

SUMMARY
Tobacco smoking has been correlated with a lower incidence of Alzheimer's disease (AD). This negative correlation has been attributed to nicotine's properties. However, the undesired side‐effects of nicotine and the absence of clear evidence of positive effects of this drug on the cognitive abilities of AD patients have decreased the enthusiasm for its therapeutic use. In this review, we discuss evidence showing that cotinine, the main metabolite of nicotine, has many of the beneficial effects but none of the negative side‐effects of its precursor. Cotinine has been shown to be neuroprotective, to improve memory in primates as well as to prevent memory loss, and to lower amyloid‐beta (Aβ)) burden in AD mice. In AD, cotinine's positive effect on memory is associated with the inhibition of Aβ aggregation, the stimulation of pro‐survival factors such as Akt, and the inhibition of pro‐apoptotic factors such as glycogen synthase kinase 3 beta (GSK3β). Because stimulation of the α7 nicotinic acetylcholine receptors (α7nAChRs) positively modulates these factors and memory, the involvement of these receptors in cotinine's effects are discussed. Because of its beneficial effects on brain function, good safety profile, and nonaddictive properties, cotinine may represent a new therapeutic agent against AD.
Keywords: Amyloid‐β peptide, Alzheimer's disease, Memory, Tobacco
Alzheimer's disease: The Cholinergic System as a Therapeutic Target
Alzheimer's disease (AD) is a progressive neurodegenerative disorder and the most common cause of dementia [1, 2]. The disease is characterized by extracellular accumulation of senile plaques, mainly composed of aggregated forms of the amyloid‐beta peptide (Aβ), as well as intracellular accumulation of neurofibrillary tangles of the microtubule‐associated protein tau [1]. These neuropathological changes are associated with structural brain abnormalities, inflammation, and cognitive impairment such as impairment of working memory in AD patients. The progressive loss of memory in AD patients correlates with increased levels of Aβ and the deterioration of the cholinergic system in the brain. The degenerative process involves a sequence of pathological events, including early degeneration of the cerebral basal forebrain and subsequent deterioration of the cortical cholinergic system [3, 4]. This deterioration commonly includes a reduction in the levels of acetylcholine (ACh) and α3, α4, and α7 nicotinic ACh receptors (nAChRs) as well as a decrease in the activity of choline acetyltranferase in the brain [5].
Of these nAChRs, the α7 receptors are considered to be ideal therapeutic targets for several neurological conditions, including AD, schizophrenia, and Parkinson's disease (PD) as well as tobacco addiction [6, 7]. The α7nAChR is a homomeric pentamer that has a high permeability to calcium (PCa:PNa≈ 10), and undergoes rapid and reversible desensitization and pronounced inward rectification [8]. The α7 subunit is highly expressed in the cortex, hippocampus, and hypothalamus [8], and has also been suggested to have functionally important expression in nonneuronal tissues such as cells of the immune system [9]. There is evidence suggesting that Aβ, which accumulates in the brain of AD patients, has a high affinity for the α7 receptors [10], acting as both an agonist [11] and an antagonist [12] at these receptors. Based on this evidence, it has been proposed that positive modulators of these receptors may be neuroprotective against Aβ toxicity and stimulate learning and memory [13].
Current therapies for AD improve the function of the cholinergic system; acetylcholinesterase inhibitors reduce the clearance and increase the availability of acetylcholine in the brain [14, 15, 16]. Another current therapeutic approach aims to decrease glutamate excitotoxicity by blocking the N‐methyl D‐aspartate (NMDA) glutamate receptor using the receptor antagonist memantine. Unfortunately, these therapies only slightly ameliorate cognitive deficits in AD patients and show only short‐term effectiveness [17, 18, 19, 20, 21].
A negative correlation between tobacco use and the incidence of AD has been reported [22]. In research into components of tobacco that may enhance cholinergic function, nicotine (3‐[1‐methyl‐2‐pyrrolidinyl] pyridine), an alkaloid derived from tobacco, has been extensively investigated. Nicotine binds to Aβ, blocking its aggregation into fibrils and is thereby neuroprotective [23]. Nicotine also diminishes AD pathology in animal models of the disease [24, 25]. However, clinical studies that aimed to determine the efficacy of nicotine against AD pathology have not shown a significant effect of nicotine in enhancing memory [26, 27] but rather a clear positive effect on attention in AD [27] and PD patients [25, 28]. The positive effect of nicotine on attention has been mostly attributed to its agonistic stimulation of the nAChRs, which plays an important role in mediating memory and attention processes [29, 30, 31]. It is likely that nicotine's effect on cognitive abilities may be counteracted by the nicotine‐induced desensitization of the receptor. However, the failure of nicotine to improve memory in AD patients, its inherent toxicity, and the fact that it induces tachyphylaxis and addiction have discouraged its use in the clinical arena [32].
New evidence suggests that cotinine ([5S]‐1‐methyl‐5‐[3‐pyridyl]‐pyrrolidin‐2‐one), the main metabolite of nicotine, has similar beneficial properties against AD pathology as nicotine but does not have the adverse side‐effects of nicotine. Specifically, it has been shown that cotinine prevented working and reference memory loss in a mouse model of AD (Tg6799) and prevented Aβ aggregation in vitro as well as plaque deposition in vivo[33].
Based on this evidence, this review discusses the potential of cotinine as an agent to prevent or treat AD.
Cotinine
In humans, more than 80% of nicotine is metabolized to cotinine by cytochrome P450 2A6 (CYP2A6) [34] and cytochrome P450 2A5 (CYP2A5) [35] enzymes [36]. The physiologically active form of cotinine, the (‐)‐isomer, accumulates in the body as a result of tobacco exposure. The metabolic rate of cotinine synthesis is determined by one's genetic background. For instance, it has been shown that individuals expressing a shorter form of CYP2A6 (i.e., CYP2A6*4) produce lower levels of cotinine [37]. The expression of different CYP2A6 variants may explain differences in the metabolism of cotinine in people of different ethnicities [38]. For example, CYP2A forms with low‐enzymatic activity are represented differently in different ethnic groups, as observed in about 9.1% in white and 21.9% in black populations [38]. Cotinine, is mostly metabolized by the liver to its major metabolites, trans‐3’‐hydroxycotinine and its glucuronide [39, 40, 41]. Ethnicity affects the clearance of cotinine, with African Americans showing a lower average clearance of cotinine than Caucasians [42]. In addition to genetic factors, food consumption can also influence cotinine production [38], as grapefruit juice has been shown to inhibit the activity of CYP2A6 [43, 44].
