

Summary
Background
Defining the impact of diabetes and related risk factors on brain cognitive function is critically important for patients with diabetes.
Aims
To investigate the alterations in hippocampal serine/threonine kinases signaling in the early phase of type 1 and type 2 diabetic rats.
Methods
Early experimental diabetes mellitus was induced in rats with streptozotocin or streptozotocin/high fat. Changes in the phosphorylation of proteins were determined by immunoblotting and immunohistochemistry.
Results
Our data showed a pronounced decrease in the phosphorylation of Ca2+/calmodulin‐dependent protein kinase II (CaMKII) in the hippocampi of both type 1 and type 2 diabetic rats compared with age‐matched control rats. Unexpectedly, we found a significant increase in the phosphorylation of synapsin I (Ser 603) and GluR1 (Ser 831) in the same experiment. In addition, aberrant changes in hippocampal protein kinase C (PKC) and protein kinase A (PKA) signaling in type 1 and type 2 diabetic rats were also found. Moreover, PP1α and PP2A protein levels were decreased in the hippocampus of type 1 diabetic rats, but significantly up‐regulated in type 2 diabetic rats.
Conclusions
The disturbance of CaMKII/PKA/PKC phosphorylation in the hippocampus is an early change that may be associated with the development and progression of diabetes‐related cognitive dysfunction.
Keywords: Diabetes, Brain, Hippocampus, CaMKII, Phosphorylation, PKC, PKA
Introduction
Accumulating evidence indicates that the brain is susceptible to damage as a result of diabetes and that the impact of diabetes‐related risk factors on brain structure and function is notably important to patients with diabetes 1, 2, 3. Lower scores in global cognitive function have been recognized as a complication of diabetes mellitus in patients 4, 5. However, the precise in vivo intracellular molecular mechanisms underlying diabetes‐mediated deficits in learning and memory have not been fully elucidated.
Protein kinases have a wide distribution in the brain and play a major role in learning and the formation of memory 6, 7, 8. Ca2+/calmodulin‐dependent protein kinase II (CaMKII) activity is essential for both long‐term potentiation (LTP) induction and maintenance 9, 10. In addition, calcium/phospholipid‐dependent kinase C (protein kinase C, PKC) 11 and cyclic AMP‐dependent kinase A (protein kinase A, PKA) 12 play pivotal roles in hippocampal LTP induction. We recently determined that hippocampal LTP was impaired in olfactory bulbectomized mice through N‐methyl‐D‐aspartate (NMDA) receptor hypofunction with a concomitant decrease in CaMKII autophosphorylation and PKC phosphorylation, but without changes in PKA and ERK activity 7, 8.
The present study addresses the relationship between diabetes mellitus and changes in cognition with a particular focus on how alterations in serine/threonine kinase signaling may function in the early phase of diabetes mellitus. Our study suggests that the disturbance of CaMKII/PKA/PKC phosphorylation in the hippocampus is an early change that might contribute to the development and progression of diabetes‐related cognitive dysfunction.
Material and Methods
Animals
Male Sprague–Dawley (SD) rats weighing 200–230 g were obtained from Zhejiang Medical Animal Centre (Hangzhou, China). Rats were housed under climate‐controlled conditions with a 12‐h light/dark cycle and provided with standard food and water. An acclimation period of at least 1 week was provided before initiating experimental protocols. Type 1 diabetes was induced by a single injection of freshly prepared streptozotocin (STZ, 60 mg/kg; intraperitoneal; Sigma, St. Louis, MO, USA); control rats were injected only with vehicle (citrate buffer). Before diabetes induction, the animals were weighed and fasted for 12 h. The concentration of blood glucose was repeatedly monitored by test strips (Accu‐Chek Active, Roche®, Indianapolis, IN, USA) every week after STZ injection. Blood glucose levels were measured. Only subjects with glucose levels >16.67 mmol/L were considered diabetic. Type 2 diabetic rats were fed the high‐fat diet and treated as previously described 13. Briefly, glucose and insulin levels were measured in the same way, and the insulin sensitivity index (ISI) was calculated as ln(FBG × FINS)−1. Diabetes was induced by multiple low‐dose streptozotocin injections (30 mg/kg; Sigma) in rats with insulin resistance 13. All of the animals from these groups were sacrificed by decapitation 4 weeks (type 1 diabetes) or 8 weeks (type 2 diabetes) after the induction of diabetes. The brains of all the animals were quickly removed and frozen. All experimental protocols and animal handling procedures were performed in accordance with the National Institutes of Health (NIH, USA) guidelines for the care and use of laboratory animals and were approved by the Committees for Animal Experiments in Zhejiang University in China.
