

SUMMARY
Objective: To assess the impact of comorbid autism spectrum disorders (ASD) on the response to second‐generation antipsychotics (SGA) in pediatric bipolar disorder (BPD). Methods: Secondary analysis of identically designed 8‐week open‐label trials of SGA monotherapy (risperidone, olanzapine, quetiapine, ziprasidone, or aripiprazole) in youth with BPD. Results: Of the 151 BPD subjects 15% (n= 23) met criteria for comorbid ASD. There were no differences in the rate of antimanic response (YMRS change ≥30% or CGI‐Improvement ≤2: 65% vs. 69%; P= 0.7) in the presence of comorbid ASD. Conclusion: No difference observed in the rate of antimanic response or tolerability to SGA monotherapy in the presence of ASD comorbidity.
Keywords: Autism spectrum disorder (ASD), bipolar disorder (BPD), comorbid
Introduction
An emerging literature suggests comorbidity between autism spectrum disorders (ASD) and bipolar disorder (BPD). Case reports of severe periodic mood disturbances highly suggestive of mania have been described in individuals with ASD for years [1, 2, 3, 4, 5, 6, 7]. A high incidence of BPD has also been reported in first‐degree relatives of children with ASD [8]. Using data from a large clinical population, Wozniak et al. [9] examined the symptomatic and diagnostic overlap between ASD and BPD in a large outpatient pediatric psychiatric sample and found that 11% of the youth diagnosed with BPD had comorbid ASD and 21% of patients diagnosed with ASD had comorbid BPD. These researchers noted striking homology in the phenotypic features of these disorders regardless of the comorbidity with the other disorder.
Despite this overlap, there is a paucity of information on the treatment of youth presenting with comorbid ASD and BPD. In an early review of the extant literature on children with comorbid ASD and BPD, Lainhart et al. [10] found that conventional neuroleptics (haloperidol, chlorpromazine, thioridazine) and traditional mood stabilizers (lithium, carbamazepine) were minimally effective for the treatment of mania in children with ASD. In contrast, recent double‐blind, placebo‐controlled studies demonstrated that the second‐generation antipsychotics (SGA) risperidone was well tolerated and efficacious in treating target symptoms of irritability and disruptive, aggressive, and self‐injurious behaviors in youth with ASD [11, 12], a spectrum of symptoms highly suggestive of BPD.
In recent years, large randomized clinical trials (RCT) have documented the thymoleptic effects of SGAs in the treatment of youth with BPD, which have led to U.S. Food and Drug Administration (FDA) approval of olanzapine, aripiprazole, and risperidone as monotherapy agents for the treatment of acute mania in youth [13, 14, 15, 16, 17]. We have also documented improvement in affective symptoms in secondary analyses of an RCT of youth with subaverage IQ and disruptive behavior disorders [18], suggesting that SGAs may have therapeutic utility in the management of affective symptoms in youth. Consistent with these findings, our group has shown an improvement in manic symptoms in youth with BPD in open label, prospective trials of olanzapine [19, 20], risperidone [19], ziprasidone [21], and aripiprazole [22] monotherapies.
In this series of open‐label trials in pediatric BPD, we did not exclude youth with comorbid ASD. Thus the aim of this study was to estimate the impact of comorbid ASD on the rate of response to SGA monotherapy in youth with BPD. We hypothesized that the rate of ASD comorbidity in our sample of youth with BPD would be consistent with the literature. Additionally, we hypothesized that the antimanic response to SGAs would not be affected by the comorbid presence of ASD.
Methods
This is a secondary analysis of identically designed open‐label trials of SGA monotherapy (risperidone, olanzapine, quetiapine, ziprasidone, or aripiprazole) for the treatment of BPD in children and adolescents. These trials were conducted from 1999 to 2005. With the exception of the risperidone and olanzapine trials, which were conducted concurrently, all trials were conducted consecutively with minimal overlap between them. At no time was there an overlap of more than two SGA trials. The decision to assign a subject to a particular SGA trial was based on the subject's prior treatment with the trial medications. If the subjects had never been treated with either of the SGA trial medications available at a given time, they were assigned randomly to one of the concurrent trials. Subjects with a history of treatment with one of these trial medications were assigned to the other trial. Subjects who had a history of treatment with both medications were not eligible for either available treatment trial. Each study consisted of an 8‐week, open‐label treatment with flexible dosing to optimal treatment response.