Moreover, cotinine crosses the blood–brain barrier [45, 46] and is almost completely absorbed when administered orally. Despite its structural similarities with nicotine (Figure 1), cotinine has distinct pharmacological properties. Cotinine is 100 times less toxic than nicotine, has a longer half‐life (20–24 h vs. 2 h, respectively), and is not addictive in humans [47].
Figure 1.
Comparison of the structures of cotinine and nicotine.
The first studies showing some behavioral effects were performed in different species of animals. A seminal study performed by Yamamoto et al., showed that intravenously administered cotinine slightly increased in EEG activity and behavioral arousal in cats with a minor decrease in blood pressure [48]. In another study, Risner et al. [49] reported the behavioral effects of nicotine compared with those of its metabolites, nornicotine and cotinine, in beagle dogs and squirrel monkeys. Subjects responded under a multiple fixed‐interval (FI) 300‐seconds, fixed‐ratio (FR) 30 response schedule of food presentation. In the dogs, cotinine (0.01–10.0 mg/kg i.m.) produced only dose‐dependent decreases in rates of responding during both FI and FR components. In the squirrel monkeys, however, cotinine (0.1–3.0 mg/kg i.m.) increased responding during FI components; a high dose of 30.0 mg/kg decreased responding during both FI and FR components. These studies initially suggested that cotinine may have behavioral effects. More recently, we and other laboratories have found that cotinine is a memory enhancer in various animal models of disease. For example, cotinine improved the working memory performance of adult rhesus monkeys (Macaca mulatta) in the delayed matching‐to‐sample task (DMTS). Also, we recently published evidence showing that cotinine (5 mg/kg) decreased anxiety and enhanced the extinction of contextual fear memory after fear conditioning in wild type mice [50]. In addition, in the transgenic (Tg) mouse model of AD (Tg6799), long‐term treatment with cotinine prevented working and reference memory impairment, as tested in three different learning and memory tasks, including circular platform, cognitive interference, and radial arm water maze tests. The doses effective in the Tg6799 mice were within the same range as those that improved working memory in primates [51, 52]. In addition to its mnemonic qualities, cotinine has anti‐Aβ aggregation properties that add to its value as a new treatment for AD [33].
The pro‐cognitive effect of cotinine in the Tg6799 mice may be explained by the reduction in the level of the aggregated forms of Aβ, including Aβ plaques and oligomeric forms of the peptide, in the hippocampus and cortex [33]. An advantage of cotinine is that it is not only an anti‐Aβ aggregation compound but also a molecule that stimulates signaling pathways that support memory and brain homeostasis. For instance, cotinine induced the activation of the pro‐survival protein kinase B (Akt)/glycogen synthase kinase 3β (GSK3β) pathway in the brains of both Tg6799 and wild type control mice, suggesting that the activation of these factors is independent from its effect on Aβ aggregation [33]. Interestingly, the Akt/GSK3β pathway is stimulated by the α7 receptor, has roles inhibiting neuronal cell death, and is involved in promoting neuronal synaptic plasticity and long‐term potentiation [53, 54, 55]. Therefore, α7 receptors and the Akt/GSK3β pathway are considered therapeutic targets for improving memory and attention in individuals with various neurological disorders, including AD [13, 56].
Nicotinic Acetylcholine Receptors (nAChRs) as a Therapeutic Target against Alzheimer's disease
Nicotinic acetylcholine receptors are cationic, ligand‐gated channels that mediate fast neurotransmission in the central and peripheral nervous systems [57]. The α7 and α4β2 nAChR subtypes are the most abundant of the nicotinic receptors and are fundamental for mediating working memory and attention in mammals. These receptors are localized throughout the cortex, hippocampus, amygdala, hypothalamus, striatum, and other regions involved in these cognitive processes [13]. Because decreased levels of these receptors have been found in AD brains [58], this reduction is considered to explain, at least in part, the cognitive deficits in AD.
The α7 receptors are very important for mediating sensory gating, attention and learning, and memory, making them an ideal target to improve these cognitive functions. However, these receptors are susceptible to agonist‐induced desensitization, a characteristic that complicates the use of agonists as therapeutic agents. The desensitization can also explain why many acetylcholinesterase inhibitors, which increase the synaptic levels of ACh, as well as drugs acting as agonists of the α7 receptors only induce modest and short‐term therapeutic effects [59].
An alternative approach to treating memory and attention deficits that does not cause the tachyphylaxis induced by α7 nAChR agonists is the use of nonagonist positive modulators of these receptors [60], including partial agonists such as S24795 and GTS‐21[61] or positive allosteric modulators (PAMs) [62] such as PNU‐120596 [63, 64] and galantamine [65, 66]. For example, PAMs can facilitate ACh neurotransmission by binding to receptor regions other than the active site, changing the receptor conformation, and in some cases preventing agonist‐induced desensitization [13].
In an attempt to improve the cognitive abilities of AD patients with this new approach, modulators of the α7 receptors, such as 3‐(2,4‐dimethoxybenzylidene) anabaseine (GTS‐21 or DMXB‐A) [56, 67, 68], and many others, are under development or are currently being investigated in clinical trials [56, 69, 70, 71].
Cotinine as a Modulator of the Nicotinic Acetylcholine Receptors
Cotinine is weak agonist of the α7 receptor, and whether this receptor is the main target of cotinine is still controversial [72]. However, the possibility that cotinine is an allosteric modulator of this receptor needs to be further explored.
Alternatively, to explain the beneficial effects of cotinine on cognitive abilities, it has been postulated that cotinine desensitizes nAChRs located on inhibitory GABAergic neurons of the hippocampus, provoking the activation of the excitatory glutamate receptors in this region of the brain, and thereby stimulating cognitive abilities [73, 74]. This hypothesis is interesting; however, direct evidence that cotinine desensitizes the hippocampal α7 receptor is still needed. Contrary to this idea, new evidence shows that chronic treatment with cotinine stimulates the Akt/GSK3β pathway in the hippocampus and cortex of AD and control littermate mice [33]. Because the Akt/GSK3β pathway is located downstream of the α7 nAChR, it is unlikely that desensitization of the α7 nAChR, which is highly expressed in these brain regions, could lead to the marked activation of these signaling pathway. It is feasible that instead of desensitizing α7 nAChRs, cotinine may act as a PAM of the human α7 nAChR. The positive modulation of these receptors would explain the positive effects of cotinine not only on learning and memory but also in reversing apomorphine‐induced deficits in prepulse inhibition of acoustic startle in rats [75], a process modulated by the α7 nAChR.
