ischemic injury, we proved that apocynin was protective against ischemic injury by testing neurological scores, infarct volumes, and ROS and MDA production. These findings were consistent with previous studies in which inhibition of NADPH oxidase‐mediated protective effects during ischemia 14, 15, as well as the study of apocynin‐reducing lipid peroxidation and oxidative DNA damage through the inhibition of NADPH oxidase 16. We also added TBCA, a novel regulator of NADPH oxidase 16, which could reverse the protective effect of TAT‐M9. These results indicated that the anti‐oxidative effect of TAT‐M9 may at least partially occur through the suppression of NADPH oxidase. From another perspective, we used apocynin pretreatment before TAT‐M9 and detected that the two drugs showed no synergy effect compared with TAT‐M9 along, which indicated that the underlying mechanism for TAT‐M9 might be related to inhibition of NADPH oxidase. Furthermore, our preliminary experiments investigating the expression of Prdx2, as well as the level of Akt and ERK1/2, showed similar results to the in vitro studies (the results were not expressed in the current paper). All these together suggest that the anti‐oxidative effect of TAT‐M9 could be through multiple pathways in vivo.
The present study suggests that the novel region of Nogo‐A may be a promising treatment for cerebral ischemia‐reperfusion injury, although many shortcomings and challenges still remain. Regretfully, we did not explain exactly how TAT‐M9 was associated with NADPH oxidase. Whether the connection was direct or not was still unclear. We presumed that TAT‐M9 might affect the MAPK signaling pathways through inhibition of Rac1 or the ERK1/2 signaling pathway as in the previous in vitro study ERK1/2 was proved to be involved in the TAT‐M9 protection 11 and either Rac1 or ERK1/2 was a classic downstream signal for amino‐Nogo‐A 10, 35. Then, the signal might target NADPH oxidase phox subunit for protective effect 34. Further study is needed to precisely determine the NADPH oxidase signaling pathways. On the other hand, the pharmacokinetics and side effects of TAT‐M9 still need further exploring. Moreover, little is known regarding the therapeutic time windows for TAT‐M9. Whether TAT‐M9 delivery at a few hours after stroke is still protective or not is not studied in the current study. For future clinical translation, the full therapeutic window still remains to be characterized.
In conclusion, the present report showed that intraperitoneal injection of TAT‐M9 results in efficacious delivery to the brain. TAT‐M9 administration attenuated cerebral ischemic injury by inhibiting NADPH oxidase‐mediated oxidative damage and neuronal cell apoptosis in mice. These results suggest that TAT‐M9 may have a potential application in treatment for cerebrovascular diseases.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by the Major Program of National Natural Science Foundation of China (Grant 30930091, 30970936), the Program for Changjiang Scholars and Innovative Research Team in University (Beijing, China. Grant 2010CXTD01) and the National Natural Science Foundation of China (Grants 81072888, 81071060, 81035375, 81272801).
The first two authors contributed equally to this work.
References
- 1. Niizuma K, Endo H, Chan PH. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J Neurochem 2009;109:133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ikeda Y, Long MD. The molecular basis of brain injury and brain edema: the role of oxygen free radicals. Neurosurgery 1990;27:1–11. [DOI] [PubMed] [Google Scholar]
- 3. Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab 2001;21:2–14. [DOI] [PubMed] [Google Scholar]
- 4. Wang Q, Sun AY, Simonyi A, et al. Neuroprotective mechanisms of curcumin against cerebral ischemia‐induced neuronal apoptosis and behavioral deficits. J Neurosci Res 2005;82:138–148. [DOI] [PubMed] [Google Scholar]
- 5. Chen SD, Lin TK, Yang DI, et al. Protective effects of peroxisome proliferator‐activated receptors gamma coactivator‐1alpha against neuronal cell death in the hippocampal CA1 subfield after transient global ischemia. J Neurosci Res 2010;88:605–613. [DOI] [PubMed] [Google Scholar]
- 6. Rastogi L, Godbole MM, Ray M, et al. Reduction in oxidative stress and cell death explains hypothyroidism induced neuroprotection subsequent to ischemia/reperfusion insult. Exp Neurol 2006;200:290–300. [DOI] [PubMed] [Google Scholar]
- 7. Papadopoulos CM, Tsai SY, Alsbiei T, O'Brien TE, Schwab ME, Kartje GL. Functional recovery and neuroanatomical plasticity following middle cerebral artery occlusion and IN‐1 antibody treatment in the adult rat. Ann Neurol 2002;51:433–441. [DOI] [PubMed] [Google Scholar]
- 8. Seymour AB, Andrews EM, Tsai SY, et al. Delayed treatment with monoclonal antibody IN‐1 1 week after stroke results in recovery of function and corticorubral plasticity in adult rats. J Cereb Blood Flow Metab 2005;25:1366–1375. [DOI] [PubMed] [Google Scholar]