All study procedures were reviewed and approved by the subcommittee for human subjects of our institution. All subjects’ parents or guardians signed written informed consent forms and children older than 7 years of age signed written assent forms.
Subjects
Male and female subjects, 4–17 years of age were included in the trial. Each subject met criteria for bipolar I disorder or BPD NOS and were currently displaying manic, hypomanic, or mixed symptoms (with or without psychotic features) according to the DSM‐IV based on clinical assessment and confirmed by structured diagnostic interview (Kiddie Schedule of Affective Disorders and Schizophrenia Epidemiological Version). BPD not otherwise specified (NOS) was defined as having severe mood disturbance, which meets DSM‐IV Criteria A for BPD but fewer elements in criteria B (at least 2 items with elevated mood and 3 with irritable mood) or having episodes of less than 1 week in duration (lasting at least 2 days). As per the aforementioned definition of subthreshold mania, that is, BPD‐NOS, the diagnosis of BPD‐II is subsumed in the BPD‐NOS diagnostic category.
We excluded subjects with any serious, unstable medical illness including hepatic, renal, gastroenterologic, respiratory, cardiovascular, endocrinologic, neurologic, immunologic, or hematologic disease. Excluded were subjects with a full‐scale IQ of less than 70, pregnancy or lactation, or prior exposure to trial medication. Subjects with DSM‐IV substance (except nicotine or caffeine) dependence within past 6 months, history of schizophrenia or other primary psychotic disorder, or who were judged to be at serious suicidal risk were also excluded from participation. Other psychiatric comorbidities were not considered exclusionary.
No patient was entered into the study if they were adequately stabilized on an antimanic therapy. Mood stabilizers, anticonvulsants, and other neuroleptic therapy were not allowed during this study. Antidepressants were exclusionary to the study.
Administration of Study Medications
In each trial, the final dose of study medication was determined according to response and tolerability. In each 8‐week trial, subjects were seen at weekly intervals and received a prescription for study medication, as described below. Medication could be lowered at any visit, per clinician decision.
Risperidone was initiated at an open‐label dose of 0.25 mg/day for children ≤12 years and 0.5 mg/day for older youth to be increased weekly to a maximum does of 2.0 mg/day for ≤12 and up to 4.0 mg/day for older youth [23]. Olanzapine was initiated at an open‐label dose of 1.25 mg/day to be increased weekly to a possible maximum of 10 mg/day for children ≤6 years [19] and was initiated at 2.5 mg per day, with weekly increases of 2.5–5 mg to a maximum possible dose of 20 mg allowed for the study. For this secondary analysis, a subsample of olanzapine data was drawn from an open‐label trial of olanzapine augmentation with topiramate to study the antimanic response and treatment‐emergent weight gain in youth with BPD [24]. For those subjects receiving olanzapine plus topiramate, topiramate was initiated at a dose of 25 mg per day, with weekly increases of 25 mg per day up to the maximum dose of 100 mg per day [24]. Aripiprazole was initiated 5 mg/day to a maximum possible dose of 15 mg/day in 5 mg increments every 2 weeks for children ≤12 years and was initiated at 10 mg/day increased in 5 mg increments on a biweekly basis to a maximum dose of 20 mg/day for older youth. Ziprasidone was initiated at 1 mg/kg to be increased to 1.5 mg/kg by week 2 and up to 2 mg/kg by week 3. Total daily dose could not exceed 160 mg administered twice daily. Quetiapine was initiated at 50 mg/day to be increased to maximum possible dose of 400 mg/day in 25 mg increments.
Administration of Concomitant Medications
Given the strong overlap of juvenile BPD with attention deficit hyperactivity disorder, the psychostimulants methylphenidate hydrochloride, dextroamphetamine sulfate, and mixed amphetamine salts were allowed during the study if, in the clinician's judgment, it was in the best interest of the patient to continue this treatment or if the patient did not wish to stop stimulant treatment and only if the patient had been on a stable dose for at least 30 days. However, none of the subjects in this study had also been prescribed a stimulant. If extrapyramidal symptoms (EPS) occurred, benztropine mesylate was allowed in doses of up to a maximum of 2 mg/day. The use of the benzodiazepine lorazepam was permitted during the study in doses of 2 mg or less per day. Nonpharmacological treatments such as individual, family, or group therapy were allowed if they were in place before the subject joined the study and if the therapy regimen remained the same throughout the study.
