

Summary
Aims
Stromal cell‐derived factor 1 (SDF‐1) is critical for neural progenitor cell (NPC) migration after ischemia for nerve repair, but how hypoxic induction of SDF‐1 is regulated has not been fully addressed. Here, we examined the regulation of SDF‐1 hypoxic induction by the transcription factors nuclear factor‐κB (NF‐κB) and hypoxic inducible factor 1α (HIF‐1α) in astrocytes.
Methods and Results
Stromal cell‐derived factor‐1 in astrocyte‐conditioned medium (ACM) collected from hypoxic astrocytes induced a time‐ and dose‐dependent increase in NPC migration using chemotaxis assay. The increase in NPC migration correlated with increased SDF‐1 production in astrocytes by real‐time PCR and ELISA assays. Astrocytes produced SDF‐1 time‐dependently upon 3% O 2 treatment, which was associated with increased levels of NF‐κB and HIF‐1α using Western blot analysis. Anti‐HIF‐1α compound, 3‐(5′‐hydroxymethyl‐2′‐furyl)‐1‐benzylindazole (YC‐1) and NF‐κB inhibitor pyrrolidine dithiocarbamate (PDTC), decreased hypoxic induction of SDF‐1, and PDTC pretreatment cancelled HIF‐1α expression as well, thus NPC migration induced by ACM was decreased accordingly. Moreover, lentiviurs siRNA for NF‐κB p65 abrogated induction of HIF‐1α and SDF‐1 under hypoxia in astrocytes.
Conclusions
Hypoxic induction of SDF‐1 is reliant upon NF‐κB and HIF‐1α. There is a cross‐talk between HIF‐1α and NF‐κB, both HIF‐1α and SDF‐1 are downstream targets of NF‐κB in hypoxia condition.
Keywords: Hypoxic inducible factor 1α, Hypoxia, Migration, Neural progenitor cell, nuclear factor‐κB, Stromal cell‐derived factor‐1
Introduction
Ischemia induces a robust neuroinflammatory response that includes marked changes in the gene‐expression profiles 1 and is characterized as hypoxia by a decreased oxygen tension within cells. Migration is a critical process for stem cells recruitment into target area for tissue repairing. Stromal cell‐derived factor‐1 (SDF‐1), a member of the CXC chemokine 2, regulates neuronal migration and axonal path‐finding in the developing nervous system and guides neuroblast migration and vasculogenesis in response to ischemic injury 3, 4. Especially, SDF‐1 is upregulated in the lesion, recruiting bone marrow‐derived stem cells and neural progenitor cells (NPC) toward the damaged tissue after cerebral ischemia 5, 6, 7 for nerve repair. We recently reported that SDF‐1 regulates NPC proliferation 8 and reactive astrocytes showed significant increase in SDF‐1 expression 9. Consequently, the regulation of SDF‐1 expression in ischemia represents a promising therapeutic strategy to improve nerve repair.
Hypoxic inducible factor 1α (HIF‐1α), a master regulator of the cellular response to hypoxia, is precisely regulated by the cellular oxygen concentration 10, maintaining tissue integrity and homeostasis in ischemia. In nonhypoxic conditions, HIF‐1α is subject to ubiquitination, via the von Hippel Lindau protein E3 ubiquitin ligase, and undergoes subsequent rapid proteasomal degradation 10, 11. It is evidenced that multiple genes are activated by HIF‐1α, including glycolytic enzymes, vascular endothelial growth factor and the proapoptotic proteins, functioning in both adaptive and pathological processes 12. At the same time, HIF‐1α expression and activity are critically regulated by many factors. Nuclear factor‐κB (NF‐κB) is a critical transcriptional activator of HIF‐1α and that basal NF‐κB activity is required for HIF‐1α protein accumulation under hypoxia in cultured cells, liver, and brain of hypoxia animals 13. Hypoxia also induces the nuclear translocation of NF‐κB, which precedes HIF‐1α accumulation 13, 14. NF‐κB activation is controlled by IκB kinase (IKK), mainly IKK‐β, needed for phosphorylation‐induced degradation of IκB inhibition in response to infection and inflammation 14, 15. All the collected data implicate that a cross‐talk may exist among SDF‐1, HIF‐1α, and NF‐κB. Therefore, we hypothesized that SDF‐1 produced by astrocytes under hypoxia condition was regulated by HIF‐1α and NF‐κB, and both SDF‐1 and HIF‐1α were activated by NF‐κB as the downstream targets.
In this study, we demonstrated that SDF‐1 from hypoxic astrocytes was responsible for NPCs migration, and hypoxic induction of SDF‐1 was both HIF‐1α and NF‐κB dependent. Knock‐down of NF‐κB expression abrogated increased HIF‐1α expression, followed by SDF‐1 hypoxic induction was abolished too, which led to a decreased NPC migration accordingly. Therefore, both SDF‐1 and HIF‐1α were regulated by NF‐κB upon hypoxic response.