Positive modulation of the α7 nAChRs could also explain cotinine's positive effect on neuronal survival because the activation of Akt stimulates pro‐survival proteins (e.g., Bcl‐2 and the transcription factor cAMP responsive element‐binding protein [CREB; Ref. 11] and inhibits the pro‐apoptotic protein c‐Jun N‐terminal kinase (JNK) via the activation of the apoptosis signal‐regulating kinase (Ask) [76, 77; Figure 2). Furthermore, α7 nAChRs can favor synaptic plasticity and cognition by activating the protein kinases phosphoinositide‐3 kinase [PI3K; Refs. 78, 79], Akt, extracellular signal‐regulated kinase 1/2 (ERK1/2), and the transcription factor CREB, which participate in mediating the structural and molecular changes required for learning and memory processes [80, 81].
Figure 2.
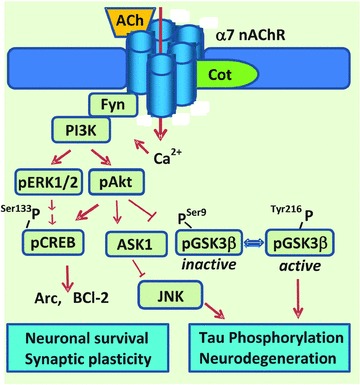
Hypothetical model of cotinine's potentiation of the α7 nAChR. The scheme represents the hypothetical positive allosteric modulation of the α7 nAChR by cotinine, and the activation of signaling pathways that are downstream of the a7 nAChR. The consequences of the activation by cotinine of components of the PI3K‐Akt‐GSK3β pathway on AD pathology are suggested. The activation of Akt by cotinine may result in the inhibition of the tau kinase GSK3β and consequently the formation of hyperphosphorylated forms of tau found in the neurofibrillary tangles (one of the neuropathological hallmarks of AD). Also, the inhibition of GSK3β may inhibit its pro‐apoptotic effect. On the other hand, the stimulation of pERK can stimulate pCREB activity and as a result stimulate the expression of Arc, a protein that participate in the remodeling of the synapses during learning and memory processes and is required for long‐term memory. CREB stimulate the expression of antiapoptotic factors such as Bcl2.
The activation of PI3K heterodimers p85/p110 by the α7 receptor is triggered by the binding of p85 (the regulatory subunit) to phospho‐tyrosyl proteins such as Fyn, which leads to the release of p110 [the catalytic subunit; Ref. 82]. Thus, PI3K stimulates the phosphorylation of Akt at residues threonine 308 and serine 473 [83]. The active form of Akt can then promote neuronal survival by stimulating CREB and Bcl‐2 activity and by inactivating Ask1 by phosphorylation at serine 83 [84].
Of equal importance, because GSK3β is considered to be one of the main tau kinases in vivo, the inhibition of GSK3β by cotinine may also prevent the abnormal phosphorylation of tau observed in AD brains and the consequent appearance of neurofibrillary tangles of hyperphosphorylated tau [85, 86, 87]. The potential inhibition of tau phosphorylation by cotinine is currently being investigated in our laboratory as a new target mechanism against AD.
Furthermore, the stimulation of both CREB and ERK1/2 by cotinine may promote the expression of activity‐regulated cytoskeleton‐associated protein (Arc, also termed Arg3.1), derived from an immediate early gene required for the consolidation of memory [88, 89, 90]. Arc expression is stimulated by brain‐derived neurotrophic factor (BDNF), serum response element [91], NMDA receptors [92], elongation factor 2 [93], CREB, and ERK1/2 [94]. This versatile protein is believed to mediate memory storage in the brain's active networks by coupling changes in neuronal activity patterns to diverse forms of synaptic plasticity [95]. The α7 receptors seem to have a clear role in mediating Arc gene expression, as an increase in levels of Arc mRNA were found in rats treated with the selective α7 receptor partial agonist SSR180711 [96]. Thus, it is through the modulation of the α7 nAChRs and its associated pathways that cotinine may exhibit its pro‐cognitive actions, giving rise to further potential implications in other pathologies that involve a downregulation of these cell signaling cascades.
Cotinine‐inhibited Aβ1–42 Aggregation
According to the updated amyloid cascade hypothesis, AD is predominantly caused by the neurotoxicity of aggregated forms of soluble Aβ[97, 98, 99, 100]. This concept, though still controversial, is supported by the fact that the level of soluble Aβ correlates better with dementia than does plaque burden in AD patients [101, 102].
In aqueous solutions, Aβ undergoes a time‐ and concentration‐dependent transition from a soluble α‐helical to an insoluble β‐sheet structure. Aβ can exist as nontoxic soluble monomers, neurotoxic oligomers and protofibrils, or as insoluble fibrils. Because only the aggregated forms of Aβ are toxic, a great deal of translational research effort has focused on investigating compounds with anti‐Aβ aggregation activity. One recent 78‐week randomized, double‐blind, placebo‐controlled phase 2 study investigated ELND005 (an oral amyloid anti‐aggregation agent) in mild to moderate AD. Therein, the anti‐aggregation approach did not show significant clinical efficacy [103]. It is possible that the small sample size (n = 166 participants) masked a beneficial effect of this drug; however, it is likely that anti‐aggregation drugs may need to target additional aspects of the pathology to achieve clinical efficacy. Studies with larger sample sizes and employing different anti‐aggregation drugs are needed.
It has been postulated that the oligomerization and fibrillation processes are pathways that can occur sequentially or independently of each other. Thus, each of these pathways can be targeted separately or simultaneously by anti‐aggregation compounds [104]. For example, some compounds such as curcumin inhibit oligomerization but not fibrillation [105], and other compounds such as o‐vanillin inhibit the formation of both oligomers and fibrils [106]. Other molecules such as the naphthalene sulfonates inhibit fibrillation but not oligomerization [107]. Few compounds inhibit both processes, and it is these that may be the more promising agents for stopping Aβ toxicity in vivo[108, 109].