Clinician Rated Assessment Scales
The severity of mania symptoms was assessed with the Young Mania Rating Scale (YMRS) [25, 26, 27]. Clinicians completed the YMRS as a measure of current symptom severity at the baseline visit and at each weekly visit of the 8‐week trial. To be included in the study, subjects must have had a score of 15 or greater on the YMRS.
At baseline, week 4, and endpoint, depression symptomatology and severity were assessed with the Children's Depression Rating Scale—Revised (CDRS‐R) [28], ADHD was assessed with the Attention Deficit Hyperactivity Disorder Rating Scale (ADHD‐RS), and psychosis or other psychiatric symptoms were assessed with the Brief Psychiatric Rating Scale (BPRS) [29]. Psychosocial functioning was assessed using the Global Assessment of Functioning Scale (GAF) [30].
To assess clinically significant severity and improvement relative to baseline, we used the NIMH Clinical Global Impression (CGI) severity (CGI‐S) and improvement (CGI‐I) scales [31] at each weekly visit. CGI‐S and CGI‐I were assessed separately for depression, mania, conduct disorder, and ADHD. The score for the CGI‐S ranges from 1 (normal, not at all ill) to 7 (among the most extremely ill patients). The score for the CGI‐I ranges from 1 (very much improved) to 7 (very much worse).
Diagnostic Assessments of Psychiatric Comorbidity
In order to assess the rates of psychiatric comorbidity, the structured diagnostic interview Kiddie—Schedule of Affective Disorders and Schizophrenia—Epidemiological Version (K‐SADS‐E) [32] was conducted with mothers and subjects 12 years and older. These interviews were administered by highly trained and supervised nonclinician interviewers. All diagnoses were reviewed by a sign‐off committee of experienced board certified child and adolescent psychiatrists chaired by the senior investigator (JB). We computed kappa coefficients of agreement by having experienced, board‐certified child and adult psychiatrists, and licensed clinical psychologists diagnose subjects from audio‐taped interviews made by the assessment staff. Based on 500 assessments from interviews of children and adults, the median kappa coefficient between raters and clinicians was 0.99 and between clinicians and the clinical review committee chaired by the senior investigator was 0.87.
As the K‐SADS‐E and other structured interviews lack a module to evaluate ASDs, we adapted DSM‐III‐R diagnostic criteria into interview format to assess this disorder. Based on this diagnostic interview, study subjects who met DSM‐III‐R diagnostic criteria for autistic disorder or pervasive developmental disorder NOS were considered comorbid for ASD.
Definition of Clinical Response (BPD)
Response was defined by having either a 30% reduction in symptoms according to the YMRS at endpoint or by having been judged as much or very much improved on the CGI‐Improvement (≤2).
Safety Assessment
Safety was assessed at each visit using spontaneous reports of treatment‐emergent adverse events, changes in vital signs, and laboratory measures. Blood pressure and weight were recorded at each visit. Prolactin, glucose, and lipid levels were obtained at baseline and posttreatment. EPS were assessed at baseline and at each weekly visit using the Simpson‐Angus Scale [33], the Barnes Akathisia Scale [34, 35], and the Abnormal Involuntary Movement Scale [36].
Statistical Analysis
Analyses were intention to treat (ITT) with the exception that subjects must have been assessed on drug for at least 1 week. A mixed‐effects model repeated measures approach was used for baseline versus endpoint (week 8 or drop visit carried forward). Models assessing symptom improvement on the primary outcome measure were adjusted for baseline YMRS score. Continuous and categorical data were tested with ANOVA and Pearson's χ2, respectively for nonlongitudinal data (i.e., demographics at baseline, prevalence of adverse effects or response at endpoint, etc.). Statistical significance was determined at P < 0.05.
