

Summary
Aim
To investigate the effect of chronic H1‐antihistamine treatment on seizure susceptibility after drug withdrawal in nonepileptic rats and to further study its relation to glutamine synthetase (GS), which is the key enzyme for glutamate metabolism and gamma aminobutyric acid (GABA) synthesis.
Methods
After drug withdrawal from a 2‐week treatment with diphenhydramine or pyrilamine, seizure susceptibility was determined by amygdaloid kindling or pentylenetetrazol model; meanwhile, the GS expression or activity was analyzed. The glutamine, glutamate, and GABA contents were measured by high‐performance liquid chromatography.
Results
Seizure susceptibility significantly increased in amygdaloid kindling and pentylenetetrazol model 10 days after drug withdrawal from a 2‐week treatment with H1‐antihistamines. Meanwhile, GS activity and expression in the cortex or hippocampus decreased simultaneously with a marked decline of glutamine and GABA content. Comparable inhibition of GS activity by methionine sulfoximine was also sufficient to increase the susceptibility, while supplementation with glutamine reversed the high susceptibility 10 days after diphenhydramine withdrawal. Moreover, the seizure susceptibility increased 10 days after diphenhydramine withdrawal in wild‐type mice but not in histidine decarboxylase knockout mice, which lack histamine.
Conclusions
Chronic H1‐antihistamine treatment produces long‐lasting increase in seizure susceptibility in nonepileptic rodents after drug withdrawal and its mechanism involves impairment of GS through blocking the action of histamine.
Keywords: Gamma aminobutyric acid, Glutamine synthetase, H1‐antihistamine, Seizure susceptibility
Introduction
The H1‐antihistamines are among the most widely used medications in the world, most of which are available “over‐the‐counter” for treating allergic reactions and motion sickness or alleviating cold symptoms. It is well known that H1‐antihistamines, such as diphenhydramine, pyrilamine, and ketotifen, have the immense potential to promote seizures in epileptic patients and animals 1, 2, 3, and large doses may induce convulsions directly 4, 5, so epileptic patients are reminded to be cautious in taking H1‐antihistamines. However, in clinical therapy, H1‐antihistamines are chronically administered at low doses to people without epilepsy. Consequently, whether chronic treatment with H1‐antihistamines at routine doses affects seizure susceptibility in nonepileptics, especially the long‐lasting effects after drug withdrawal, is worthy of study. So far, although much evidence show that H1‐antihistamines induce epileptic exacerbation owing to their antagonism of the effects of histamine in the brain 4, 6, 7, the detailed mechanism is still unclear.
Recent studies suggest that dysfunction of glutamine synthetase (GS) in astrocytes contributes to the generation and spread of seizures 8, 9, 10. Following synaptic release, glutamate is mainly taken up by the astrocytic glutamate transporter GLT‐1 or GLAST and then converted into glutamine by GS. Subsequently, the glutamine in astrocytes is transported back to the presynaptic terminals and converted to glutamate or gamma aminobutyric acid (GABA) for reuse 11. Therefore, GS is pivotal for maintaining a balance of the neurotransmitters glutamate and GABA in the brain. Impairing GS may cause accumulation of glutamate in the extracellular space, disturb neurotransmitter synthesis, and increase seizure susceptibility 10, 12, 13. In haploinsufficient GS mice, which show reduced expression/activity of GS, the susceptibility to febrile seizures increases 10.
It has been reported that the GS activity in cerebellar astrocytes can be elevated by histamine 14. We previously found that carnosine, a histamine precursor, decreases the glutamate content in the hippocampus partly by being transformed to histamine and activating H1 receptors 15, which are expressed in both astrocytes and neurons. However, the function of astrocytic H1 receptors remains unclear 16. In this study, we hypothesized that H1‐antihistamines regulate glutamate metabolism in astrocytes, especially the key enzyme GS, through blocking H1 receptors and thereby affect seizure susceptibility. We therefore investigated the effect of withdrawal from chronic H1‐antihistamine treatment on seizure susceptibility in nonepileptic rodents along with the effects on GS in astrocytes.