Western Blotting Analysis
Hippocampal samples were homogenized in buffer containing 50 mM Tris–HCl (pH 7.4), 0.5% Triton X‐100, 4 mM EGTA, 10 mM EDTA, 1 mM Na3VO4, 30 mM sodium pyrophosphate, 50 mM NaF, 100 nM calyculin A, 50 μg/ml leupeptin, 25 μg/ml pepstatin A, 50 μg/ml trypsin inhibitor, and 1 mmol/L dithiothreitol (DTT). Insoluble material was removed by a 10‐min 13,000 g centrifugation. After determining supernatant protein concentration using Bradford's solution, samples were boiled for 3 min in Laemmli's sample buffer. Samples containing equivalent amounts of protein were subjected to SDS–polyacrylamide gel electrophoresis (PAGE). Proteins were transferred to an Immobilon PVDF membrane and then blocked with TTBS solution (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, and 0.1% Tween 20) containing 5% w/v nonfat dry milk for 1 h at room temperature, and membranes were incubated overnight at 4°C with anti‐phospho‐CaMKII (1:2000) and anti‐CaMKII (1:1000) 14, anti‐phospho‐synapsin I (Ser 603) (1:1000; Thermo Scientific), antisynapsin I 14, anti‐phospho‐GluR1 (Ser 831), and anti‐phospho‐GluR1 (Ser 845) (1:1000; Millipore, Billerica, MA, USA), anti‐GluR1, anti‐NMDAR1, anti‐PKC (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐phospho‐PKC (Ser 657), anti‐phospho‐NMDAR1 (Ser 896), and anti‐phospho‐MARCKS (1;1000; Millipore, Billerica, MA, USA), and anti‐β‐actin (1:10000; Sigma). Bound antibodies were visualized using the enhanced chemiluminescence detection system (Amersham Life Science, Buckinghamshire, UK) and analyzed semiquantitatively using the National Institutes of Health Image program.
Immunohistochemistry
Rats were anesthetized and transcardially perfusion‐fixed with 4% paraformaldehyde in phosphate‐buffered saline (PBS). The serial 35‐μm slice sections were prepared using a vibratome (Leica VT1000 A). For immunolabeling, slices were probed with anti‐phospho‐CaMKII (1:500) 14, anti‐phospho‐synapsin I (Ser 603) (1:500; Thermo Scientific), anti‐phospho‐NMDAR1 (Ser 896) (1:500; Millipore, Billerica, MA, USA), and anti‐β‐tubulin (1:500; Sigma) overnight at 4°C. After washing, the sections were incubated with Alexa 488 anti‐rabbit IgG or Alexa 594 anti‐mouse IgG (1:300; Molecular Probes, Eugene, OR, USA) in TNB buffer. Immunofluorescence was visualized using a Zeiss LSM 510 confocal microscope. The relative fluorescence intensity of phospho‐CaMKII, phospho‐synapsin I (Ser 603), and phospho‐NMDAR1 (Ser 896) in hippocampus was quantified by using the software package MATLAB R2011b (Mathworks, USA). The 3D filled plots were processed by using ImageJ v1.45 with the accompanied “Interactive 3D Surface Plot” plug‐in. The statistical and plotting software package Graphpad Prism v5.0 (GraphPad Software, Inc., La Jolla, CA, USA) was used to perform t‐test.
Statistical Analysis
The significance of differences between different groups was made using the unpaired Student's t‐test. Differences were considered significant at P < 0.05. All data are expressed as the mean ± SEM.
Results
Decreased Phosphorylation of CaMKII in the Hippocampal Region in Type 1 and Type 2 Diabetic Rats
As shown in Figure 1A, the serum glucose levels remain significantly increased 30 days after streptozotocin (60 mg/kg) injection (Figure 1A). In addition, the T2DM rat model was verified by examining the insulin sensitivity index (ISI) (Figure 1B) and serum glucose levels (Figure 1C). In the present study, diabetic rats showed significant decreases in body weight compared with control rats (Figure S1). Cognitive impairment and hippocampal atrophy have been described in patients with type 1 and type 2 diabetes mellitus 4, 5. Multifunctional CaMKII is the most abundant kinase isoform in the brain and is particularly enriched in neurons 15. Due to its unique properties regarding activation and Ca2+ sensitivity, CaMKII has long been considered a central player in memory functions 8, 9. Furthermore, we examined whether diabetes induced changes in the hippocampal phosphorylation of CaMKII. Constitutively active CaMKII was examined with a specific antibody (α‐/β‐CaM kinase II) 7, 14 that recognizes the autophosphorylation of Thr 286/287. Both α (50‐kDa) and β (60‐kDa) isoforms of CaMKII are neuron specific and are highly abundant in the rat brain 16. Our data demonstrated a significant reduction in CaMKII (Thr 286/287) phosphorylation in the hippocampal region of diabetic rats (Figure 1D,E). By contrast, the western blot analysis showed increased total CaMKII protein levels in type 1 (Figure 1D,E) and type 2 (Figure 2A,B) diabetic rats.
Figure 1.