- 9. Cheatwood JL, Emerick AJ, Schwab ME, Kartje GL. Nogo‐A expression after focal ischemic stroke in the adult rat. Stroke 2008;39:2091–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kilic E, ElAli A, Kilic U, et al. Role of Nogo‐A in neuronal survival in the reperfused ischemic brain. J Cereb Blood Flow Metab 2010;30:969–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mi YJ, Hou B, Liao QM, et al. Amino‐Nogo‐A antagonizes reactive oxygen species generation and protects immature primary cortical neurons from oxidative toxicity. Cell Death Differ 2012;10:1038–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Suh SW, Shin BS, Ma H, et al. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann Neurol 2008;64:654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kahles T, Luedike P, Endres M, et al. NADPH oxidase plays a central role in blood‐brain barrier damage in experimental stroke. Stroke 2007;38:3000–3006. [DOI] [PubMed] [Google Scholar]
- 14. Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia‐reperfusion. J Cereb Blood Flow Metab 2009;29:1262–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoshioka H, Niizuma K, Katsu M, et al. NADPH oxidase mediates striatal neuronal injury after transient global cerebral ischemia. J Cereb Blood Flow Metab 2011;31:868–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim GS, Jung JE, Niizuma K, Chan PH. CK2 is a novel negative regulator of NADPH oxidase and a neuroprotectant in mice after cerebral ischemia. J Neurosci 2009;29:14779–14789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Q, Peng Y, Chen S, et al. Pretreatment with electroacupuncture induces rapid tolerance to focal cerebral ischemia through regulation of endocannabinoid system. Stroke 2009;40:2157–2164. [DOI] [PubMed] [Google Scholar]
- 18. Hata R, Mies G, Wiessner C, et al. A reproducible model of middle cerebral artery occlusion in mice: hemodynamic, biochemical, and magnetic resonance imaging. J Cereb Blood Flow Metab 1998;18:367–375. [DOI] [PubMed] [Google Scholar]
- 19. Tatlisumak T, Takano K, Carano RA, Miller LP, Foster AC, Fisher M. Delayed treatment with an adenosine kinase inhibitor, GP683, attenuates infarct size in rats with temporary middle cerebral artery occlusion. Stroke 1998;29:1952–1958. [DOI] [PubMed] [Google Scholar]
- 20. Ashwal S, Tone B, Tian HR, Cole DJ, Pearce WJ. Core and penumbral nitric oxide synthase activity during cerebral ischemia and reperfusion. Stroke 1998;29:1037–1047. [PubMed] [Google Scholar]
- 21. Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissue by thiobarbituric acid reaction. Anal Biochem 1979;95:351–358. [DOI] [PubMed] [Google Scholar]
- 22. Wang Q, Gou X, Xiong L, et al. Trans‐activator of transcription‐mediated delivery of NEP1‐40 protein into brain has a neuroprotective effect. Aneshthesiology 2008;108:1071–1080. [DOI] [PubMed] [Google Scholar]
- 23. Lu Q, Wainwright MS, Harris VA, et al. Increased NADPH oxidase‐derived superoxide is involved in the neuronal cell death induced by hypoxia‐ischemia in neonatal hippocampal slice culture. Free Radic Biol Med 2012;53:1139–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hong H, Zeng JS, Kreulen DL, Kaufman DI, Chen AF. Atorvastatin protects against cerebral infarction via inhibition of NADPH oxidase‐derived superoxide in ischemic stroke. Am J Physiol Heart Circ Physiol 2006;291:H2210–H2215. [DOI] [PubMed] [Google Scholar]
- 25. The Atlas of Heart Disease and Stroke. World Health Organization web site. Available at: http://www.who.int/cardiovascular_diseases/resources/atlas/en/. 2011.
- 26. California Acute Stroke Pilot Registry (CASPR) Investigators . Prioritizing interventions to improve rates of thrombolysis for ischemic stroke. Neurology 2005;64:654–659. [DOI] [PubMed] [Google Scholar]
- 27. Shuaib A, Lees KR, Lyden P, et al. NXY‐059 for the treatment of acute ischemic stroke. N Engl J Med 2007;357:562–571. [DOI] [PubMed] [Google Scholar]
- 28. Spillmann AA, Bandtlow CE, Lottspeich F, Keller F, Schwab ME. Identification and characterization of a bovine neurite growth inhibitor (bNI‐220). J Biol Chem 1997;273:19283–19293. [DOI] [PubMed] [Google Scholar]
- 29. Lennon SV, Martin SJ, Cotter TG. Dose‐dependent induction of apoptosis in human tumour cell lines by widely diverging stimuli. Cell Prolif 1991;24:203–214. [DOI] [PubMed] [Google Scholar]
- 30. Bedard K, Krause KH. The NOX family of ROS‐generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245–313. [DOI] [PubMed] [Google Scholar]
- 31. Babior BM. NADPH oxidase. Curr Opin Immunol 2004;16:42–47. [DOI] [PubMed] [Google Scholar]
- 32. Wang Q, Tompkins KD, Simonyi A, Korthuis RJ, Sun AY, Sun GY. Apocynin protects against global cerebral ischemia–reperfusion‐induced oxidative stress and injury in the gerbil hippocampus. Brain Res 2006;1090:182–189. [DOI] [PubMed] [Google Scholar]
- 33. Ostrowski RP, Tang J, Zhang JH. Hyperbaric oxygen suppresses NADPH oxidase in a rat subarachnoid hemorrhage model. Stroke 2006;37:1314–1318. [DOI] [PubMed] [Google Scholar]
- 34. Yoo BK, Choi JW, Han BH, Kim WK, Kim HC, Ko KH. Role of MAPK/ERK1/2 in the glucose deprivation‐induced death in immunostimulated astroglia. Neurosci Lett 2005;376:171–176. [DOI] [PubMed] [Google Scholar]
- 35. Deng K, Gao Y, Cao Z, et al. Overcoming amino‐Nogo‐induced inhibition of cell spreading and neurite outgrowth by 12‐O‐tetradecanoylphorbol‐13‐acetate‐type tumor promoters. J Biol Chem 2010;285:6425–6433. [DOI] [PMC free article] [PubMed] [Google Scholar]