We and others have previously demonstrated that cotinine can inhibit both Aβ oligomerization and fibrillation [33]. Seminal studies have shown that cotinine binds to Aβ with high affinity (Ka= 0.1 nM) [110] and inhibits its fibrillation in vitro[23]. Recently, an atomic force microscopy analysis of Aβ1–42 fibrillation under conditions that favor fibrillation, such as high temperature (37°C) and high concentrations of the peptide (millimolar range), confirmed that cotinine inhibits Aβ1–42 aggregation in vitro, decreasing the average number and length of fibrils [33]. Further analysis of the effect of cotinine on Aβ1–42 oligomerization using dot blot techniques showed that cotinine inhibited Aβ1–42 oligomerization in vitro[111]. Consistent with a reduction in Aβ toxicity by inhibiting the aggregation of the peptide into the toxic species, we found that cotinine protected cultured cortical neurons against Aβ1–42 toxicity [111]. This anti‐aggregation effect seems to underlie the reduction in Aβ oligomers and plaques observed in the brains of AD Tg6799 mice treated with cotinine. This decrease paralleled an overall improvement of reference and working memories in the Tg AD mice [112].
The interaction of cotinine with the full‐length Aβ1–42 monomer at the atomic level was elucidated using molecular docking and molecular dynamics (MD) simulations. After the simulation, analysis of the most representative structure indicated that cotinine may interact with the histidine (His) 6, tyrosine (Tyr) 10, and His14 residues of Aβ1–42. The interaction of cotinine with His6, Tyr10, and His14 residues of Aβ1–42 induces important structural changes in the peptide that seem to play a key role in reducing its aggregation. This analysis also indicated that the interaction with cotinine greatly influences the phenylalanine 20 to methionine 35 region of the full‐length Aβ1–42 monomer [112]. This segment contains the loop (amino acids 24–28) and second hydrophobic domain (amino acids 29–35) both of which regions have been implicated in the aggregation process [113]. These results suggest that the interaction of cotinine with key residues in Aβ1–42 may induce critical changes in its secondary structure, inhibiting its aggregation.
Feasibility of using Cotinine as a Pharmacological Therapy against AD
Clinical studies assessing the effect of cotinine on the progression of AD have not yet been performed. However, the pharmacokinetic profiles and safety of orally and intravenously administered cotinine have been previously investigated in humans [47, 114, 115, 116, 117, 118, 119]. A seminal study indicated that doses of up to 1800 mg of cotinine per day during a 4‐day period were well tolerated in humans [114]. Later, another study investigated the safety and efficacy of several oral doses of cotinine fumarate, up to 160 mg per day during a period of 10 days, as an aid for tobacco cessation in abstinent cigarette smokers [47]. At the doses tested, cotinine did not show efficacy in reducing tobacco consumption but did show that, unlike nicotine, its metabolite did not elicit withdrawal effects, addictive behavior, or any negative cardiovascular effects, such as increasing heart rate or systolic or diastolic blood pressures [47]. The multiple mechanisms of action of cotinine and its good safety profile in humans make this drug an ideal candidate for preventing, delaying, or halting AD pathology.
Conclusions
AD is characterized by deterioration of the cholinergic system, including loss of cholinergic neurons and downregulation of nAChRs in the brain [120]. Nicotine itself shows limited use in the clinic because of its toxicity and addictive properties. However, cotinine prevents the memory loss and inhibits the amyloid burden in the brain in a mouse model of AD. Cotinine also inhibits the aggregation of Aβ peptides (considered the main cause of the pathology), activates the pro‐survival enzyme Akt, and inhibits the pro‐apoptotic factor GSK3βin vivo.
Conveniently, cotinine has almost a 10‐fold longer half‐life than nicotine and a good safety profile in humans. Cotinine has shown memory enhancing properties not only in mice, but in monkeys as well, indicating that its beneficial actions are not restricted to rodents. The new information about the effects of cotinine in the brain gives us a better understanding of cotinine's potential for the treatment of AD and opens a new avenue in the search for therapies for this devastating disease.
Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the Bay Pines VA Healthcare System and the James A. Haley Veterans’ Hospital. The contents do not represent the views of the Department of Veterans Affairs or the United States Government. This work was also supported by grants from the James and Esther King Biomedical Research Program DOH 1KG03. We are also grateful to the Bay Pines Foundation, Inc. The authors appreciate the editorial help of Mr. J. Alex Grizzell.
References
- 1. Braak H, de Vos RA, Jansen EN, Bratzke H, Braak E. Neuropathological hallmarks of Alzheimer's and Parkinson's diseases. Prog Brain Res 1998;117:267–285. [DOI] [PubMed] [Google Scholar]
- 2. Mott RT, Hulette CM. Neuropathology of Alzheimer's disease. Neuroimaging Clin N Am 2005;15:755–765, ix. [DOI] [PubMed] [Google Scholar]
- 3. Mufson EJ, Counts SE, Perez SE, Ginsberg SD. Cholinergic system during the progression of Alzheimer's disease: Therapeutic implications. Exp Rev Neurother 2008;8:1703–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jacobs RW, Duong T, Scheibel AB. Immunohistochemical analysis of the basal forebrain in Alzheimer's disease. Mol Chem Neuropathol 1992;17:1–20. [DOI] [PubMed] [Google Scholar]
- 5. Nordberg A. Nicotinic receptor abnormalities of Alzheimer's disease: therapeutic implications. Biol Psychiatr 2001;49:200–210. [DOI] [PubMed] [Google Scholar]