Results
One hundred fifty‐one subjects were enrolled in identically designed open‐label trials of SGA treatment for pediatric BPD (aripiprazole, N= 9, 9.3 ± 4.9 mg/day; quetiapine, N= 18, 270.5 ± 161.9 mg/day; risperidone, N= 50, 1.4 ± 0.67 mg/day; olanzapine, N= 53, 8.5 ± 4.2 mg/day; and ziprasidone, N= 21, 56.2 ± 34.4 mg/day) for which a concomitant diagnosis of ASD was available. Subjects were predominantly male (65%, N= 98) and the mean age was 9.1 ± 3.0 years.
Fifteen percent (N= 23) of participating subjects met criteria for comorbid ASD on structured diagnostic interview. There was no difference in the rate of ASD across treatment trials (aripiprazole, 22%, N= 2; quetiapine, 17%, N= 3; risperidone, 14%, N= 7; olanzapine, 19%, N= 10; and ziprasidone, 5%, N= 1; P= 0.6). There were no statistically significant differences in age (8.2 ± 2.5 vs. 9.2 ± 3.1; P= 0.2) or sex (males: 52% vs. 67%; P= 0.2) between BPD + ASD and BPD−ASD subjects. The majority of subjects (83%, N= 126) met criteria for BPD‐I, with no difference in the number of BPD−NOS subjects with or without comorbid ASD (17%, N= 4 vs. 16%, N= 21 respectively; P= 0.9).
To facilitate comparisons of doses between different treatment studies, dose at endpoint was standardized (mean of 0, standard deviation of 1.0) within each treatment arm so that the relative dose assigned to the BPD + ASD and BPD − ASD groups could be collapsed for all subjects. The difference observed in the average dose of SGA received by subjects in the BPD + ASD and BPD − ASD groups was not statistically significant (standardized mean of 0.27 ± 1.3 vs. −0.03 ± 0.95 respectively; P= 0.2).
As illustrated in Table 1, the BPD + ASD group was clinically and statistically significantly more impaired at study entry according to the YMRS, BPRS, and GAF. There were no differences in rate of response, as measured by various definitions including a 50% reduction in YMRS or our a priori definition of response (YMRS change of ≥30% or CGI‐Improvement ≤2) (Figure 1). Likewise, the mean change in YMRS total score, corrected for increased YMRS scores at baseline in the BPD + ASD group, was not statistically significantly different in youth with or without comorbid ASD (−14.6 ± 12.5 vs. −12.7 ± 10.8; P= 0.412).
Table 1.
Clinical correlates of youth with bipolar disorder in the context of comorbidity with ASD
| BPD+ASD (N = 23) | BPD (N = 128) | F(1,153) | P‐value | |
|---|---|---|---|---|
| YMRS | 35.4 ± 6.9 | 28.4 ± 8.2 | 15.7 | <0.001 |
| CDRS | 41.9 ± 12.4 | 39.9 ± 12.2 | 0.5 | 0.5 |
| BPRS | 48.6 ± 11.7 | 42.5 ± 13.4 | 4.3 | 0.04 |
| ADHD‐RS | 37.5 ± 11.6 | 34.0 ± 11.9 | 1.7 | 0.2 |
| GAF | 48.5 ± 5.1 | 51.2 ± 6.2 | 6.4 | 0.01 |
Figure 1.
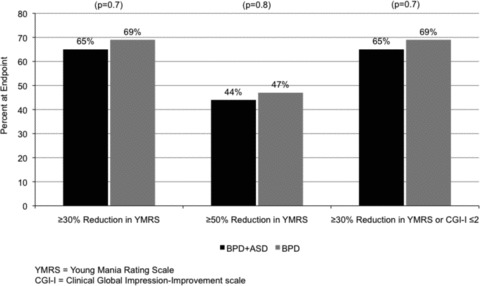
Antimanic response to SGA monotherapy in youth with bipolar disorder in the context of comorbidity with ASD.