Materials and Methods
Animals
The animal experiments were carried out on adult (8 weeks old) male Sprague–Dawley rats, male wild‐type mice (WT; C57BL/6N strain, 25–28 g), and histidine decarboxylase knockout mice (HDC‐KO; C57BL/6N strain, 25–28 g). All animals were maintained under a 12‐h light–dark cycle in the Experimental Animal Center of Zhejiang University. Food and water were available ad libitum. All experiments were carried out in accordance with the ethical guidelines of the Zhejiang University Animal Experimentation Committee and were in complete compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
H1‐Antihistamine Administration and Seizure Susceptibility in Adult Rats
Adult rats were given the diphenhydramine (10 or 20 mg/kg; Sigma‐Aldrich, St. Louis, MO, USA), pyrilamine (10 mg/kg; Sigma‐Aldrich), or saline intraperitoneally once daily for 2 weeks, and then the experiments were carried out as follows:
Sample Collection
Ten days after drug withdrawal, the cerebral cortex, hippocampus, and thalamus were isolated for the determination of GS activity by a kit (Jiancheng, Nanjing, China) or protein expression analysis, as well as measurement of the glutamine, glutamate, and GABA content by high‐performance liquid chromatography (HPLC).
Immunohistochemistry
The rats were deeply anesthetized with chloral hydrate (400 mg/kg) and perfused transcardially with saline and buffered 4% paraformaldehyde, 10 days after drug withdrawal. The dissected brains were then cut into 10‐μM coronal sections and immunostained for glial fibrillary acidic protein (GFAP). In brief, the sections were first incubated with 3% bovine serum albumin in PBS for 30 min and then with rabbit anti‐GFAP IgG (1:200; Millipore, Billerica, MA, USA) in PBS containing 0.3% Triton X‐100 overnight. After washing 3 times for 10 min with PBS, sections were incubated sequentially in Cy3‐conjugated secondary antisera (1:200; Beyotime, Jiangsu, China) for 1 h at room temperature. All images were captured by DP Controller and analyzed with Image J software (US National Institute of Health, Bethesda, MD, USA).
Kindling
After 2–3 days of withdrawal, rats were mounted in a stereotaxic apparatus (Stoelting, Wood Dale, IL, USA) under chloral hydrate anesthesia (400 mg/kg), and electrodes were implanted into the right basolateral amygdala (AP: −2.4 mm, L: −4.8 mm, V: −8.8 mm) 17. After surgery, rats were allowed 7–8 days of recovery. Kindling stimulation of the amygdala was performed 10 days after drug withdrawal as described previously 15, 18, 19. Stimuli were applied through a constant‐current stimulator (SEN‐7203, SS‐202J; Nihon Kohden, Tokyo, Japan), and the electroencephalogram (EEG) at the amygdala was recorded with a digital amplifier (Neuroscan System, Charlotte, NC, USA). One day before kindling stimulation, the afterdischarge threshold was determined by a 1‐second, 60‐Hz monophasic square‐wave stimulation of 1 milliseconds per pulse. The stimulus intensity began at 50 μA and was then increased by 20 μA steps every 30 min. The minimal intensity that produced at least 5 seconds of afterdischarge was designated as the afterdischarge threshold for that animal and was used for daily stimulation. All rats were stimulated daily for 22 days. Seizure severity was classified according to Racine's scale 20: (0) no convulsion; (1) facial movement; (2) head nodding; (3) unilateral forelimb clonus; (4) bilateral forelimb clonus and rearing; and (5) bilateral forelimb clonus and rearing and falling.
Pentylenetetrazol Model
One, 2, or 10 days after withdrawal from a 2‐week diphenhydramine treatment, rats were given 40 mg/kg pentylenetetrazol (PTZ) intraperitoneally once, and then the seizure behavior was observed for 30 min using the following scale 21: (0) no convulsion; (1) twitching of vibrissae and pinnae; (2) motor arrest with pronounced body twitching or head nodding; (3) motor arrest with generalized myoclonic jerks; (4) clonic seizure with rearing while the rat remains on its feet; (5) tonic‐clonic seizure with loss of postural control; and (6) multiple occurrence of stage 5 seizures. To determine the seizure susceptibility after glutamine supplementation, 10 days after diphenhydramine withdrawal, the rats were administered 2 g/kg glutamine or saline by gavage. Two and a half hours later, PTZ injection was carried out as above.