Materials and Methods
Materials
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), trypsin, neurobasal medium, DMEM/F12, and B27 supplement were provided by Gibco (Grand Island, NY, USA). Basic fibroblast growth factor (bFGF), epidermal growth factor (EGF), N‐acetylcysteine, poly‐d‐lysine, 3‐(5′‐hydroxymethyl‐2′‐furyl)‐1‐benzylindazole (YC‐1), pyrrolidine dithiocarbamate (PDTC), Hoechst 33342, antinestin, and anti‐β‐actin antibodies were obtained from Sigma‐Aldrich (St. Louis, MO, USA). Recombinant SDF‐1α protein, anti‐SDF‐1 monoclonal antibody, biotinylated goat anti‐SDF‐1 antibody, and neutralization antibody for SDF‐1 were purchased from R&D Systems (Minneapolis, MN, USA). Chemotaxis chambers were purchased from Neuro Probe (Gaithersburg, MD, USA). Anti‐MAP2 and anti‐HIF‐1α polyclonal antibodies were from Millipore (Bedford, MA, USA), and anti‐GFAP antibody was purchased from Dako Corp (Carpinteria, CA, USA). Anti‐NF‐κB p65 and anti‐TATA box binding protein (TBP) antibodies were purchased from Abcam (Cambridge, MA, USA). AlexaFluro 488 goat anti‐mouse IgG and AlexaFluro 594 goat anti‐rabbit IgG were purchased from Molecular Probes (Eugene, OR, USA). Horseradish peroxidase‐conjugated (HRP) secondary antibodies for Western blot were from Santa Cruz Biotech (Santa Cruz, CA, USA). Nuclear Extraction Kit (NE‐PER), M‐PER Protein Extraction Buffer, and enhanced chemiluminescent solution (ECL) were obtained from Pierce (Rockford, IL, USA).
Mouse NPC, Neuron, and Astrocyte Culture
Primary cultures of NPCs and cortical neurons were prepared from the brain of E15–E16 C57 mouse embryos (obtained from the Experimental Animal Center of the Fourth Military Medical University, China). Briefly, dissociated brain tissue from E15–E16 mouse was incubated with 0.125% trypsin in Ca2+ and Mg2+‐free Hank's balanced salt solution at 37°C for 10 min. Then, the cortex were washed in DMEM supplemented with 10% FBS to stop trypsin activity and further dissociated by trituration. For NPC culture, single cells were cultured in substrate‐free tissue culture flasks and grown in suspension as spheres in neurosphere initiation medium (NPIM), which consisted of neurobasal medium supplemented with 2% B27, 20 ng/mL bFGF, 20 ng/mL EGF, and 60 ng/mL N‐acetylcysteine. Cells were passaged at 5 days intervals as previously described 8, and half of the medium was changed every other day. The purity of NPC was assessed by immunocytochemical staining using antibody to its mark, nestin, revealing that this culture procedure yielded more than 95% NPCs. NPCs of passage 3–12 were used for migration assay.
For neurons culture, the dissociated single cell suspension was cultured on poly‐d‐lysine coated plates in neurobasal medium supplemented with 2% B27, 0.5 mM glutamine, 100 U/mL penicillin, and 100 U/mL streptomycin. It took 10 days for reincubation, the time required for maturation of cortical neurons and half of the medium was changed every 2 days. The cells were characterized by immunocytochemical staining with anti‐MAP2 antibody, revealing that this culture procedure yielded more than 95% neurons. Neurons seeded at a density of 3 × 105 cells/well in a 24‐well plate were treated with 3% O2 for 1, 2, 4, 8, 12, 24, and 48 h. The supernatants were collected at each time point and subjected to ELISA assay for SDF‐1 production, and protein samples were harvested to check the expression changes of interested proteins by Western blot assay.
Mouse cultures of astrocytes were prepared from the cortex of postnatal C57BL/6 mice (obtained from the Experimental Animal Center of Fourth Military Medical University, China) as previously described 9 with modifications. Cells were cultured at a density of 2 × 107 cells/150 cm2 in DMEM supplemented with 10% FBS, 50 U/mL penicillin, and 50 mg/mL streptomycin at 37°C in a humidified atmosphere containing 95% air and 5% CO2. The adherent astrocytes were quaked with 180 rpm rate for 16 h after 10 days culture and then digested by 0.25% trypsin. The cell suspension was cultured under the same condition, and the purity of astrocyte was assessed by immunocytochemical staining using antibody to its mark, glial fibrillary acidic protein (GFAP). This process yielded a culture of >95% pure astrocytes, and no microglia, oligodendrocytes, and macrophage were detected using their specific marks respectively. All the studies utilizing animal subjects were performed in full compliance with the Fourth Military Medical University ethical guidelines.
Astrocytes were seeded into 24‐well plates at a density of 3 × 105 cells/well. Cells were treated with 3% O2 for 1, 2, 4, 8, 12, 24, and 48 h. To determine the roles of HIF‐1α and NF‐κB in SDF‐1 induction under hypoxia, cells were pretreated with 1 mM YC‐1, the inhibitor for HIF‐1α and 1 mM PDTC, the inhibitor for NF‐κB p65 for 2 h before hypoxic treatment, respectively. To further confirm their roles in SDF‐1 induction under hypoxia, astrocytes were infected with NF‐κB p65siRNA lentivirus for 48 h then subjected to hypoxic injury as before. The supernatants were collected for SDF‐1 production by ELISA assay. Cell‐free culture supernatants were collected as astrocyte‐conditioned medium (ACM) from the astrocytes after different treatments at desire time points.
Migration Assays
In vitro chemotaxis assays were performed using Transwell cell chambers to detect NPC migration following the manufacturer's instruction. 3 × 104 NPCs in 50 μL were seeded onto Chemotaxis filters (5.7‐mm, 8‐μm pore; Neuro Probe) in NPIM. ACM collected from astrocytes which were subjected to either 3% O2 treatment, YC‐1 or PDTC pretreatment were added to the lower chamber. Culture medium alone from the same condition was used as control. Chemotaxis chambers were incubated at 37°C in 5% CO2 for 0, 30 min, 1, and 2 h. After the desire migration periods, nonmigrating cells were completely wiped from the top surface of the membrane. We measured migrating cells adhering to the undersurface of the filters by Hoechst 33342 staining and quantified with Image‐ProPlus software. Counts were expressed as total cell number per field. Results are indicative of four independent experiments.