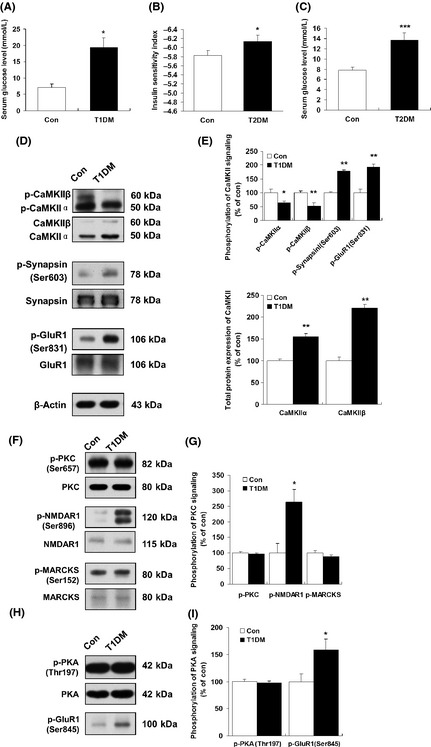
Decreased phosphorylation of CaMKII in the hippocampal region in type 1 diabetic rats. (A) Changes in blood glucose 30 days after STZ injection in type 1 diabetic rats. (B) Measurement of insulin sensitivity index in type 2 diabetic rats. (C) Changes in blood glucose 60 days after STZ/high‐fat treatment in type 2 diabetic rats. *P < 0.05 versus control animals. ***P < 0.001 versus control animals. Immunoblotting was carried out using antibodies recognizing phospho‐ or total protein. (D) The changes in CaMKII (Thr 286/287), synapsin I (Ser 603), and GluR1 (Ser 831) phosphorylation in the hippocampal region in type 1 diabetic rats were detected by western blots. (E) Quantitative analysis of relative CaMKII (Thr 286/287), synapsin I (Ser 603), and GluR1 (Ser 831) phosphorylation in type 1 diabetic rats was performed by densitometry. *P < 0.05; **P < 0.01 versus control rat (n = 5). (F) Representative western blots of PKC (Ser 657), N‐methyl‐D‐aspartate 1 (NMDAR1) (Ser 896), and Myristoylated alanine‐rich C‐kinase substrate (MARCKS) (Ser 152) phosphorylation. (G) Quantitative analysis of relative PKC (Ser 657), NMDAR1 (Ser 896), and MARCKS (Ser 152) phosphorylation in type 1 diabetic rats performed by densitometry. *P < 0.05 versus control rat (n = 5). (H) The expression of phospho‐PKA (Thr 197) and phospho‐GluR1 (Ser 845) in control and type 1 diabetic rats. (I) The results of (H) were quantitated by densitometry. *P < 0.05 versus control rat (n = 5). β‐Actin was used to ensure equal protein loading in each lane.
Figure 2.
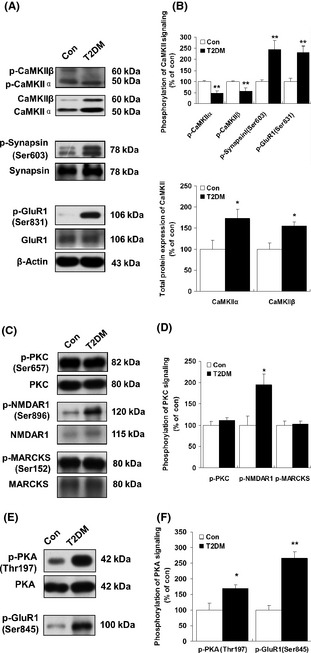
Changes in hippocampal phospho‐CaMKII in type 2 diabetic rats. (A) Hippocampal phospho‐CaMKII (Thr 286/287), phospho‐synapsin I (Ser 603), and phospho‐GluR1 (Ser 831) total proteins in type 2 diabetic rats were determined by western blotting (see Materials and methods). (B) The results of (A) were quantitated by densitometry. (C) Representative western blots of PKC (Thr 657), N‐methyl‐D‐aspartate 1 (NMDAR1) (Ser 896), and Myristoylated alanine‐rich C‐kinase substrate (MARCKS) (Ser 152) phosphorylation in control and type 2 diabetic rats. (D) The results of (C) were quantitated by densitometry. (E) Representative western blots of PKA (Thr 197) and GluR1 (Ser 845) phosphorylation in control and type 2 diabetic rats. (F) The results of (E) were quantitated by densitometry. β‐Actin was used to ensure equal protein loading in each lane. Data are expressed as percentage of values of control animals (mean ± SEM, n = 5). *P < 0.05;**P < 0.01 versus control animals. T2DM, type 2 diabetes mellitus.
Subcellularly, CaMKII is localized to the dendrites and even more specifically to the postsynaptic densities of excitatory synapses 17. Here, we employed immunohistochemistry to analyze the phospho‐CaMKII (Thr 286/287) protein in hippocampal CA1 pyramidal neurons. Whereas the CA1 pyramidal neurons from nondiabetic control rats largely showed strongly labeled phospho‐CaMKII (Thr 286/287)‐immunoreactive neurons in the somata and dendrites of neurons, the CA1 pyramidal neurons from type 1 (Figure 3A,B) and type 2 (Figure 4A,B) diabetic rats showed weak staining compared with controls. No significant changes in β‐tubulin‐ and DAPI‐positive staining were observed in hippocampus between diabetic and control rats.