As illustrated in Figure 2, the rate of spontaneously reported side effects did not differ between groups, with the exception of slurred speech or teary eyes (both <5%) reported higher in ASD subjects. In both groups, the most commonly reported side effects were sedation, symptoms of common cold, increased appetite, gastrointestinal (GI) complaints, headache, and agitation. In all, EPS were observed in three subjects with BPD, two subjects treated with olanzapine, and one subject treated with aripiprazole. During the course of these trials, two subjects with BPD in the olanzapine trial received benztropine mesylate adjunctively for the management of EPS. There was a statistically significant increase in weight relative to baseline associated with 8 weeks of SGA monotherapies (2.4 ± 2.7 kg; F(3, 150) = 26.2, P < 0.001). The two BPD groups (BPD − ASD and BPD + ASD) were similar in the mean weight gain (BPD − ASD = 2.5 ± 2.8 kg vs. BPD + ASD = 2.1 ± 1.8 kg; F = 0.33, P= 0.6).
Figure 2.

Adverse effects associated with SGA monotherapy in youth with bipolar disorder in the context of comorbidity with ASD.
The difference in the rate of drop out observed between BPD + ASD and BPD − ASD groups was not statistically significant (43%, n= 10 vs. 26%, N= 33, P= 0.08).
Discussion
This study examined whether the comorbidity of ASD in youth with BPD moderates the therapeutic effects of SGAs. In this study, comorbidity with ASD was present in 15% of the youth with BPD. Although BPD youth with comorbid ASD were slightly younger and more likely to be female, this difference was not statistically significant. The rate of comorbidity between BPD and ASD identified in this sample of subjects is consistent with the rate reported by Wozniak et al. [9] in youth referred to an outpatient psychopharmacology clinic. These investigators found that 11% of youth satisfying criteria for BPD on structured diagnostic interviews were also diagnosed with ASD.
The increased functional impairment related to comorbid ASD in pediatric BPD was reflected in the significantly more impaired baseline YMRS, BPRS, and GAF scores in the BPD youth with ASD. This is consistent with the previous report of youth with comorbid BPD and ASD who were significantly more impaired on Child Behavior Checklist as compared to youth with BPD or ASD alone [9]. However, the increased severity associated with BPD and ASD comorbidity did not impact the relative rate of response. Sixty‐five percent of the youth with comorbid BPD and ASD showed a ≥30% improvement in YMRS or a CGI‐I ≤2, while 69% of those youth with BPD without comorbid ASD also demonstrated this level of improvement. These results suggest that the rate of antimanic response to SGAs in youth with BPD is not adversely impacted by a comorbidity with ASD.
In general, SGA monotherapies were well tolerated by youth with BPD. Although not statistically significant, BPD youth with comorbid ASD required a higher dose of SGA on average (numerically but not statistically significant; P= 0.2). A trend toward significance was observed in the rate of drop out, which was higher among BPD youth with versus without ASD (43% vs. 26%; P= 0.08). There was no difference in the reported rates of adverse effects, including EPS and weight gain, in the presence of ASD with the exception of slurred speech and teary eyes that were experienced at a higher frequency by a minority (<5%) of BPD participants with ASD. Importantly, none of the BPD participants with ASD experienced EPS, which is consistent with low rates of EPS similar to placebo reported with risperidone therapy in youth with autistic disorder with marked irritability/aggression [37]. Though there was significant weight gain (2.4 ± 2.7 kg) associated with SGA monotherapy, the difference in mean weight gain was not significant between the BPD youth with and without comorbid ASD (2.1 ± 1.8 kg vs. 2.5 ± 2.8 kg, respectively).
Clearly, the results of this study need to be considered preliminary due to its uncontrolled nature. This was an open study; therefore, the assessments were not blind to treatment. Children and adolescents were primarily ascertained for BPD and the diagnosis of ASD was assigned by structured diagnostic interview. Thus, this study did not employ full clinical evaluation of ASD or standardized assessments of ASD symptomatology. Finally, since the group sizes among the available treatment studies differ widely, this secondary analysis is not adequately powered to compare the antimanic response among the various SGAs.
Conclusion
Despite these limitations, this study suggests that ASD is a common comorbidity in pediatric BPD and is associated with significant increase in functional impairment. Although we did not find any evidence of an effect of ASD comorbidity on the antimanic response in youth with BPD, this result needs to be replicated and validated through controlled trials in larger samples.