Transfection of siRNA Targeting NF‐κB p65 Subunit
A double‐stranded siRNA (sense, 5′‐GCCCUAUCCCUUUACGUCA‐3′, antisense 5′‐UGACGUAAAGGGAUAGGGC‐3′), which encoded amino acid residues 347 and 353 of the NF‐κB p65 subunit, was designed to target the NF‐κB p65 subunit (p65 siRNA). Lentivirus provided a convenient vector by which to integrate RNAi expression constructs. Pantropic VSV‐G‐pseudotyped viral particles containing the RNA copy of the lentivector expression construct can be efficiently used to deliver and stably express effector and reporter sequences in a wide range of mammalian target cells. In this study, we packaged HIV‐based lentivector expression construct in VSV‐G‐pseudotyped viral particles by cotransfecting 293T producer cells with a lentivector expression construct and Packaging Plasmid Mix. At the end, we collected viral particles and determined the titer. The green fluorescent protein (GFP) expression construct as an internal positive control in all of experiments. Two days postdissociation, astroctytes were infected with p65siRNA lentivirus at multiplicity of infection 16 from 0.1, 1, 10, to 100. A nonspecific control siRNA was also transfected at the same concentration as control. The cells were incubated for 24 h, the culture media was refreshed, and the cells were cultured for a further 24 h. At 24‐h postinfection, cells were incubated with Hoechst 33342 for nuclear staining and then infected and total cells were counted. The silencing effect of protein expression was confirmed by Western blot analysis.
RNA Extraction and TaqMan Real‐Time RT‐PCR
Cells were incubated in 3% O2 for different time points, and total cellular mRNA was extracted using Rneasy Mini Kit (Qiagen, Valencia, CA, USA). The real‐time quantification of SDF‐1 mRNA was performed using SYBR Green I dye (Applied Biosystems, Foster City, CA, USA) with gene‐specific primers as following:
Mouse SDF‐1 forward: 5′‐GCTCTGCATCAGTGACGG TA‐3′
Mouse SDF‐1 reverse: 5′‐TAATTACGGGTCAATGCACA‐3′
SYBR Green I, double‐stranded DNA binding dye, was detected by using the laser‐based ABI Prism 7700 Sequence Detection System (Applied Biosystems). PCR amplification was performed by using an optical 96‐well reaction plate and caps. The final reaction mixture of 25 μL consisted of 200 nM each primer, 1× SYBR Green PCR Master Mix (Applied Biosystems) containing a reference dye, and cDNA at the following conditions: 50°C for 2 min, 95°C for 10 min, followed by 40 cycles at 95°C for 15 second, and 60°C for 1 min. The cDNAs were prepared from each RNA sample by using a TaqMan Reverse Transcription Kit. SDF‐1 mRNA levels were determined and standardized with GAPDH internal control. Standards and samples were run in triplicate.
ELISA Assay
To assess the secretion of SDF‐1, cell‐free supernatant from neurons and astrocytes cultures after different treatment was collected and tested in triplicate for soluble SDF‐1 by enzyme‐linked immunosorbent assay (ELISA). Mouse‐specific Duoset ELISA development system kits (R&D Systems) was used according to the manufacturer's instructions.
Western Blot Analysis
To study the effects of hypoxia on the expression of HIF‐1α and NF‐κB, cells were treated for various time points (0, 1, 2, 4, 8, 12, 24, 48 h) in 3% O2 culture condition. For experiments utilizing inhibitors, cells were pretreated with YC‐1 (1 mM) or PDTC (1 mM) for 2 h followed by the same hypoxia stimulation. To determine the roles of NF‐κB in SDF‐1 production and HIF‐1α expression, NF‐κB p65siRNA transfection was carried out 24–48 h before hypoxia treatment.
After each treatment, cells were rinsed twice with PBS, then nuclear protein was harvested using Nuclear Extraction Kit or total protein was obtained by M‐PER Protein Extraction Buffer containing 1× protease inhibitor cocktail. Cell proteins were quantified by a BCA Kit (Pierce, Rockford, IL, USA) and separated on 10% polyacrylamide gel followed by transfer onto an Immun‐Blot PVDF membrane. The membrane was blocked for 1 h with 5% nonfat milk in Tris–phosphate buffer containing 0.05% Tween 20 (TBS‐T). It was further incubated overnight at 4°C with primary antibodies including anti‐HIF‐1α polyclonal antibody (1:100), anti‐NF‐κB polyclonal antibody (1:500), anti‐TBP antibody (1:1000), and anti‐β‐actin monoclonal antibody (1:10,000). After five washes with TBS‐T, 5 min for each, membranes were further incubated with HRP‐conjugated secondary antibodies for 1–2 h and followed by four TBS‐T washes. The target protein signal was detected and digitized using ECL 17 and Image J program.
Data Analysis
Data were presented as means ± SD. Statistical comparisons were performed using a t‐test or one‐way ANOVA. Significance was considered to be <0.05. To account for any donor‐specific differences, all experiments were performed from at least three donors.
Results
Astrocyte‐Conditioned‐Medium‐Mediated NPC Migration
Increased NPC migration was evidenced in ischemia, which is characterized as low oxygen tension, but the mechanisms remain unclear. Primary mouse astrocytes were cultured in 10% FBS, and the purity of astrocyte was determined by anti‐GFAP immunocytochemical staining as showed in Figure 1A. NPCs were cultured in NPIM, and the purity of NPC was determined by antinestin immunocytochemical staining as showed in Figure 1A. The purities of astrocytes and NPCs were over 95%. Astrocytes of passage 3–12 were dissociated and subjected to 3% O2 for 12 h, then cell‐free supernatant was collected as ACM. The migration of NPC induced by ACM was measured by chemotaxis assay. The result showed that ACM induced NPC migration time‐dependently, peaked at 2 h, as showed in Figure 1B. The migration of NPC was also determined by different percentage of ACM (0%, 25%, 50%, 100%). It showed that ACM induced NPC migration dose‐dependently as illustrated in Figure 1C. The medium from the same culture condition alone served as control. The data showed no NPC migration happened under control medium stimulation.