- 6. Rahman S, Lopez‐Hernandez GY, Corrigall WA, Papke RL. Neuronal nicotinic receptors as brain targets for pharmacotherapy of drug addiction. CNS Neurol Disord-Dr 2008;7:422–41. [DOI] [PubMed] [Google Scholar]
- 7. Mihailescu S, Drucker‐Colin R. Nicotine, brain nicotinic receptors, and neuropsychiatric disorders. Arch Med Res 2000;31:131–144. [DOI] [PubMed] [Google Scholar]
- 8. Seguela P, Wadiche J, Dineley‐Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain alpha 7: A nicotinic cation channel highly permeable to calcium. J Neurosci 1993;13:596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang H, Yu M, Ochani M, et al Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003;421:384–388. [DOI] [PubMed] [Google Scholar]
- 10. Wang HY, Lee DH, D’Andrea MR, Peterson PA, Shank RP, Reitz AB. beta‐Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Bio Chem 2000;275:5626–5632. [DOI] [PubMed] [Google Scholar]
- 11. Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. Beta‐amyloid activates the mitogen‐activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci 2001;21:4125–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abbott JJ, Howlett DR, Francis PT, Williams RJ. Abeta(1–42) modulation of Akt phosphorylation via alpha7 nAChR and NMDA receptors. Neurobiol Aging 2008;29:992–1001. [DOI] [PubMed] [Google Scholar]
- 13. Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP. Nicotinic receptors: Allosteric transitions and therapeutic targets in the nervous system. Nat Rev 2009;8:733–750. [DOI] [PubMed] [Google Scholar]
- 14. Nordberg A. Mechanisms behind the neuroprotective actions of cholinesterase inhibitors in Alzheimer disease. Alzheimer Dis Assoc Disord 2006;20(2 Suppl 1):S12–8. [DOI] [PubMed] [Google Scholar]
- 15. Munoz‐Torrero D. Acetylcholinesterase inhibitors as disease‐modifying therapies for Alzheimer's disease. Curr Med Chem 2008;15:2433–2455. [DOI] [PubMed] [Google Scholar]
- 16. Nikolov R. Alzheimer's disease therapy–an update. Drug News Perspect 1998;11:248–255. [PubMed] [Google Scholar]
- 17. Tsuno N. Donepezil in the treatment of patients with Alzheimer's disease. Exp Rev Neurother 2009;9:591–598. [DOI] [PubMed] [Google Scholar]
- 18. Umegaki H, Itoh A, Suzuki Y, Nabeshima T. Discontinuation of donepezil for the treatment of Alzheimer's disease in geriatric practice. Int Psychogeriatr / IPA 2008;20:800–806. [DOI] [PubMed] [Google Scholar]
- 19. Raina P, Santaguida P, Ismaila A, et al Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann Intern Med 2008;148:379–397. [DOI] [PubMed] [Google Scholar]
- 20. Gongadze N, Antelava N, Kezeli T, Okudjava M, Pachkoria K. The mechanisms of neurodegenerative processes and current pharmacotherapy of Alzheimer's disease. Georgian Med News 2008;155:44–48. [PubMed] [Google Scholar]
- 21. Santoro A, Siviero P, Minicuci N, et al Effects of donepezil, galantamine and rivastigmine in 938 Italian patients with Alzheimer's disease: A prospective, observational study. CNS drugs 2010;24:163–76. [DOI] [PubMed] [Google Scholar]
- 22. Graves AB, van Duijn CM, Chandra V, et al Alcohol and tobacco consumption as risk factors for Alzheimer's disease: A collaborative re‐analysis of case‐control studies. EURODEM Risk Factors Research Group. Int J Epidemiol 1991;20(Suppl 2):S48–S57. [DOI] [PubMed] [Google Scholar]
- 23. Salomon AR, Marcinowski KJ, Friedland RP, Zagorski MG. Nicotine inhibits amyloid formation by the beta‐peptide. Biochemistry 1996;35:13568–13578. [DOI] [PubMed] [Google Scholar]
- 24. Shim SB, Lee SH, Chae KR, et al Nicotine leads to improvements in behavioral impairment and an increase in the nicotine acetylcholine receptor in transgenic mice. Neurochem Res 2008;33:1783–1788. [DOI] [PubMed] [Google Scholar]
- 25. Vidal C. Nicotinic receptors in the brain. Molecular biology, function, and therapeutics. Mol Chem Neuropathol 1996;28:3–11. [DOI] [PubMed] [Google Scholar]
- 26. Howe MN, Price IR. Effects of transdermal nicotine on learning, memory, verbal fluency, concentration, and general health in a healthy sample at risk for dementia. Int Psychogeriatr / IPA. 2001;13:465–475. [DOI] [PubMed] [Google Scholar]
- 27. White HK, Levin ED. Four‐week nicotine skin patch treatment effects on cognitive performance in Alzheimer's disease. Psychopharmacology 1999;143:158–165. [DOI] [PubMed] [Google Scholar]
- 28. Kelton MC, Kahn HJ, Conrath CL, Newhouse PA. The effects of nicotine on Parkinson's disease. Brain Cogn 2000;43:274–282. [PubMed] [Google Scholar]
- 29. Lukas RJ, Changeux JP, Le Novere N, et al International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev 1999;51:397–401. [PubMed] [Google Scholar]
- 30. Jensen AA, Frolund B, Liljefors T, Krogsgaard‐Larsen P. Neuronal nicotinic acetylcholine receptors: Structural revelations, target identifications, and therapeutic inspirations. J Med Chem 2005;48:4705–4745. [DOI] [PubMed] [Google Scholar]
- 31. Linert W, Bridge MH, Huber M, Bjugstad KB, Grossman S, Arendash GW. In vitro and in vivo studies investigating possible antioxidant actions of nicotine: Relevance to Parkinson's and Alzheimer's diseases. Biochim Biophys Acta 1999;1454:143–152. [DOI] [PubMed] [Google Scholar]
- 32. Olincy A, Stevens KE. Treating schizophrenia symptoms with an alpha7 nicotinic agonist, from mice to men. Biochem Pharmacol 2007;74:1192–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Echeverria V, Zeitlin R, Burgess S, et al Cotinine reduces Amyloid‐β aggregation and improves memory in Alzheimer's disease mice. J Alzheimer's Dis 2011;24:817–835. [DOI] [PubMed] [Google Scholar]
- 34. Lewis DF, Dickins M, Lake BG, Eddershaw PJ, Tarbit MH, Goldfarb PS. Molecular modelling of the human cytochrome P450 isoform CYP2A6 and investigations of CYP2A substrate selectivity. Toxicology 1999;133:1–33. [DOI] [PubMed] [Google Scholar]
- 35. Visoni S, Meireles N, Monteiro L, Rossini A, Pinto LF. Different modes of inhibition of mouse Cyp2a5 and rat CYP2A3 by the food‐derived 8‐methoxypsoralen. Food Chem Toxicol 2008;46:1190–1195. [DOI] [PubMed] [Google Scholar]