Methionine Sulfoximine (MSO) Administration and Seizure Susceptibility in Adult Rats
To determine the seizure susceptibility after treatment with the GS inhibitor, adult rats were injected with 7.5, 10, or 15 mg/kg MSO (Sigma‐Aldrich), and 24 h later, the cortical and hippocampal GS activity was measured or the susceptibility to lithium–pilocarpine‐induced seizures was determined. Briefly, rats received intraperitoneal injections of lithium chloride (3 mEq/kg) followed by pilocarpine (40 mg/kg) 4 h later. The latency to the first behavioral seizure (forelimb clonus, rearing, and loss of balance) after pilocarpine administration, referred to as the seizure onset time (SOT), was recorded 22, 23.
H1‐Antihistamine Administration and Maximal Electroshock (MES) or PTZ Seizure Susceptibility in HDC‐KO and WT Mice
Both HDC‐KO and WT mice were treated with 40 mg/kg diphenhydramine or saline intraperitoneally once daily for 2 weeks. Ten days after drug withdrawal, the MES threshold and PTZ‐induced seizures were tested.
The threshold for MES seizures was determined by a rodent shocker (Hugo Sachs Elektronik, March‐Hugstetten, Germany), which delivered a constant current (0.2 seconds, 50 Hz) through a pair of ear‐clip electrodes. The endpoint was tonic extension of the hindlimbs. The stimulus current intensity began at 5 mA and was varied by an “up‐and‐down” method 24. That is, the current was lowered 1 mA if the preceding shock caused tonic hindlimb extension or was raised 1 mA if not.
For PTZ‐induced seizures, mice were given single dose of 60 mg/kg PTZ intraperitoneally, and then the seizure behavior was observed for 30 min using the following scale 25: (0) no response; (1) ear and facial twitching; (2) myoclonic body jerks; (3) clonic forelimb convulsions; (4) generalized clonic convulsions and turning onto the side; (5) generalized clonic‐tonic convulsions; and (6) death within 30 min.
Western Blot Analysis
Total proteins were purified using tissue protein extraction reagents (Kangchen, Shanghai, China). Protein samples were separated on 12% SDS–polyacrylamide gels and then electrotransferred onto a nitrocellulose membrane. After blocking with 5% fat‐free milk, the membranes were incubated with the antibodies for mouse anti‐GS (1:600; Millipore) or mouse anti‐glyceraldehyde‐3‐phosphate dehydrogenase (anti‐GAPDH, 1:5000; Kangchen) at 4°C overnight. After repeated washing, the membranes were reacted with IRDye 700 anti‐mouse molecular probe or IRDye 800 anti‐rabbit molecular probe (Odyssey; LI‐COR, Lincoln, NE, USA) for 2 h. Images were acquired with the Odyssey infrared imaging system and analyzed by Odyssey software.
Analysis of Glutamate, Glutamine, and GABA Concentrations by HPLC
Samples were deproteinized with 0.4 mol/L perchloric acid and centrifuged at 15,000 × g for 20 min at 4°C. Then, the supernatant was removed and filtered through a 0.22‐μm polyvinylidene difluoride membrane. Analysis of glutamate and GABA in the supernatant was performed by HPLC as reported in our previous study 15, and the analysis method for glutamine was the same as for glutamate. The HPLC was controlled and the data acquired and analyzed using CoulArray® software. All of the equipment was from ESA (Chelmsford, MA, USA).
Data Analysis
Data are presented as mean ± SEM. Statistical analysis was performed with SPSS 11.5 for Windows (SPSS Inc., Chicago, IL, USA). Comparisons of the data of PTZ seizure behavior were made with the nonparametric Mann–Whitney U‐test. Seizure progression during amygdaloid kindling was analyzed by two‐way analysis of variance (ANOVA) followed by the LSD test. Other analyses used one‐way ANOVA followed by the LSD or Dunnett's T3 post hoc test (where equal variances were not assumed) for multiple comparisons, or the two‐tailed t‐test for 2 groups, to calculate the significance. P < 0.05 was considered statistically significant.