Figure 1.
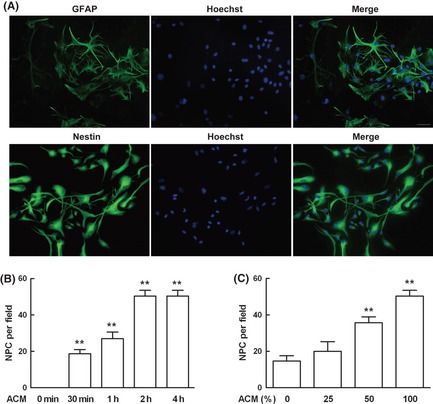
Astrocyte conditioned medium (ACM) mediated neural progenitor cell (NPC) migration. (A) The purities of mouse astrocytes and NPC were identified by immunostaining with their marks glial fibrillary acidic protein (GFAP) and nestin, respectively, and nuclei were counterstained with Hoechst 33342. This culture system yielded more than 95% astrocytes and NPCs. (B) Astrocytes of passage 3–12 were dissociated and subjected to 3% O 2 for 12 h, supernatant was collected as ACM and NPC migration induced by ACM was measured by chemotaxis assay. ACM mediated NPC migration in a time‐dependent manner. (C) ACM induced NPC migration in a dose‐dependent manner. Each value represented the mean ± SD of four independent experiments (n = 4 experiments, **P < 0.01 vs. control group).
SDF‐1 in ACM under Hypoxia is Responsible for NPC Migration
Stromal cell‐derived factor‐1 is considered critical both for brain development and for nerve repair after ischemia, and then we checked the production of SDF‐1 in astrocytes after exposure to hypoxia. To establish whether the migration of NPC was due primarily to SDF‐1 production in hypoxic astrocytes, we neutralized ACM with SDF‐1‐specific antibody that harvested from astrocytes exposure to 3% O2 for 12 h. Then, the migration of NPC induced by ACM was reassessed. The data demonstrated that the migration of NPC mediated by ACM time‐ and dose‐dependently was abrogated by neutralized antibody for SDF‐1, whereas an isotype control IgG had no effect (Figure 2A,B). We further checked SDF‐1 mRNA induction in astrocytes under hypoxia. Mouse astrocytes at passage 3–12 were subjected to 3% O2 for different time points, and real‐time PCR assay was used to measure SDF‐1 mRNA induction. SDF‐1 mRNA induction increased markedly 2 h after hypoxia treatment and peaked at 12 h, lasting for 48 h in astrocytes upon hypoxia stimulation (Figure 2C). At the same time, the supernatant at different time points was harvested to determine SDF‐1 secretion after hypoxia. SDF‐1 induction increased markedly after 12 h hypoxic treatment compared with control (Figure 2D).
Figure 2.
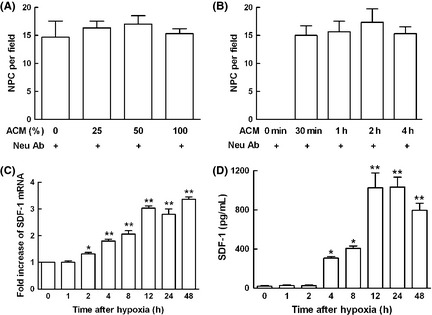
Hypoxic induction of stromal cell‐derived factor 1 (SDF‐1) was responsible for astrocyte conditioned medium (ACM)‐mediated neural progenitor cell (NPC) migration. (A) ACM was collected after exposure to 3% O 2 for 12 h, and NPC migration induced by ACM was measured by chemotaxis assay. The effect of NPC migration induced by ACM dose‐dependently was abolished by neutralized antibody for SDF‐1. (B) The effect of NPC migration mediated by ACM time‐dependently was abolished by neutralized antibody for SDF‐1. (C) Astrocytes of passage 3–12 were dissociated and subjected to 3% O 2 for different time points, cells were collected and real‐time PCR assay was used to measure SDF‐1 mRNA induction, GAPDH served as internal control. Hypoxia increased SDF‐1 mRNA expression time‐dependently. (D) The supernatant of cultured astrocytes at different time points were collected to determine SDF‐1 secretion after hypoxia by ELISA assay. SDF‐1 production increased time‐dependently upon hypoxia stimulation in mouse astrocytes. Each value represented the mean ± SD of three independent experiments (n = 3 experiments, *P < 0.05, **P < 0.01 vs. control group).
Based on the observation that ACM induced NPC migration, we then asked whether other cell types besides astrocytes were involved in SDF‐1 production under hypoxia stimulation, because all type of cells were insulted upon ischemia injury. We cultured neurons from embryonic mice and the purity of neurons was determined by immunocytochemical staining with anti‐MAP2 (neuronal marker). This culture system yielded more than 95% neurons (data not shown). Mouse neurons cultured for 10 days subjected to 3% O2 as before, SDF‐1 mRNA induction was measured by real‐time PCR assay. Comparing SDF‐1 mRNA between neurons and astrocytes, the data showed that there were nearly no changes in neurons (data not shown) after hypoxia treatment, therefore, the changes of SDF‐1 in neurons were not checked in the following experiments.