- 36. Donato MT, Viitala P, Rodriguez‐Antona C, et al CYP2A5/CYP2A6 expression in mouse and human hepatocytes treated with various in vivo inducers. Drug Metabol Dispos 2000;28:1321–1326. [PubMed] [Google Scholar]
- 37. Yamanaka H, Nakajima M, Nishimura K, et al Metabolic profile of nicotine in subjects whose CYP2A6 gene is deleted. Eur J Pharm Sci 2004;22:419–425. [DOI] [PubMed] [Google Scholar]
- 38. Nakajima M, Fukami T, Yamanaka H, et al Comprehensive evaluation of variability in nicotine metabolism and CYP2A6 polymorphic alleles in four ethnic populations. Clin Pharmacol Ther 2006;80:282–297. [DOI] [PubMed] [Google Scholar]
- 39. Caldwell WS, Greene JM, Byrd GD, et al Characterization of the glucuronide conjugate of cotinine: A previously unidentified major metabolite of nicotine in smokers’ urine. Chem Res Toxicol 1992;5:280–285. [DOI] [PubMed] [Google Scholar]
- 40. Ghosheh O, Hawes EM. N‐glucuronidation of nicotine and cotinine in human: formation of cotinine glucuronide in liver microsomes and lack of catalysis by 10 examined UDP‐glucuronosyltransferases. Drug Metabol Dispos 2002;30:991–996. [DOI] [PubMed] [Google Scholar]
- 41. Kuehl GE, Murphy SE. N‐glucuronidation of trans‐3’‐hydroxycotinine by human liver microsomes. Chem Res Toxicol 2003;16:1502–1506. [DOI] [PubMed] [Google Scholar]
- 42. Bramer SL, Kallungal BA. Clinical considerations in study designs that use cotinine as a biomarker. Biomarkers 2003;8:187–203. [DOI] [PubMed] [Google Scholar]
- 43. Hukkanen J, Jacob P, 3rd , Benowitz NL. Effect of grapefruit juice on cytochrome P450 2A6 and nicotine renal clearance. Clinical Pharmacol Thera 2006;80:522–530. [DOI] [PubMed] [Google Scholar]
- 44. Tassaneeyakul W, Guo LQ, Fukuda K, Ohta T, Yamazoe Y. Inhibition selectivity of grapefruit juice components on human cytochromes P450. Arch Biochem Biophys 2000;378:356–363. [DOI] [PubMed] [Google Scholar]
- 45. Riah O, Courriere P, Dousset JC, Todeschi N, Labat C. Nicotine is more efficient than cotinine at passing the blood‐brain barrier in rats. Cell Mol Neurobiol 1998;18:311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lockman PR, McAfee G, Geldenhuys WJ, Van der Schyf CJ, Abbruscato TJ, Allen DD. Brain uptake kinetics of nicotine and cotinine after chronic nicotine exposure. J Pharmacol Exp Ther 2005;314:636–642. [DOI] [PubMed] [Google Scholar]
- 47. Hatsukami DK, Grillo M, Pentel PR, Oncken C, Bliss R. Safety of cotinine in humans: Physiologic, subjective, and cognitive effects. Pharmacol Biochem Behav 1997;57:643–650. [DOI] [PubMed] [Google Scholar]
- 48. Yamamoto KI, Domino EF. Nicotine‐induced EEG and behavioral arousal. Int J Neuropharmacol 1965;4:359–373. [DOI] [PubMed] [Google Scholar]
- 49. Risner ME, Goldberg SR, Prada JA, Cone EJ. Effects of nicotine, cocaine and some of their metabolites on schedule‐controlled responding by beagle dogs and squirrel monkeys. J Pharmacol Exp Ther 1985;234:113–119. [PubMed] [Google Scholar]
- 50. Zeitlin R, Patel S, Solomon R, Tran J, Weeber EJ, Echeverria V. Cotinine enhances the extinction of contextual fear memory and reduces anxiety after fear conditioning. Behav Brain Res 2012; 228(2):284–293. [DOI] [PubMed] [Google Scholar]
- 51. Terry AV, Jr. , Hernandez CM, Hohnadel EJ, Bouchard KP, Buccafusco JJ. Cotinine, a neuroactive metabolite of nicotine: Potential for treating disorders of impaired cognition. CNS Drug Rev 2005;11:229–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Buccafusco JJ, Terry AV, Jr . A reversible model of the cognitive impairment associated with schizophrenia in monkeys: Potential therapeutic effects of two nicotinic acetylcholine receptor agonists. Biochem Pharmacol 2009;78:852–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mizuno M, Yamada K, Takei N, et al Phosphatidylinositol 3‐kinase: a molecule mediating BDNF‐dependent spatial memory formation. Mol Psychiatry 2003;8:217–224. [DOI] [PubMed] [Google Scholar]
- 54. Sui L, Wang J, Li BM. Role of the phosphoinositide 3‐kinase‐Akt‐mammalian target of the rapamycin signaling pathway in long‐term potentiation and trace fear conditioning memory in rat medial prefrontal cortex. Learn Mem 2008;15:762–776. [DOI] [PubMed] [Google Scholar]
- 55. Peineau S, Taghibiglou C, Bradley C, et al LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 2007;53:703–717. [DOI] [PubMed] [Google Scholar]
- 56. Kem WR. The brain alpha7 nicotinic receptor may be an important therapeutic target for the treatment of Alzheimer's disease: Studies with DMXBA (GTS‐21). Behav Brain Res 2000;113:169–181. [DOI] [PubMed] [Google Scholar]
- 57. Levin ED. Nicotinic receptor subtypes and cognitive function. J Neurobiol 2002;53:633–640. [DOI] [PubMed] [Google Scholar]
- 58. Jurgensen S, Ferreira ST. Nicotinic receptors, amyloid‐beta, and synaptic failure in Alzheimer's disease. J Mol Neurosci 2010;40:221–229. [DOI] [PubMed] [Google Scholar]
- 59. Howland RH. Alternative drug therapies for dementia. J Psychosoc Nurs Mental Health Ser 2011;49:17–20. [DOI] [PubMed] [Google Scholar]
- 60. Faghih R, Gfesser GA, Gopalakrishnan M. Advances in the discovery of novel positive allosteric modulators of the alpha7 nicotinic acetylcholine receptor. Recent Pat CNS Drug Discov 2007;2:99–106. [DOI] [PubMed] [Google Scholar]
- 61. Lopez‐Hernandez GY, Thinschmidt JS, Morain P, et al Positive modulation of alpha7 nAChR responses in rat hippocampal interneurons to full agonists and the alpha7‐selective partial agonists, 4OH‐GTS‐21 and S 24795. Neuropharmacology 2009;56:821–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lopez‐Hernandez G, Placzek AN, Thinschmidt JS, et al Partial agonist and neuromodulatory activity of S 24795 for alpha7 nAChR responses of hippocampal interneurons. Neuropharmacology 2007;53:134–144. [DOI] [PubMed] [Google Scholar]