Results
H1‐Antihistamines Increased Seizure Susceptibility After Withdrawal in Adult Rats
The seizure susceptibility of rats was investigated by amygdaloid kindling acquisition and the PTZ seizure model 22, 23, 26. In adult rats, the effect of chronic H1‐antihistamine treatment on amygdaloid kindling seizures was determined 10 days after withdrawal from a 2‐week treatment with either diphenhydramine or pyrilamine (Figure 1A–C). The afterdischarge thresholds were significantly lowered by a 2‐week 20 mg/kg diphenhydramine (from 174.3 ± 16.2 to 130.0 ± 5.3 μA) or 10 mg/kg pyrilamine treatment after drug withdrawal (Figure 1A). Furthermore, both diphenhydramine and pyrilamine facilitated the progression of behavioral seizure stages after withdrawal during 22 days of kindling stimulation (Figure 1B). Diphenhydramine at 10 mg/kg promoted kindling to stages 2 and 3 compared with controls, while diphenhydramine at 20 mg/kg and pyrilamine at 10 mg/kg promoted kindling to stages 2–5 compared with controls after drug withdrawal (Figure 1C). Because overdose of H1‐antihistamines induces seizures directly, to rule out the possibility of a chemical kindling effect in our paradigm, the animal behavior and EEG were monitored simultaneously when H1‐antihistamines were administered. There was no epileptiform behavior or EEG discharge even after the dose of diphenhydramine reached 40 mg/kg and only occurred when the dose reached 100 mg/kg (Figure S1). Moreover, no behavioral difference was observed between the control and diphenhydramine groups at 20 mg/kg/day after prolonged treatment, even up to 40 days. Furthermore, 2‐week treatment with 20 mg/kg diphenhydramine significantly increased stage in PTZ model 1, 5 or 10 days after drug withdrawal (Figure 1D), which indicated a persistent increased seizure susceptibility after drug withdrawal from H1‐antihistamine treatment.
Figure 1.
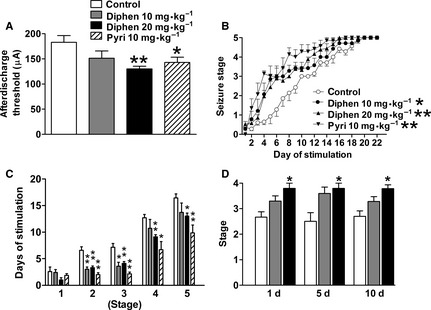
H1‐antihistamines increases seizure susceptibility after withdrawal in rats. Adult rats were treated with 10 or 20 mg/kg/day diphenhydramine (diphen) or 10 mg/kg/day pyrilamine (pyri) for 2 weeks, The afterdischarge threshold (A) of amygdala was tested on the 10th day after drug withdrawal and then 22 days of daily kindling stimulation was followed from the next day. The behavioral stage of seizures (B) and the number of stimulations required to reach each seizure stage (C) during kindling acquisition were recorded. One, 5, or 10 days after withdrawal from a 2‐week treatment with 10 or 20 mg/kg/day diphenhydramine (diphen), the pentylenetetrazol‐induced seizure behavioral stage was measured (D). Values are expressed as mean ± SEM. n = 7–9 (A–C); n = 5–9 (D). *P < 0.05, **P < 0.01 compared with control group.
Effects of Diphenhydramine on MES and PTZ‐Induced Seizures After Withdrawal in HDC‐KO and WT Mice
To investigate whether the promotive effect of H1‐antihistamines on seizure susceptibility was through blocking the action of histamine, we compared the MES threshold and PTZ‐induced seizure behavior between HDC‐KO mice, which have a low level of histamine in the brain 27, and WT adult mice 10 days after withdrawal from a 2‐week treatment with 40 mg/kg diphenhydramine (this dose corresponds to 20 mg/kg in rats 28). We found the MES threshold in WT mice was significantly lowered by the previous chronic treatment of diphenhydramine, while in HDC‐KO mice there was no such difference (Figure 2A). As expected, the seizure behavioral stage induced by PTZ was more severe in the diphenhydramine‐treated group in WT but not in HDC‐KO mice (Figure 2B). Furthermore, PTZ induced a higher seizure behavioral stage in HDC‐KO mice compared with WT mice both without diphenhydramine treatment (Figure 2B), which also revealed the higher seizure susceptibility after deficiency of endogenous histamine in the brain.
Figure 2.