Figure 3.
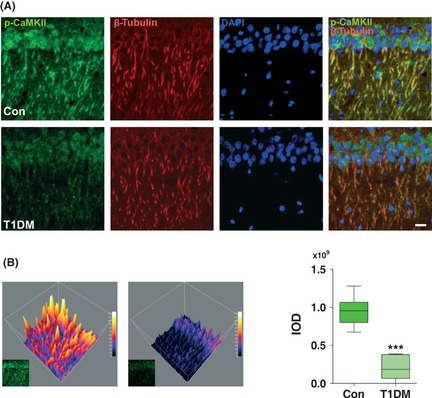
Changes in immunostaining of phospho‐CaMKII in hippocampus of type 1 diabetic rats. (A) Representative image of hippocampal CA1 pyramidal neurons stained with anti‐phospho‐CaMKII and β‐tubulin. Markedly decreased phospho‐CaMKII staining was observed by confocal microscopy in the hippocampus of type 1 diabetic rats. DAPI counterstaining indicates nuclear localization (blue). Scale bar = 50 μm. (B) Visualization of grayscale intensities of phospho‐CaMKII through 3D mesh plots (Left). The integrated optical density (IOD) determined by Graphpad Prism v5.0 according to the staining intensity of phospho‐CaMKII (Right). ***P < 0.001 versus control (n = 6 slices from three animals for each group). T1DM, type 1 diabetes mellitus.
Figure 4.
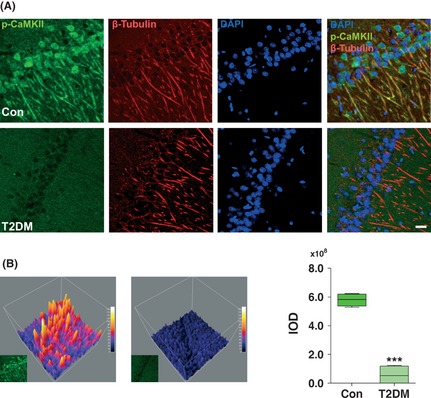
Changes in immunohistochemical expression of phospho‐CaMKII in the hippocampus of type 2 diabetic rats. (A) Immunohistochemical localization of phospho‐CaMKII was examined in the hippocampal CA1 region. Markedly decreased phospho‐CaMKII staining was observed by confocal microscopy in the hippocampus of type 2 diabetic rat. DAPI counterstaining indicates nuclear localization (blue). Scale bar = 50 μm. (B) The representative 3D filled surface plots (Left) indicate the grayscale intensities of phospho‐CaMKII in control and T2DM rats. The integrated optical density (IOD) determined by Graphpad Prism v5.0 according to the staining intensity of phospho‐CaMKII (Right). ***P < 0.001 versus control (n = 6 slices from three animals for each group). T2DM, type 2 diabetes mellitus.
Increased Phosphorylation of GluR1 (Ser 831) and Synapsin I (Ser 603) in the Hippocampus in Type 1 and Type 2 Diabetic Rats
CaMKII serves as the synaptic tag that marks a synapse for long‐term modification. After activation, CaMKII phosphorylates a variety of substrates, such as synapsin I, MAP‐2, and several types of glutamate receptors 18, 19. Therefore, we evaluated synapsin I (Ser 603) and GluR1 (Ser 831) phosphorylation by immunoblotting with phospho‐specific antibodies. Unexpectedly, in the present study, no decrease in synapsin I (Ser 603) and GluR1 (Ser 831) phosphorylation was observed in the same context in either type 1 or type 2 diabetes (Figures 1D,E and 2A,B). This result suggests that synapsin I (Ser 603) and GluR1 (Ser 831) phosphorylation may play a complementary response in the pathogenesis of diabetes. As shown in Figure 5A,B, immunoreactivity for the phosphorylation of synapsin I (Ser 603) was present in the presynapse of neurons (Figure 5A,B). Coinciding with the immunoblot data, our immunohistochemical staining demonstrated a significantly increased and intense immunolabeling of synapsin I (Ser 603) (Figure 5A) in hippocampal pyramidal neurons in type 1 diabetic rats. No significant changes in β‐tubulin‐ and DAPI‐positive staining were observed in the same context.
Figure 5.