Hypoxia‐Inducible Factor 1 Affected SDF‐1 Expression under Hypoxia in Mouse Astrocytes
Our result showed that astrocytes activation manifesting increased SDF‐1 production upon hypoxic stimulation. Then, we asked that how SDF‐1 production was regulated in this situation in case to truly understand the mechanisms involved and may shed light on potential therapy targets. Among bodies of regulative elements under hypoxia, transcription factor HIF‐1α plays a central role during pathophysiology process in case of survival and homeostasis, regulating the target genes expression adapting to the insult environment. Therefore, we next examined whether the hypoxic effect upon induction of SDF‐1 was dependent on HIF‐1α transcription.
Mouse astrocytes subjected to 3% O2 for different time points and protein samples were harvested, HIF‐1α expression was determined by Western blot. HIF‐1α expression increased at 1 h upon hypoxia, peaked at 2 h, and lasted 24 h as showed in Figure 3A,B. To further determine the effects of HIF‐1α on SDF‐1 induction, astrocytes were pretreated with 1 mM YC‐1, the inhibitor for HIF‐1α for 2 h, and then subjected to hypoxia for 24 h, which was the peak time for SDF‐1 secretion upon hypoxia stimulation. SDF‐1 mRNA induction was measured by real‐time PCR. The increased SDF‐1 mRNA was abolished by YC‐1 pretreatment upon hypoxia stimulation as showed in Figure 3C. Accordingly, the migration of NPC induced by ACM for 2 h, which was collected from astrocytes after exposure to 3% O2 for 12 h, was decreased as showed in Figure 3D. This data implicated that SDF‐1 production was HIF‐1α‐dependent under hypoxia.
Figure 3.
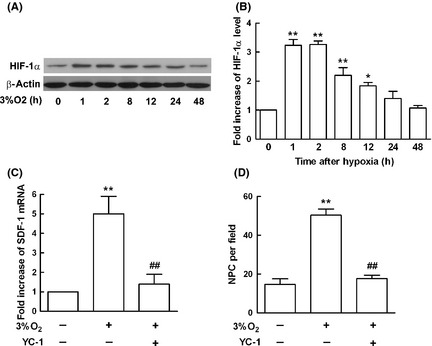
Hypoxia‐inducible factor 1α (HIF‐1α) affected Stromal cell‐derived factor‐1 (SDF‐1) expression under hypoxia in mouse astrocytes. (A) Mouse astrocytes were subjected to 3% O 2, and protein samples were harvested at different time points. Western blot analysis showed the expression levels of HIF‐1α, and β‐actin served as internal control. (B) HIF‐1α levels in astrocytes increased time‐dependently after exposure to 3% O 2 compared with the control. (C) To determine the effects of HIF‐1α on SDF‐1 production, astrocytes were pretreated with YC‐1 (1 mM), the inhibitor for HIF‐1α for 2 h, and then subjected to hypoxia for 12 h, which was the peak time for SDF‐1 mRNA expression upon hypoxia stimulation. Real‐time PCR results showed YC‐1 decreased SDF‐1 mRNA induction markedly, GAPDH served as internal control. (D) The migration of neural progenitor cell (NPC) mediated by astrocyte conditioned medium (ACM) for 2 h, which was collected from astrocytes after exposure to 3% O 2 for 12 h, was abolished by YC‐1 pretreatment in astrocytes. Each value represented the mean ± SD of three independent experiments (n = 3 experiments, ** P < 0.01 vs. control group, ## P < 0.01 vs. 3% O 2 group).
NF‐κB p65 was Responsible for SDF‐1 Production upon Hypoxia Stimulation in Astrocytes
Nuclear factor‐κB is an ubiquitous transcription factor known to be activated by a wide variety of stimuli, which, in turn, regulates the expression of inflammatory and oncogenic genes. Therefore, we next examined whether hypoxic induction of SDF‐1 occurred specifically through an NF‐κB‐dependent pathway. Mouse astrocytes were subjected to 3% O2 for different time points as before, and nuclear protein samples were extracted. The most common form of NF‐κB is a dimer of p65 and p50, which is often referred to simply as NF‐κB. The levels of p65 increased significantly upon hypoxia stimulation by Western blot assay as showed in Figure 4A, TBP served as nuclear protein loading control. To further determine the effects of p65 on SDF‐1 induction, astrocytes were pretreated with PDTC (1 mM), the inhibitor of p65 for 2 h before exposure to hypoxia for 12, 24, and 48 h. The secretion of SDF‐1 was determined using ELISA assay. The increase in SDF‐1 induction upon hypoxia stimulation was abolished after PDTC pretreatment (Figure 4B). ACM was collected from different treatments, and the migration of NPC was reassessed. The migration of NPC induced by ACM for 2 h, which was collected from astrocytes after exposure to 3% O2 for 12 h, was decreased in PDTC‐pretreated group as showed in Figure 4C. This result showed that NF‐κB seemed to play a critical role in hypoxic induction of SDF‐1.
Figure 4.