Conflict of Interest
Dr. Gagan Joshi has received the Ethel DuPont Warren Fellowship Award 2005–2006 and the Pilot Research Award from the American Academy of Child and Adolescent Psychiatry 2005. He is a reviewer and member of the National Institute of Mental Health Editorial Board. He has received CME sponsored support from McNeil Pediatrics (CME sponsored by SynerMed Communications). He was supported by Shire as a member of the national advisory board for the year 2009. Dr. Joshi has received research support from Bristol Myers Squibb and Glaxo Smith Kline (Site PI for Multi‐centered Trials). Dr. Joshi has received research support as a co‐investigator for clinical trials sponsored by Abbott, Bristol Myers Squibb, Cephalon, Eli Lilly & Co., Johnson & Johnson, McNeil, Merck, New River, Novartis, Organon, Otsuka, Takeda, Pfizer, and Shire Pharmaceuticals.
Dr. Joseph Biederman is currently receiving research support from the following sources: Elminda, Janssen, McNeil, Next Wave Pharmaceuticals, and Shire. In 2011, Dr. Biederman gave a single unpaid talk for Juste Pharmaceutical Spain, and received honoraria from the MGH Psychiatry Academy for a tuition‐funded CME course. In 2010, Dr. Biederman received a speaker's fee from Fundación Dr. Manuel Camelo A.C., provided single consultations for Shionogi Pharma Inc. and Cipher Pharmaceuticals Inc., and received honoraria from the MGH Psychiatry Academy for a tuition‐funded CME course.
Dr. Janet Wozniak received research support from McNeil, Shire, Janssen, and Johnson & Johnson. She was a speaker for Primedia/MGH Psychiatry Academy. Her spouse, John Winkelman, MD, PhD, received research support from GlaxoSmithKline and served on consultant/advisory boards for Impax Laboratories, Pfizer, UCB, Zeo Inc., and Sunivion.
Dr. Robert Doyle received honoraria for presentations from Shire, Novartis, and McNeil. He has been on advisory boards for McNeil, Novartis, and Shire pharmaceutical companies.
Dr. Paul Hammerness has participated in CME activities/professional talks supported by Ortho‐McNeil Janssen and Shire and served on the advisory board for Shire. He has participated, as an investigator/principal investigator, in research studies funded by the following pharmaceutical companies/companies: Cephalon, Eli Lilly, Elminda Ltd, GlaxoSmithKline, Johnson & Johnson, McNeil, Merck, New River, Novartis, Ortho‐McNeil Janssen, Pfizer, Shire, Takeda. Dr. Hammerness has also received honoraria from commercial entities supporting the MGH Psychiatry Academy, http://www.mghcme.org.
Dr. Eric Mick has received research support from Ortho‐McNeil Janssen Scientific Affairs, Pfizer, Shire Pharmaceuticals, and has been an advisory board member for Shire Pharmaceuticals.
All other authors have no conflicts of interest. [Correction added on 13 June 2011, after first online publication: Conflict of Interest was inserted.]
Acknowledgments
This work was supported by grants from industry (AstraZeneca, Bristol‐Myers Squibb, Janssen LP, Pfizer Inc) and the Stanley Medical Research Institute to Dr Biederman. We also wish to acknowledge the generous support from the Norma Fine Pediatric Psychopharmacology Fellowship Fund and members of the MGH Pediatric Psychopharmacology Council.