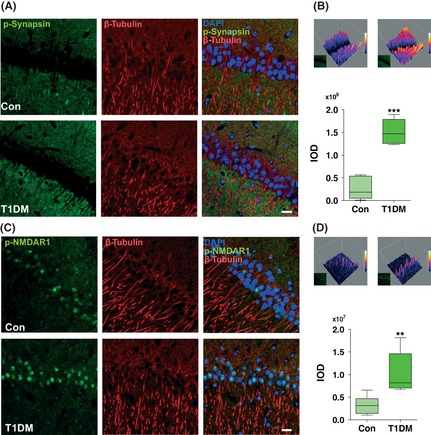
The STZ treatment induced aberrant immunostaining of phospho‐synapsin I (Ser 603) and phospho‐NMDAR1 (Ser 896) in type 1 diabetic rats. Immunohistochemical localization of phospho‐synapsin I (Ser 603) (A, B) and phospho‐NMDAR1 (Ser 896) (C, D) was examined in the hippocampus CA1 region. Markedly increased phospho‐synapsin I (Ser 603) (A) and phospho‐NMDAR1 (Ser 896) (C) stainings were observed by confocal microscopy in the hippocampus of type 1 diabetic rats. DAPI counterstaining indicates nuclear localization (blue). Scale bar = 50 μm. The 3D filled surface plots (Upper) indicate the grayscale intensities of phospho‐synapsin I (Ser 603) (B) and phospho‐NMDAR1 (Ser 896) (D). The integrated optical density (IOD) determined by Graphpad Prism v5.0 according to the staining intensity of proteins (Lower). **P < 0.001; ***P < 0.001 versus control (n = 6 slices from three animals for each group). T1DM, type 1 diabetes mellitus.
Effect of Diabetes on the Phosphorylation of PKC Signaling in the Hippocampal Region
As shown previously, the activation of PKC leads to the translocation of conventional isoforms to the plasma membrane and primarily serves to phosphorylate potential substrates 11. Immunoblot analysis showed that neither the levels of phosphorylated PKC nor those of total PKC were affected in the hippocampus of either type 1 (Figure 3F,G) or type 2 (Figure 2C,D) diabetic rats compared with controls. High‐affinity substrates of PKC include Myristoylated alanine‐rich C‐kinase substrate (MARCKS) (Ser 152) and NMDAR1 (Ser 896), which show regional specificity within the mammalian brain 7. To evaluate the physiological relevance of PKC activation following diabetes further, we next assessed the phosphorylation of NMDAR1 (Ser 896) and MARCKS (Ser 152) by immunoblotting with phospho‐specific antibodies. We found that phosphorylation of NMDAR1 (Ser 896) was significantly increased in both type 1 and type 2 diabetes, whereas PKC (Ser 657) phosphorylation remained unchanged in the hippocampal region (Figures 3F,G and 2C,D). Consistently, the increased immunostaining of phospho‐NMDAR1 (Ser 896) in the cytoplasm of CA1 pyramidal neurons of T1DM rats was also observed when compared with control rats (Figure 5C,D). In addition, no significant changes were observed in either total MARCKS protein or phospho‐MARCKS (Ser 152) in the same context (Figures 1F and 2C).
Changes in the Phosphorylation of PKA in the Hippocampus in Diabetic Rats
Protein kinase A‐dependent phosphorylation of the GluR1 (Ser 845) subunit plays an essential role in the regulation of α‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐propionic acid (AMPA) receptors. PKA catalyzes GluR1 phosphorylation at Ser 845, thereby increasing AMPA channel conductance 20. Our study shows that diabetes had no effects on total PKA protein expression (Figure 1H,I). Moreover, in the type 1 diabetic rat hippocampal region, no significant change was observed in the expression of PKA phosphorylation (Thr 197) compared to controls (Figure 1H,I). Conversely, in the type 2 diabetic rats' hippocampal regions, we found a significant increase in the expression of PKA phosphorylation (Thr 197) (Figure 2E,F). The phosphorylation of GluR1 (Ser 845) was measured in the same tissue extracts used for the determination of phosphorylated proteins in this study. Notably, we found that the phosphorylation of GluR1 (Ser 845) was dramatically increased in the hippocampi of both type 1 (Figure 1H,I; 158.73 ± 19.70% of controls, P < 0.05) and type 2 (Figure 2E,F; 266.33 ± 20.73% of controls, P < 0.01) diabetic rats. These results suggest a link between the increased phosphorylation of both PKA (Thr 197) and GluR1 (Ser 845) in the hippocampus in the early diabetic state.
Disturbance of PP1 and PP2A Proteins in the Hippocampal Region in Diabetic Rats
We were particularly interested in the levels of phosphoprotein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A), which belong to a class of phosphatases known as protein serine/threonine phosphatases 21, 22. Thus, we sought to confirm PP1α and PP2A hippocampal expression in both type 1 and type 2 diabetic rats by immunoblotting. As shown in Figure 6, we found a significant decrease in PP1α (62.82 ± 4.24% of controls, P < 0.01) and PP2A (70.84 ± 7.61% of controls, P < 0.05) protein levels in the hippocampus of type 1 diabetic rats compared with control rats (Figure 6A,B). In addition, a significant increase in serum alanine transaminase (ALT) and blood urea nitrogen (BUN) was observed in high‐fat‐fed type 2 diabetic rats (Figure S2). In contrast to type 1 diabetic rats, western blot analysis of hippocampal tissue lysates showed an evident and significant increase in PP1α and PP2A in the hippocampus of type 2 diabetic rats (Figure 7A,B).