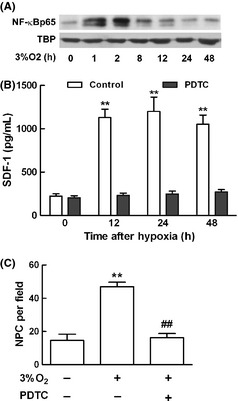
NF‐κB p65 was responsible for stromal cell‐derived factor‐1 (SDF‐1) production upon hypoxia stimulation in astrocytes. (A) Mouse astrocytes were subjected to 3% O 2 culture condition for different time points, and nuclear protein samples were extracted. Western blot analysis showed the expression levels of p65 and TBP served as nuclear protein loading control. (B) To determine the effects of p65 on SDF‐1 production, astrocytes were pretreated with pyrrolidine dithiocarbamate (PDTC) (1 mM), the inhibitor for p65 for 2 h, and then subjected to hypoxia for 12, 24, and 48 h to check SDF‐1 secretion using ELISA assay. PDTC pretreatment abolished SDF‐1 hypoxic induction. (C) The migration of neural progenitor cell (NPC) mediated by astrocyte conditioned medium (ACM) for 2 h, which was collected from astrocytes after exposure to 3% O 2 for 12 h, was abolished by PDTC pretreatment in astrocytes. Each value represented the mean ± SD of three independent experiments (n = 3 experiments, **P < 0.01 vs. control group, ## P < 0.01 vs. 3% O 2 group). NF‐κB, nuclear factor‐κB.
The Production of SDF‐1 in Astrocytes under Hypoxia was through NF‐κB/HIF‐1α Signaling Pathways
Nuclear factor‐κB is a critical transcriptional activator of HIF‐1α and that basal NF‐κB activity is required for HIF‐1α protein accumulation under hypoxia in cultured cells, liver, and brain of hypoxia animals 13. All the collected data in the present study showed that hypoxic induction of SDF‐1 was dependent on both HIF‐1α and NF‐κB, and it was reported that NF‐κB selectively augmented HIF‐1α signaling during hypoxia 18, and then, we asked whether there was a cross‐talk between HIF‐1α and NF‐κB. We first constructed siRNA lentivirus targeting NF‐κB p65. Using this siRNA delivery system, mouse astrocytes at passage 3–12 were infected with NF‐κB p65siRNA and control siRNA (Figure 5A). Using fluorescence microscope, we found over 90% cells were GFP positive at MOI 10 and there are no toxicity after infection. NF‐κB p65 expression was knocked down over 75% efficiently by Western blot assay as showed in Figure 5B accordingly, and β‐actin was used as internal control. Based on this system, we further explored the expression changes of HIF‐1α and SDF‐1 in astrocytes under hypoxia. Astrocytes in which NF‐κB p65 knocked down were subjected to 3% O2 for 1, 2, and 8 h, HIF‐1α expression decreased significantly compared with control cells (Figure 5C). Furthermore, the supernatant of astrocytes was harvested after exposure to 3% O2 for 12, 24, and 48 h to determine SDF‐1 production. The hypoxic induction of SDF‐1 in astrocytes was also abrogated after NF‐κB p65 was knocked down by p65siRNA as showed in Figure 5D.
Figure 5.
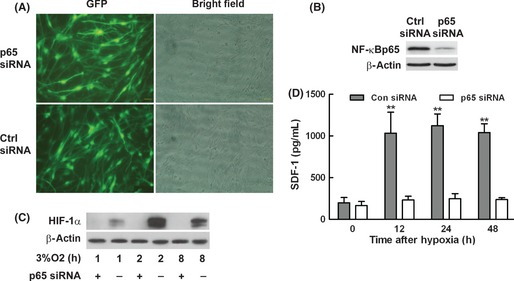
The induction of stromal cell‐derived factor‐1 (SDF‐1) in astrocytes under hypoxia was through nuclear factor‐κB (NF‐κB)/Hypoxic inducible factor 1α (HIF‐1α) signaling pathways. (A) Mouse astrocytes of passage 3–12 were infected with p65siRNA lentivirus and control siRNA, and the efficiency of infection was determined as GFP by fluorescence microscope compared to bright field. More than 90% of astrocytes were infected with p65siRNA lentivirus and control siRNA. (B) Western blot results showed the level of NF‐κB was knocked down by p65siRNA and β‐actin served as internal control. (C) The expression changes of HIF‐1α in astrocytes at different time points after p65 gene knocked down under hypoxia was determined by Western blot assay. (D) The supernatant of astrocytes after hypoxia was collected, and SDF‐1 secretion was determined by ELISA assay. Hypoxic induction of SDF‐1 in astrocytes was abrogated after cells were infected with p65siRNA. Each value represented the mean ± SD of three independent experiments (n = 3 experiments, **P < 0.01 vs. control group).
In conclusion, we have demonstrated that the induction of SDF‐1 in astrocytes is NF‐κB/HIF‐1α dependent under hypoxia, knock‐down of NF‐κB p65 resulted in both HIF‐1α and SDF‐1 production abolished. These results may enhance our understanding of the molecular mechanisms of astrocyte activation and provide a potential site of pharmacological or genetic intervention for the treatment of central nervous system (CNS) disorders associated with cerebral ischemia.
Discussion
The data presented above suggest that the migration of NPC is mediated by SDF‐1, which is relying on both HIF‐1α and NF‐κB, and HIF‐1α is regulated by NF‐κB as a target gene upon hypoxia. This is illustrated by the presence of inhibitors and p65 siRNA, implicating that there is a cross‐talk between HIF‐1α and NF‐κB in the regulation of chemokine SDF‐1 production upon hypoxia insult.
The hypoxic response is an ancient stress response triggered by low ambient oxygen within cells and occurs under several pathophysiological situations including inflammation, cancer, and ischemia 19. Ischemic brain damage, caused by stroke and trauma, induced a number of inflammatory mediators expression in the brain 20, 21. Astrocytes play critical roles in health and disease, acting as active partners with neurons in two‐way communication that alters neuronal physiology and survival 22, 23, 24. The influence of the astrocytes on nerve repair during ischemia is complex, involving both adaptive and pathological functions.