- 63. Sattelle DB, Buckingham SD, Akamatsu M, et al Comparative pharmacology and computational modelling yield insights into allosteric modulation of human alpha7 nicotinic acetylcholine receptors. Biochem Pharmacol 2009;78:836–843. [DOI] [PubMed] [Google Scholar]
- 64. Hu M, Gopalakrishnan M, Li J. Positive allosteric modulation of alpha7 neuronal nicotinic acetylcholine receptors: Lack of cytotoxicity in PC12 cells and rat primary cortical neurons. Br J Pharmacol 2009;158:1857–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gauthier S, Scheltens P. Can we do better in developing new drugs for Alzheimer's disease? Alzheimers Dement 2009;5:489–491. [DOI] [PubMed] [Google Scholar]
- 66. Coyle J, Kershaw P. Galantamine, a cholinesterase inhibitor that allosterically modulates nicotinic receptors: Effects on the course of Alzheimer's disease. Biol Psychiatr 2001;49:289–299. [DOI] [PubMed] [Google Scholar]
- 67. Briggs CA, McKenna DG, Piattoni‐Kaplan M. Human alpha 7 nicotinic acetylcholine receptor responses to novel ligands. Neuropharmacology 1995;34:583–590. [DOI] [PubMed] [Google Scholar]
- 68. Arendash GW, Sengstock GJ, Sanberg PR, Kem WR. Improved learning and memory in aged rats with chronic administration of the nicotinic receptor agonist GTS‐21. Brain Res 1995;674:252–259. [DOI] [PubMed] [Google Scholar]
- 69. Sakata M, Wu J, Toyohara J, et al Biodistribution and radiation dosimetry of the alpha7 nicotinic acetylcholine receptor ligand [11C]CHIBA‐1001 in humans. Nucl Med Biol 2011;38:443–448. [DOI] [PubMed] [Google Scholar]
- 70. Toyohara J, Hashimoto K. alpha7 Nicotinic receptor agonists: Potential therapeutic drugs for treatment of cognitive impairments in Schizophrenia and Alzheimer's disease. Open Med Chem J 2010; 4:37–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Freedman R, Olincy A, Buchanan RW, et al Initial phase 2 trial of a nicotinic agonist in schizophrenia. Am J Psychiatry 2008;165:1040–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Riah O, Dousset JC, Bofill‐Cardona E, Courriere P. Isolation and microsequencing of a novel cotinine receptor. Cell Mol Neurobiol 2000;20:653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Buccafusco JJ, Shuster LC, Terry AV, Jr . Disconnection between activation and desensitization of autonomic nicotinic receptors by nicotine and cotinine. Neurosci Lett 2007;413:68–71. [DOI] [PubMed] [Google Scholar]
- 74. Buccafusco JJ, Beach JW, Terry AV, Jr . Desensitization of nicotinic acetylcholine receptors as a strategy for drug development. JPharmacol Exp Ther 2009;328:364–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Buccafusco JJ, Terry AV, Jr . The potential role of cotinine in the cognitive and neuroprotective actions of nicotine. Life Sci 2003;72:2931–2942. [DOI] [PubMed] [Google Scholar]
- 76. Sekine Y, Takeda K, Ichijo H. The ASK1‐MAP kinase signaling in ER stress and neurodegenerative diseases. Curr Mol Med 2006;6:87–97. [DOI] [PubMed] [Google Scholar]
- 77. Tobiume K, Matsuzawa A, Takahashi T, et al ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2001;2:222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bandaru SS, Lin K, Roming SL, Vellipuram R, Harney JP. Effects of PI3K inhibition and low docosahexaenoic acid on cognition and behavior. Physiol Behav 2009; 100(3):239–244. [DOI] [PubMed] [Google Scholar]
- 79. Buckingham SD, Jones AK, Brown LA, Sattelle DB. Nicotinic acetylcholine receptor signalling: Roles in Alzheimer's disease and amyloid neuroprotection. Pharmacol Rev 2009;61:39–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Villarreal JS, Barea‐Rodriguez EJ. ERK phosphorylation is required for retention of trace fear memory. Neurobiol Learn Mem 2006;85:44–57. [DOI] [PubMed] [Google Scholar]
- 81. Wood MA, Kaplan MP, Park A, et al Transgenic mice expressing a truncated form of CREB‐binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn Mem 2005;12:111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Renzoni DA, Pugh DJ, Siligardi G, et al Structural and thermodynamic characterization of the interaction of the SH3 domain from Fyn with the proline‐rich binding site on the p85 subunit of PI3‐kinase. Biochemistry 1996;35:15646–15653. [DOI] [PubMed] [Google Scholar]
- 83. Alessi DR, James SR, Downes CP, et al Characterization of a 3‐phosphoinositide‐dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 1997;7:261–269. [DOI] [PubMed] [Google Scholar]
- 84. Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal‐regulating kinase 1. Mol Cell Biol 2001;21:893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhou XW, Winblad B, Guan Z, Pei JJ. Interactions between glycogen synthase kinase 3beta, protein kinase B, and protein phosphatase 2A in tau phosphorylation in mouse N2a neuroblastoma cells. J Alzheimers Dis 2009;17:929–937. [DOI] [PubMed] [Google Scholar]
- 86. Mandelkow EM, Drewes G, Biernat J, et al Glycogen synthase kinase‐3 and the Alzheimer‐like state of microtubule‐associated protein tau. FEBS letters 1992;314:315–321. [DOI] [PubMed] [Google Scholar]
- 87. Hernandez F, de Barreda EG, Fuster‐Matanzo A, Goni‐Oliver P, Lucas JJ, Avila J. The role of GSK3 in Alzheimer disease. Brain Res Bull 2009;80:248–250. [DOI] [PubMed] [Google Scholar]
- 88. Ploski JE, Pierre VJ, Smucny J, et al The activity‐regulated cytoskeletal‐associated protein (Arc/Arg3.1) is required for memory consolidation of pavlovian fear conditioning in the lateral amygdala. J Neurosci 2008;28:12383–12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bramham CR, Worley PF, Moore MJ, Guzowski JF. The immediate early gene arc/arg3.1: Regulation, mechanisms, and function. J Neurosci 2008;28:11760–11767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gusev PA, Cui C, Alkon DL, Gubin AN. Topography of Arc/Arg3.1 mRNA expression in the dorsal and ventral hippocampus induced by recent and remote spatial memory recall: Dissociation of CA3 and CA1 activation. J Neurosci 2005;25:9384–9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Pintchovski SA, Peebles CL, Kim HJ, Verdin E, Finkbeiner S. The serum response factor and a putative novel transcription factor regulate expression of the immediate‐early gene Arc/Arg3.1 in neurons. J Neurosci 2009;29:1525–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bloomer WA, VanDongen HM, VanDongen AM. Arc/Arg3.1 translation is controlled by convergent N‐methyl‐D‐aspartate and Gs‐coupled receptor signaling pathways. J Biol Chem 2008;283:582–92. [DOI] [PubMed] [Google Scholar]