Figure 6.
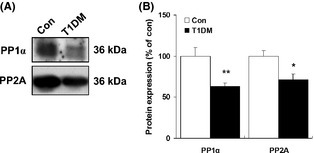
Changes in expression of hippocampal PP1α and PP2A in type 1 diabetic rats. (A) Representative western blots of PP1α and PP2A in the hippocampal region in type 1 diabetic rats. (B) Quantitative analysis of relative PP1α and PP2A in type 1 diabetic rats was performed by densitometry. Immunoblotting with an anti‐β‐actin antibody shows equal amounts of loaded protein in each lane. Data are expressed as percentage of values of control animals (mean ± SEM, n = 5). *P < 0.05; **P < 0.01 versus control rat. T1DM, type 1 diabetes mellitus.
Figure 7.
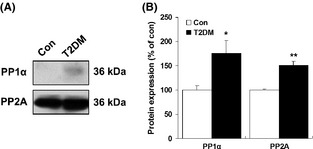
Western blot analysis of hippocampal PP1α and PP2A in type 2 diabetic rats. (A) Representative western blots of PP1α and PP2A in the hippocampal region of type 2 diabetic rats. (B) Quantitative analysis of relative levels of PP1α and PP2A in type 2 diabetic rats. β‐Actin was used to ensure equal protein loading in each lane. Data are expressed as percentage of values of control animals (mean ± SEM, n = 5). *P < 0.05; **P < 0.01 versus control rat. T2DM, type 2 diabetes mellitus.
Discussion
Defining the impact of diabetes and related risk factors on brain structure and function is critically important for patients with diabetes 1, 2, 3. However, few studies have been conducted on the effects of diabetes on serine/threonine kinase signaling in the brain in the early phases of diabetes mellitus. We investigated the biochemical alterations in phosphorylation of CaMKII, PKC, and PKA signaling in the hippocampi of type 1 and type 2 diabetic rats. Our study suggests that disturbances of CaMKII/PKA/PKC phosphorylation in the hippocampus are early changes that may contribute to the development and progression of diabetes‐related cognitive dysfunction.
Both type 1 and type 2 diabetes mellitus are associated with derangements in the regulation of intracellular Ca2+ 23. Biochemical and biophysical research has revealed that the induction and maintenance of LTP require the activation of protein kinases 6, 7, 8. In this study, we investigated alterations in the phosphorylation of CaMKII in the hippocampi of type 1 and type 2 diabetic rats. Consistent with previous reports 24, we also found that total CaMKIIα protein levels increased. Furthermore, our data showed an evident and significant reduction in the phosphorylation of CaMKII (Thr 286/287) in the hippocampi of both type 1 and type 2 diabetic rats. Both the AMPA receptor and synapsin I phosphorylation sites are known to be preferentially phosphorylated by CaMKII 25, 26. CaMKII (Thr 286/287) autophosphorylation and GluR1 phosphorylation function as molecular switches underlying LTP and explicit memory 27. In hippocampal LTP, CaMKII activation is closely associated with stably increased phosphorylation of the GluR1 and synapsin I 9. Unexpectedly, we observed a significant increase in the phosphorylation of synapsin I (Ser 603) and GluR1 (Ser 831), which are substrates of CaMKII in presynaptic and postsynaptic regions, respectively, of the hippocampus. The reasons for the different effects of diabetes on the protein expressions of CaMKII and its potential substrates in the hippocampus are presently unknown. It is likely that the increase in phosphorylation of synapsin I (Ser 603) and GluR1 (Ser 831) in type 1 diabetic rats was secondary to PP1α and PP2A inactivation, as the PP1α and PP2A protein levels were significantly decreased in the type 1 diabetic rats.
Because impaired LTP in the hippocampus is correlated with reduced PKC activities, these observations led us to evaluate whether changes in the phosphorylation of PKC, which is required for the activation of PKC kinase, are associated with the pathological process of diabetes 7. In our study, STZ (type 1 diabetes) or STZ/high‐fat (type 2 diabetes) treatment did not alter hippocampal PKC phosphorylation until after at least 4 weeks in type 1 diabetic rats and 8 weeks in type 2 diabetic rats. Postsynaptically, PKC may act instructively by directly phosphorylating AMPA receptors in the hippocampus 11. This effect could be enhanced through the liberation of calmodulin from neurogranin 28. Unexpectedly, our study demonstrated that NMDAR1 (Ser 896) phosphorylation dramatically increased both STZ (60 mg/kg) and STZ‐/high‐fat‐treated animals. Whether a complementary response loop exists between kinases/phosphatases and increased phosphorylation of NMDAR1 (Ser 869) in the hippocampus of diabetic rats remains to be observed. Because experimental animals in this study are in the early phases of diabetes mellitus, we postulate that a significant complementary response arises against diabetes‐related risk factors.