In the CNS, chemokines are produced by astrocytes and other cell types in response to hypoxia/ischemia. Chemokines play central roles in the inflammatory process by forming a chemo‐attractant gradient that attract inflammatory cells (neutrophils, monocytes, and macrophages) in blood to transmigrate across the blood–brain barrier into the brain 25, 26. Chemokine SDF‐1 has been evidenced widely increased during ischemia and mediates NPC migration 3, 4, while the regulation of SDF‐1 hypoxic production remains elusive.
Hypoxia is controlled by HIF‐1, which is a heterodimeric transcription factor composed of a constitutively expressed HIF‐1β (ARNT) subunit and an oxygen‐regulated HIF‐1α subunit. HIF‐1α is susceptible to oxygen‐dependent degradation under normoxia conditions through the hydroxylation of proline molecules by prolyl hydroxylases 27. HIF‐1α subunit is accumulated during hypoxia by posttranslational stabilization, but undergoes a rapid oxygen‐dependent degradation via the ubiquitin‐proteasome pathway upon reoxygenation. HIF‐1‐mediated induction of hypoxia‐sensitive genes takes place at functionally essential HIF‐1‐binding sites to response in both adaptive and pathological processes 12. As a transcription factor, HIF‐1α exerted its effects through transcriptional upregulation of chemokine SDF‐1 during hypoxia in this study. It seemed that SDF‐1 production was initiated by HIF‐1α, while HIF‐1α was not necessary during the whole process. Details of the mechanisms whereby the activation and degradation of HIF‐1α mRNA translation remain to be elucidated.
Upregulation of inflammatory genes by hypoxia/ischemia may be regulated by different transcription factors at different stages of the inflammation, including HIF‐1α and NF‐κB 28, 29, 30. NF‐κB is the collective name for a transcription factor that exists as either a hetero‐ or homodimer, which consists of subunits RelA (p65), RelB, cRel, p50 and its precursor p105 (NF‐κB1), p52 and its precursor p100 (NF‐κB2) 31. It is clear that NF‐κB directs the synthesis of cytokines and chemokines which induce the migration of immune effector cells. There have been several studies demonstrating the cross‐talk between the NF‐κB and HIF‐1 signaling pathways, including shared target genes. Rius 13 showed that NF‐κB was a critical transcriptional activator of HIF‐1α, and basal NF‐κB activity is required for the accumulation of HIF‐1α in cultured cells under conditions of hypoxia as well as in the liver and brain of hypoxic animals 13. This evidence provides us the concept of NF‐κB and HIF‐1α engaging in a positive enhancement loop under conditions of hypoxia and inflammation. van Uden 32 showed that the individual NF‐κB members have differential effects on HIF‐1α mRNA levels 32. Furthermore, the presence of an NF‐κB site within the HIF‐1α promoter upstream of the transcriptional start site provides further evidence of the link between these two crucially important transcription factors 33. The binding of NF‐κB p50 and p65 to the HIF‐1α promoter in response to hypoxia gives evidence that NF‐κB regulates HIF‐1α via a transcriptional mechanism 34 and indicates that the basal level of HIF‐1α mRNA is directly modulated by NF‐κB.
The interactions between NF‐κB inflammatory mediators and hypoxia have been investigated in different cell types. HIF‐1α‐dependent upregulation of NF‐κB p65 and IKKα occurred in neutrophils, and this led to the conclusion that HIF‐1α‐dependent NF‐κB signaling is of critical importance in the hypoxic response in neutrophils 19. We demonstrated that the expression of both HIF‐1α and NF‐κB in astrocytes was increased dramatic upon hypoxia insult in this study and the hypoxic induction of both HIF‐1α and SDF‐1 was regulated by NF‐κB upon hypoxic insults. However, it should be noted that these observations in this in vitro study may not be entirely extrapolated to the in vivo situations because the in vitro and in vivo responses of astrocytes to hypoxia/ischemia may not be identical. Further in vivo studies are needed to validate the in vitro observations.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported in part by research grants from National Natural Science Foundation of China, No. 81101457, No. 81070063, and Scientific Research Foundation for the Returned Overseas Chinese Scholars No. HG3402.
The first two authors contributed equally to this study.
References
- 1. Iadecola C, Anrather J. The immunology of stroke: From mechanisms to translation. Nat Med 2011;17:796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Imitola J, Raddassi K, Park KI, et al. Directed migration of neural stem cells to sites of CNS injury by the stromal cell‐derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci U S A 2004;101:18117–18122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stumm R, Hollt V. CXC chemokine receptor 4 regulates neuronal migration and axonal pathfinding in the developing nervous system: Implications for neuronal regeneration in the adult brain. J Mol Endocrinol 2007;38:377–382. [DOI] [PubMed] [Google Scholar]
- 4. Guyon A, Nahon JL. Multiple actions of the chemokine stromal cell‐derived factor‐1alpha on neuronal activity. J Mol Endocrinol 2007;38:365–376. [DOI] [PubMed] [Google Scholar]
- 5. Stumm RK, Rummel J, Junker V, et al. A dual role for the SDF‐1/CXCR4 chemokine receptor system in adult brain: Isoform‐selective regulation of SDF‐1 expression modulates CXCR4‐dependent neuronal plasticity and cerebral leukocyte recruitment after focal ischemia. J Neurosci 2002;22:5865–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thored P, Arvidsson A, Cacci E, et al. Persistent production of neurons from adult brain stem cells during recovery after stroke. Stem Cells 2006;24:739–747. [DOI] [PubMed] [Google Scholar]