- 93. Park S, Park JM, Kim S, et al Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR‐LTD. Neuron 2008;59:70–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Waltereit R, Dammermann B, Wulff P, et al Arg3.1/Arc mRNA induction by Ca2+ and cAMP requires protein kinase A and mitogen‐activated protein kinase/extracellular regulated kinase activation. J Neurosci 2001;21:5484–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Plath N, Ohana O, Dammermann B, et al Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 2006;52:437–444. [DOI] [PubMed] [Google Scholar]
- 96. Kristensen SE, Thomsen MS, Hansen HH, Timmermann DB, Hay‐Schmidt A, Mikkelsen JD. The alpha7 nicotinic receptor agonist SSR180711 increases activity regulated cytoskeleton protein (Arc) gene expression in the prefrontal cortex of the rat. Neurosci letters 2007;418:154–158. [DOI] [PubMed] [Google Scholar]
- 97. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science 2002;297:353–356. [DOI] [PubMed] [Google Scholar]
- 98. Ferreira ST, Vieira MN, De Felice FG. Soluble protein oligomers as emerging toxins in Alzheimer's and other amyloid diseases. IUBMB life 2007;59:332–345. [DOI] [PubMed] [Google Scholar]
- 99. Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer's amyloid beta‐peptide. Nat Rev Mol Cell Biol 2007;8:101–112. [DOI] [PubMed] [Google Scholar]
- 100. Echeverria V, Berman DE, Arancio O. Oligomers of beta‐amyloid peptide inhibit BDNF‐induced arc expression in cultured cortical Neurons. Curr Alzheimer Res 2007;4:518–521. [DOI] [PubMed] [Google Scholar]
- 101. Rowan MJ, Klyubin I, Wang Q, Hu NW, Anwyl R. Synaptic memory mechanisms: Alzheimer's disease amyloid beta‐peptide‐induced dysfunction. Biochem Soc Trans 2007;35(Pt 5):1219–1223. [DOI] [PubMed] [Google Scholar]
- 102. Weisman D, Hakimian E, Ho GJ. Interleukins, inflammation, and mechanisms of Alzheimer's disease. Vitam Horm 2006;74:505–530. [DOI] [PubMed] [Google Scholar]
- 103. Salloway S, Sperling R, Keren R, et al A phase 2 randomized trial of ELND005, scyllo‐inositol, in mild to moderate Alzheimer disease. Neurology 2011;77:1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Necula M, Kayed R, Milton S, Glabe CG. Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J Bio Chem 2007;282:10311–10324. [DOI] [PubMed] [Google Scholar]
- 105. Yang F, Lim GP, Begum AN, et al Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Bio Chem 2005;280:5892–5901. [DOI] [PubMed] [Google Scholar]
- 106. De Felice FG, Vieira MN, Saraiva LM, et al Targeting the neurotoxic species in Alzheimer's disease: Inhibitors of Abeta oligomerization. Faseb J 2004;18:1366–1372. [DOI] [PubMed] [Google Scholar]
- 107. Ferrao‐Gonzales AD, Robbs BK, Moreau VH, et al Controlling {beta}‐amyloid oligomerization by the use of naphthalene sulfonates: Trapping low molecular weight oligomeric species. J Bio Chem 2005;280:34747–34754. [DOI] [PubMed] [Google Scholar]
- 108. Zhao W, Wang J, Ho L, Ono K, Teplow DB, Pasinetti GM. Identification of antihypertensive drugs which inhibit amyloid‐beta protein oligomerization. J Alzheimers Dis 2009;16:49–57. [DOI] [PubMed] [Google Scholar]
- 109. Hou Y, Aboukhatwa MA, Lei DL, Manaye K, Khan I, Luo Y. Anti‐depressant natural flavonols modulate BDNF and beta amyloid in neurons and hippocampus of double TgAD mice. Neuropharmacology 2010;58:911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Szymanska I, Radecka H, Radecki J, Kaliszan R. Electrochemical impedance spectroscopy for study of amyloid beta‐peptide interactions with (‐) nicotine ditartrate and (‐) cotinine. Biosens Bioelectron 2007;22:1955–1960. [DOI] [PubMed] [Google Scholar]
- 111. Burgess SZR, Gamble‐George J, Echeverria V. Cotinine is neuroprotective against beta‐amyloid toxicity. J Alzheimer's Assoc 2008;4:T466. [Google Scholar]
- 112. Echeverria V, Burgess S, Zeitlin R, et al Cotinine: A dual‐action drug with multiple benefits against Alzheimer's disease Alzheimer's & Dementia. J Alzheimer's Assoc 2010;6:S536–S537. [Google Scholar]
- 113. Antzutkin ON, Balbach JJ, Tycko R. Site‐specific identification of non‐beta‐strand conformations in Alzheimer's beta‐amyloid fibrils by solid‐state NMR. Biophys J 2003;84:3326–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Bowman ER, Mc KH, Jr . Studies on the metabolism of (‐)‐cotinine in the human. J Pharmacol Exp Ther 1962;135:306–311. [PubMed] [Google Scholar]
- 115. Borzelleca JF, Bowman ER, Mc KH, Jr . Studies on the respiratory and cardiovascular effects of (‐)‐cotinine. J pharmacol Exp Ther 1962;137:313–318. [PubMed] [Google Scholar]
- 116. Benowitz NL, Kuyt F, Jacob P, 3rd , Jones RT, Osman AL. Cotinine disposition and effects. Clinical Pharmacol Ther 1983;34:604–611. [DOI] [PubMed] [Google Scholar]
- 117. Benowitz NL, Sharp DS. Inverse relation between serum cotinine concentration and blood pressure in cigarette smokers. Circulation 1989;80:1309–1312. [DOI] [PubMed] [Google Scholar]
- 118. Hatsukami D, Lexau B, Nelson D, Pentel PR, Sofuoglu M, Goldman A. Effects of cotinine on cigarette selfadministration. Psychopharmacology 1998;138:184–189. [DOI] [PubMed] [Google Scholar]
- 119. Hatsukami D, Pentel PR, Jensen J, et al Cotinine: Effects with and without nicotine. Psychopharmacology 1998;135:141–150. [DOI] [PubMed] [Google Scholar]
- 120. Oddo S, LaFerla FM. The role of nicotinic acetylcholine receptors in Alzheimer's disease. J Physiol Paris 2006;99:172–179. [DOI] [PubMed] [Google Scholar]