Although PKA does not appear to alter the conductance of the AMPAR 29, substantial evidence indicates that the cAMP pathway acts through CaMKII to facilitate LTP 30. In this study, we suggest that PKA signaling is involved in the hippocampal pathological process in diabetic rats. Enhancement of synaptic transmission during LTP can result from several mechanisms that are regulated by the phosphorylation of GluR1 (Ser 845) 20. Unexpectedly, a significant increase in GluR1 (Ser 845) phosphorylation was observed in the hippocampi of type 1 and type 2 diabetic rats, suggesting a complementary response of PKA signaling during the early diabetic state.
Cellular serine/threonine phosphatases are involved in the regulation of cell homeostasis through the negative regulation of signaling pathways initiated by protein kinases. Our data highlight a difference in the expression patterns of PP1α and PP2A in the hippocampi of type 1 and type 2 diabetic rats. It appears likely that PP1α and PP2A may be associated with the aberrant increase in synapsin I (Ser 603) and GluR1 (Ser 831) phosphorylation in type 1 diabetic rats because PP1α and PP2A levels are down‐regulated in diabetic rats. This finding is consistent with the fact that PP1α, PP2A, and other intracellular factors are also potential upstream kinases for GluR1 and NMDA phosphorylation 22, 31. Future studies using pharmacological and genetic approaches should further confirm the interrelationships between GluR1/synapsin I and PP1α/PP2A. Although we cannot determine the precise role of increased PP1α and PP2A levels in type 2 diabetic rats, this effect could be partly related to the dephosphorylation of CaMKII 32.
In conclusion, we suggest that alternations in hippocampal CaMKII/PKC/PKA pathways in early experimental diabetes mellitus could be, at least in part, responsible for the detrimental postdiabetic outcomes associated with cognitive dysfunction in diabetic models. The abnormal increase in synapsin I (Ser 603), GluR1 (Ser 831), and GluR1 (Ser 845) phosphorylation may be due to the complementary response of pyramidal neurons against the progression of injury due to diabetes mellitus. Nevertheless, the overall picture regarding the role of serine/threonine kinases in the diabetic brain remains incomplete. Further studies are needed to define the extent to which changes in each of these enzymes lead to cellular dysfunction in relation to learning and memory. It is critically important to investigate further the effect of STZ or STZ/high fat on NMDA and AMPA ion channels in the various pathological stages of diabetic rats.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Figure S1. Changes of body weight in type 1 (A) and type 2 (B) diabetic rats.
Figure S2. Serum biochemical measurements of ALT and BUN in tupe 2 diabetic rat.
Acknowledgments
This work was supported in part by National Natural Science Foundations of China (81120108023, 91232705, 81202533); Zhejiang Provincial Natural Science Foundation of China (R2100281); The Zhejiang Provincial Qianjiang Talent Plan (2012R10036).
The first two authors contributed equally to this work.
References
- 1. Bree Aj, Puente EC, Daphna‐Iken D, Fisher SJ. Diabetes increases brain damage caused by severe hypoglycemia. Am J Physiol Endocrinol Metab 2009;297:E194–E201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anan F, Masaki T, Shimomura T, et al. Abdominal visceral fat accumulation is associated with hippocampus volume in non‐dementia patients with type 2 diabetes mellitus. Neuroimage 2010;49:57–62. [DOI] [PubMed] [Google Scholar]
- 3. Hershey T, Perantie DC, Wu J, Weaver PM, Black KJ, White NH. Hippocampal volumes in youth with type 1 diabetes. Diabetes 2010;59:236–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bruehl H, Wolf OT, Sweat V, Tirsi A, Richardson S, Convit A. Modifiers of Cognitive function and brain structure in middle‐aged and elderly individuals With type 2 diabetes mellitus. Brain Res 2009;1280:186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Christman AL, Vannorsdall TD, Pearlson GD, Hill‐Briggs F, Schretlen DJ. Canial volume, mild cognitive deficits, and functional limitations associated with Diabetes in a community sample. Arch Clin Neuropsychol 2010;25:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shobe J. The role of PKA, CaMKII, and PKC in avoidance conditioning: permissive or instructive? Neurobiol Learn Mem 2002;77:291–312. [DOI] [PubMed] [Google Scholar]
- 7. Moriguchi S, Han F, Shioda N, et al. Nefiracetam activation of CaM kinase II and protein kinase C mediated by NMDA and metabotropic glutamate receptors in olfactory bulbectomized mice. J Neurochem 2009;110:170–181. [DOI] [PubMed] [Google Scholar]
- 8. Li X, Han F, Liu D, Shi Y. Changes of Bax, Bcl‐2 and apoptosis in hippocampus in the rat model of post‐traumatic stress disorder. Neurol Res 2010;32:579–586. [DOI] [PubMed] [Google Scholar]