- 7. Ohab JJ, Fleming S, Blesch A, Carmichael ST. A neurovascular niche for neurogenesis after stroke. J Neurosci 2006;26:13007–13016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu Y, Peng H, Cui M, Whitney NP, Huang Y, Zheng JC. CXCL12 increases human neural progenitor cell proliferation through Akt‐1/FOXO3a signaling pathway. J Neurochem 2009;109:1157–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peng H, Erdmann N, Whitney N, et al. HIV‐1‐infected and/or immune activated macrophages regulate astrocyte SDF‐1 production through IL‐1beta. Glia 2006;54:619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia‐inducible factor 1 is a basic‐helix‐loop‐helix‐PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 1995;92:5510–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Semenza GL. HIF‐1: Mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol 2000;88:1474–1480. [DOI] [PubMed] [Google Scholar]
- 12. Bruick RK, McKnight SL. A conserved family of prolyl‐4‐hydroxylases that modify HIF. Science 2001;294:1337–1340. [DOI] [PubMed] [Google Scholar]
- 13. Rius J, Guma M, Schachtrup C, et al. NF‐kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF‐1alpha. Nature 2008;453:807–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Finnberg N, El‐Deiry WS. Activating FOXO3a, NF‐kappaB and p53 by targeting IKKs: An effective multi‐faceted targeting of the tumor‐cell phenotype? Cancer Biol Ther 2004;3:614–616. [DOI] [PubMed] [Google Scholar]
- 15. Li Q, Lu Q, Bottero V, et al. Enhanced NF‐kappaB activation and cellular function in macrophages lacking IkappaB kinase 1 (IKK1). Proc Natl Acad Sci U S A 2005;102:12425–12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moissonnier P, Reviron T, Ye JH, Horvat JC. Motoneurons of the injured spinal cord of the adult dog can grow lengthy axons into an autologous peripheral nerve graft. A retrograde axonal tracing study. Spinal Cord 1996;34:320–325. [DOI] [PubMed] [Google Scholar]
- 17. Davies AG, Pierce‐Shimomura JT, Kim H, et al. A central role of the BK potassium channel in behavioral responses to ethanol in C. elegans . Cell 2003;115:655–666. [DOI] [PubMed] [Google Scholar]
- 18. Ryan S, McNicholas WT, Taylor CT. A critical role for p38 map kinase in NF‐kappaB signaling during intermittent hypoxia/reoxygenation. Biochem Biophys Res Commun 2007;355:728–733. [DOI] [PubMed] [Google Scholar]
- 19. Walmsley SR, Print C, Farahi N, et al. Hypoxia‐induced neutrophil survival is mediated by HIF‐1alpha‐dependent NF‐kappaB activity. J Exp Med 2005;201:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rothwell NJ, Luheshi GN. Interleukin 1 in the brain: Biology, pathology and therapeutic target. Trends Neurosci 2000;23:618–625. [DOI] [PubMed] [Google Scholar]
- 21. Ransohoff RM, Tani M. Do chemokines mediate leukocyte recruitment in post‐traumatic CNS inflammation? Trends Neurosci 1998;21:154–159. [DOI] [PubMed] [Google Scholar]
- 22. Chen Y, Swanson RA. Astrocytes and brain injury. J Cereb Blood Flow Metab 2003;23:137–149. [DOI] [PubMed] [Google Scholar]
- 23. Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci 2003;26:523–530. [DOI] [PubMed] [Google Scholar]
- 24. Trendelenburg G, Dirnagl U. Neuroprotective role of astrocytes in cerebral ischemia: Focus on ischemic preconditioning. Glia 2005;50:307–320. [DOI] [PubMed] [Google Scholar]
- 25. Babcock AA, Kuziel WA, Rivest S, Owens T. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci 2003;23:7922–7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kumai Y, Ooboshi H, Takada J, et al. Anti‐monocyte chemoattractant protein‐1 gene therapy protects against focal brain ischemia in hypertensive rats. J Cereb Blood Flow Metab 2004;24:1359–1368. [DOI] [PubMed] [Google Scholar]
- 27. Salceda S, Caro J. Hypoxia‐inducible factor 1alpha (HIF‐1alpha) protein is rapidly degraded by the ubiquitin‐proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox‐induced changes. J Biol Chem 1997;272:22642–22647. [DOI] [PubMed] [Google Scholar]
- 28. Vangeison G, Carr D, Federoff HJ, Rempe DA. The good, the bad, and the cell type‐specific roles of hypoxia inducible factor‐1 alpha in neurons and astrocytes. J Neurosci 2008;28:1988–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bernaudin M, Tang Y, Reilly M, Petit E, Sharp FR. Brain genomic response following hypoxia and re‐oxygenation in the neonatal rat. Identification of genes that might contribute to hypoxia‐induced ischemic tolerance. J Biol Chem 2002;277:39728–39738. [DOI] [PubMed] [Google Scholar]
- 30. Boycott HE, Wilkinson JA, Boyle JP, Pearson HA, Peers C. Differential involvement of TNF alpha in hypoxic suppression of astrocyte glutamate transporters. Glia 2008;56:998–1004. [DOI] [PubMed] [Google Scholar]
- 31. Kemler I, Fontana A. Role of IkappaBalpha and IkappaBbeta in the biphasic nuclear translocation of NF‐kappaB in TNFalpha‐stimulated astrocytes and in neuroblastoma cells. Glia 1999;26:212–220. [PubMed] [Google Scholar]
- 32. van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia‐inducible factor‐1alpha by NF‐kappaB. Biochem J 2008;412:477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bonello S, Zahringer C, BelAiba RS, et al. Reactive oxygen species activate the HIF‐1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol 2007;27:755–761. [DOI] [PubMed] [Google Scholar]
- 34. Belaiba RS, Bonello S, Zahringer C, et al. Hypoxia up‐regulates hypoxia‐inducible factor‐1alpha transcription by involving phosphatidylinositol 3‐kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol Biol Cell 2007;18:4691–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]