References
- 1. Kurita H, Osada H, Shimizu K, Tachimori H. Bipolar disorders in mentally retarded persons with pervasive developmental disorders. J Dev Phys Disabil 2004;16:377–389. [Google Scholar]
- 2. Kerbeshian J, Burd L. Case study: Comorbidity among Tourette's syndrome, autistic disorder, and bipolar disorder. J Am Acad Child Adolesc Psychiatry 1996;35:681–685. [DOI] [PubMed] [Google Scholar]
- 3. Steingard R, Biederman J. Lithium responsive manic‐like symptoms in two individuals with autism and mental retardation. J Am Acad Child Adolesc Psychiatry 1987;26:932–935. [DOI] [PubMed] [Google Scholar]
- 4. Komoto J, Usui S, Hirata J. Infantile autism and affective disorder. J Autism Dev Disord 1984;14:81–84. [DOI] [PubMed] [Google Scholar]
- 5. Sovner R, Hurley AD. Do the mentally retarded suffer from affective illness? Arch Gen Psychiatry 1983;40:61–67. [DOI] [PubMed] [Google Scholar]
- 6. Wing L. Asperger's syndrome: A clinical account. Psychol Med 1981;11:115–129. [DOI] [PubMed] [Google Scholar]
- 7. Fombonne E, du Mazaubrun C. Prevalence of infantile autism in four French regions. Soc Psychiatry Psychiatr Epidemiol 1992;27:203–210. [DOI] [PubMed] [Google Scholar]
- 8. DeLong R, Nohria C. Psychiatric family history and neurological disease in autistic spectrum disorders. Dev Med Child Neurol 1994;36:441–448. [PubMed] [Google Scholar]
- 9. Wozniak J, Biederman J, Faraone SV, et al Mania in children with pervasive developmental disorder revisited. J Am Acad Child Adolesc Psychiatry 1997;36:1552–1559; discussion 9–60. [DOI] [PubMed] [Google Scholar]
- 10. Lainhart JE, Folstein SE. Affective disorders in people with autism: A review of published cases. J Autism Dev Disord 1994;24:587–601. [DOI] [PubMed] [Google Scholar]
- 11. Nagaraj R, Singhi P, Malhi P. Risperidone in children with autism: Randomized, placebo‐controlled, double‐blind study. J Child Neurol 2006;21:450–455. [DOI] [PubMed] [Google Scholar]
- 12. Shea S, Turgay A, Carroll A, Schulz M, Orlik H, Smith I, Dunbar F. Risperidone in the treatment of disruptive behavioral symptoms in children with autistic and other pervasive developmental disorders. Pediatrics 2004;114:e634–e641. [DOI] [PubMed] [Google Scholar]
- 13. Tramontina S, Zeni CP, Ketzer CR, Pheula GF, Narvaez J, Rohde LA. Aripiprazole in children and adolescents with bipolar disorder comorbid with attention‐deficit/hyperactivity disorder: A pilot randomized clinical trial. J Clin Psychiatry 2009;70:756–764. [DOI] [PubMed] [Google Scholar]
- 14. Findling RL, Nyilas M, Forbes RA, et al Acute treatment of pediatric bipolar I disorder, manic or mixed episode, with aripiprazole: A randomized, double‐blind, placebo‐controlled study. J Clin Psychiatry 2009;70:1441–1451. [DOI] [PubMed] [Google Scholar]
- 15. Tohen M, Kryzhanovskaya L, Carlson G, et al Olanzapine versus placebo in the treatment of adolescents with bipolar mania. Am J Psychiatry 2007;164:1547–1556. [DOI] [PubMed] [Google Scholar]
- 16. DelBello MP, Kowatch RA, Adler CM, et al A double‐blind randomized pilot study comparing quetiapine and divalproex for adolescent mania. J Am Acad Child Adolesc Psychiatry 2006;45:305–313. [DOI] [PubMed] [Google Scholar]
- 17. Haas M, Delbello MP, Pandina G, et al Risperidone for the treatment of acute mania in children and adolescents with bipolar disorder: A randomized, double‐blind, placebo‐controlled study. Bipolar Disord 2009;11:687–700. [DOI] [PubMed] [Google Scholar]
- 18. Biederman J, Mick E, Faraone SV, Wozniak J, Spencer T, Pandina G. Risperidone for the treatment of affective symptoms in children with disruptive behavior disorder: A post hoc analysis of data from a 6‐week, multicenter, randomized, double‐blind, parallel‐arm study. Clin Ther 2006;28:794–800. [DOI] [PubMed] [Google Scholar]