- 9. Fukunaga K, Miyamoto E. A working model of CaM kinase II Activity in hippocampal long‐term potentiation and memory. Neurosci Res 2000;38:3–17. [DOI] [PubMed] [Google Scholar]
- 10. Lamsa K, Irvine EE, Giese KP, Kullmann DM. NMDA receptor‐dependent long‐term potentiation in mouse hippocampal interneurons shows a unique dependence on Ca (2+)/calmodulin‐dependent kinases. J Physiol 2007;584(Pt 3):885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klann E, Chen SJ, Sweatt JD. Mechanism of protein kinase C activation during the induction and maintenance of long‐term potentiation probed using a selective peptide substrate. Proc Natl Acad Sci U S A 1993;90:8337–8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus‐base long‐term memory. Cell 1997;88:615–626. [DOI] [PubMed] [Google Scholar]
- 13. Reed MJ, Meszaros K, et al. A new rat model of type 2 diabetes: the fat‐fed, streptozotocin‐treated rat. Metabolism 2000;49:1390–1394. [DOI] [PubMed] [Google Scholar]
- 14. Zhang GS, Ye WF, Tao RR, et al. Expression profiling of Ca(2+)/calmodulin‐dependent signaling molecules in the rat dorsal and ventral hippocampus after acute lead exposure. Exp Toxicol Pathol 2012;64:619–624. [DOI] [PubMed] [Google Scholar]
- 15. Mullasseril P, Dosemeci A, Lisman JE, Griffith LC. A structural mechanism for maintaining the ‘on‐state’ of the CaMKII memory switch in the post‐synaptic density. J Neurochem 2007;103:357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brocke L, Srinivasan M, Schulman H. Developmental and regional expression of multifunctional Ca2+/calmodulin‐dependent protein kinase isoforms in rat brain. J Neurosci 1995;15:6797–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kennedy MB, Bennett MK, Erondu NE. Biochemical and immunochemical evidence that the “major postsynaptic density protein” is a subunit of a calmodulin‐dependent protein kinase. Proc Natl Acad Sci U S A 1983;80:7357–7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 2000;287:2262–2267. [DOI] [PubMed] [Google Scholar]
- 19. Han F, Ali Raie A, Shioda N, Qin ZH, Fukunaga K. Accumulation of beta‐amyloid in the brain microvessels accompanies increased hyperphosphorylated tau proteins following microsphere embolism in aged rats. Neuroscience 2008;153:414–427. [DOI] [PubMed] [Google Scholar]
- 20. Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 1996;16:1179–1188. [DOI] [PubMed] [Google Scholar]
- 21. Belmeguenai A, Hansel C. A role for protein phosphatases 1, 2A, and 2B in cerebellar long‐term potentiation. J Neurosci 2005;25:10768–10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yi KD, Simpkins JW. Protein phosphatase 1, protein phosphatase 2A, and calcineurin play a role in estrogen‐mediated neuroprotection. Endocrinology 2008;149:5235–5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Biessels GJ, ter Laak MP, Hamers FP, Gispen WH. Neuronal Ca2+ dysregulation in diabetes mellitus. Eur J Pharmacol 2002;447(2–3):201–209. [DOI] [PubMed] [Google Scholar]
- 24. Bhardwaj SK, Kaur G. Effect of diabetes on calcium/calmodulin dependent protein kinase‐II from rat brain. Neurochem Int 1999;35:329–335. [DOI] [PubMed] [Google Scholar]
- 25. Fukunaga K, Muller D, Miyamoto E. Increased phosphorylation of Ca2+/calmodulin‐dependent protein kinase II and its endogenous substrates in the induction of long‐term potentiation. J Biol Chem 1995;270:6119–6124. [DOI] [PubMed] [Google Scholar]
- 26. Derkach V, Barria A, Soderling TR. Ca2+/calmodulin‐kinase II enhances channel conductance of alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci U S A 1999;96:3269–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lisman J, Schulman H, Cline H. The molecular basis of CaMKII functions in synaptic and behavioural memory. Nat Rev Neurosci 2002;3:175–190. [DOI] [PubMed] [Google Scholar]
- 28. Fukunaga K, Miyamoto E. Current studies on a working model of CaM kinase II in hippocampal long‐term potentiation and memory. Jpn J Pharmacol 1999;79:7–15. [DOI] [PubMed] [Google Scholar]
- 29. Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP‐dependent protein kinase. J Neurosci 2000;20:89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carroll RC, Nicoll RA, Malenka RC. Effects of PKA and PKC on miniature excitatory postsynaptic currents in CA1 pyramidal cells. J Neurophysiol 1998;80:2797–2800. [DOI] [PubMed] [Google Scholar]
- 31. Johannessen M, Delghandi MP, Moens U. What turns CREB on? Cell Signal 2004;16:1211–1227. [DOI] [PubMed] [Google Scholar]
- 32. Colbran RJ. Protein phosphatases and calcium/calmodulin‐dependent protein kinase II‐dependent synaptic plasticity. J Neurosci 2004;24:8404–8409. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Changes of body weight in type 1 (A) and type 2 (B) diabetic rats.
Figure S2. Serum biochemical measurements of ALT and BUN in tupe 2 diabetic rat.