- 19. Biederman J, Mick E, Hammerness P, Harpold T, Aleardi M, Dougherty M, Wozniak J. Open‐label, 8‐week trial of olanzapine and risperidone for the treatment of bipolar disorder in preschool‐age children. Biol Psychiatry 2005;58:589–594. [DOI] [PubMed] [Google Scholar]
- 20. Frazier JA, Biederman J, Jacobs TG, et al. editors. Olanzapine in the treatment of bipolar disorder in juveniles, Chicago , Illinois : American Psychiatric Association, 2000. [Google Scholar]
- 21. Biederman J, Mick E, Spencer TJ, Dougherty M, Aleardi M, Wozniak J. A prospective open‐label treatment trial of ziprasidone monotheray in children and adolescents with bipolar disorder. Bipolar Disord in press. [DOI] [PubMed] [Google Scholar]
- 22. Biederman J. An open‐label trial of aripiprazole in Youth with bipolar disorder. Biol Psychiatry 2006;59:25S. [Google Scholar]
- 23. Biederman J, Mick E, Hammerness P, et al Open‐label, 8‐week trial of olanzapine and risperidone for the treatment of bipolar disorder in preschool‐aged children. Biolog Psychiatry 2005;58:589–594. [DOI] [PubMed] [Google Scholar]
- 24. Wozniak J, Mick E, Waxmonsky J, Kotarski M, Hantsoo L, Biederman J. Comparison of open‐label, 8‐week trials of olanzapine monotherapy and topiramate augmentation of olanzapine for the treatment of pediatric bipolar disorder. J Child Adolesc Psychopharmacol 2009;19:539–545. [DOI] [PubMed] [Google Scholar]
- 25. Young RC, Biggs JT, Ziegler VE, Meyer DA. A rating scale for mania: Reliability, validity and sensitivity. Br J Psychiatry 1978;133:429–435. [DOI] [PubMed] [Google Scholar]
- 26. Gracious BL, Youngstrom EA, Findling RL, Calabrese JR. Discriminative validity of a parent version of the Young Mania Rating Scale. J Am Acad Child Adolesc Psychiatry 2002;41:1350–1359. [DOI] [PubMed] [Google Scholar]
- 27. Youngstrom EA, Danielson CK, Findling RL, Gracious BL, Calabrese JR. Factor structure of the Young Mania Rating Scale for use with youths ages 5 to 17 years. J Clin Child Adolesc Psychol 2002;31:567–572. [DOI] [PubMed] [Google Scholar]
- 28. Emslie GJ, Rush AJ, Weinberg WA, Kowatch RA, Hughes CW, Carmody T, Rintelmann J. A double‐blind, randomized, placebo‐controlled trial of fluoxetine in children and adolescents with depression. Arch Gen Psychiatry 1997;54:1031–1037. [DOI] [PubMed] [Google Scholar]
- 29. Lachar D, Bailley SE, Rhoades HM, et al New subscales for an anchored version of the brief psychiatric rating scale: Construction, reliability, and validity in acute psychiatric admissions. Psychol Assess 2001;13:384–395. [DOI] [PubMed] [Google Scholar]
- 30. Endicott J, Spitzer RL, Fleiss JL, Cohen J. The global assessment scale. A procedure for measuring overall severity of psychiatric disturbance. Arch Gen Psychiatry 1976;33:766–771. [DOI] [PubMed] [Google Scholar]
- 31. National Institute of Mental Health . CGI (Clinical Global Impression) Scale – NIMH. Psychopharmacol Bull 1985;21:839–44. [Google Scholar]
- 32. Ambrosini PJ. Historical development and present status of the schedule for affective disorders and schizophrenia for school‐age children (K‐SADS). J Am Acad Child Adolesc Psychiatry 2000;39:49–58. [DOI] [PubMed] [Google Scholar]
- 33. Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand Suppl 1970;212:11–19. [DOI] [PubMed] [Google Scholar]
- 34. Barnes TR. A rating scale for drug‐induced akathisia. Br J Psychiatry 1989;154:672–676. [DOI] [PubMed] [Google Scholar]
- 35. Inada T, Matsuda G, Kitao Y, et al Barnes Akathisia Scale: Usefulness of standardized videotape method in evaluation of the reliability and in training raters. Int J Methods Psychiatr Res 1996;6:49–52. [Google Scholar]
- 36. Rapoport JL. DSM‐III‐R and pediatric psychopharmacology. Psychopharmacol Bull 1985;21:803–806. [PubMed] [Google Scholar]
- 37. McCracken JT, McGough J, Shah B, et al Risperidone in children with autism and serious behavioral problems. N Engl J Med 2002;347:314–321. [DOI] [PubMed] [Google Scholar]