Effects of chronic diphenhydramine treatment on maximal electroshock (MES) threshold and pentylenetetrazol (PTZ)‐induced seizures after withdrawal in histidine decarboxylase knockout (HDC‐KO) and wild‐type (WT) mice. Ten days after withdrawal from a 2‐week treatment with 40 mg/kg/day diphenhydramine (diphen), the MES threshold (A, n = 9–10) and PTZ‐induced seizure behavioral stage (B, n = 6–9) were tested in HDC‐KO and WT mice. Values are expressed as mean ± SEM. *P < 0.05, **P < 0.01 Compared with control WT mice.
Effects of Chronic H1‐Antihistamine Treatment on GS and Neurotransmitter Levels After Withdrawal in Rats
To further explore the mechanism of the effect of H1‐antihistamines, we investigated the GS activity and protein expression after withdrawal from a 2‐week H1‐antihistamine treatment in adult rats. The GS activity here represents the enzyme activity in total protein; thus, it reflects both the change in the amount of the enzyme and the activity of the individual enzyme (Figure 3A–C). We found the GS activity in cortex and hippocampus was reduced to 79.2% (P < 0.01) and 87.4% (P < 0.05) of the control level 10 days after chronic 20 mg/kg diphenhydramine treatment. Previous chronic treatment of 10 mg/kg diphenhydramine or pyrilamine also significantly decreased the GS activity in hippocampus, while in cortex pyrilamine decreased GS activity but diphenhydramine did not. For protein expression, previous chronic treatment of 20 mg/kg diphenhydramine or 10 mg/kg pyrilamine clearly decreased the GS expression both in cortex and in hippocampus, but diphenhydramine at 10 mg/kg only significantly reduced the GS expression in cortex, not hippocampus (Figure 3D–F). Also, there was no loss of astrocytes in hippocampus 10 days after withdrawal from a 2‐week diphenhydramine treatment (Figure 3G–I). In thalamus, both GS activity and expression did not change.
Figure 3.

Chronic H1‐antihistamine treatment decreases glutamine synthetase (GS) activity and expression after withdrawal in rats. Ten days after withdrawal from a 2‐week treatment with diphenhydramine (diphen) or pyrilamine (pyri), the activity (A–C) and expression (D–F) of GS in the cortex (A, D), hippocampus (B, E), and thalamus (C, F) were analyzed. Also, glial fibrillary acidic protein (GFAP)‐immunoreactive astrocytes in the hippocampus were stained (G). Enlargements of CA1, as indicated by the upper box in G1 and G4, are shown in G2 and G5. Enlargements of the dentate gyrus, as indicated by the lower box in G1 and G4, are shown in G3 and G6. Bar = 40 μm for G1 and G4, 200 μm for G2 and G5, and 100 μm for G3 and G6. (H, I) Counts of GFAP‐immunoreactive astrocytes in CA1 (H) and hilus (I). Values are expressed as mean ± SEM. n = 3–5 (A–F); n = 4–5 (H, I). *P < 0.05, **P < 0.01 Compared with control.
To further test whether comparable impairment of GS activity is sufficient to increase seizure susceptibility, we used a GS‐specific inhibitor, MSO. First, different doses of MSO were given to adult rats, followed by measuring the GS activity 24 h later. MSO at 7.5 mg/kg reduced the GS activity to 81.6% and 94.8% of control in cortex and hippocampus, respectively, without significant difference, while 10 mg/kg MSO significantly reduced the GS activity to 73.1% and 64.5% (Figure 4A,B). Because this MSO‐induced decline in GS activity was comparable to that found after H1‐antihistamine withdrawal (Figure 3A,B), MSO at 7.5 and 10 mg/kg was given to rats, followed by lithium–pilocarpine administration 24 h later. The seizure susceptibility increased as shown by the clearly shortened SOT after 10 mg/kg MSO treatment, while 7.5 mg/kg MSO shortened the SOT without significance (Figure 4C).
Figure 4.

Increased susceptibility to lithium–pilocarpine seizures after inhibition of glutamine synthetase (GS) activity. GS activity in the cortex (A) and hippocampus (B) was measured 24 h after methionine sulfoximine (MSO) administration. The seizure onset time (SOT) following lithium–pilocarpine injection was determined 24 h after MSO administration (C). Values are expressed as mean ± SEM. n = 3–6 (A, B); n = 5–7 (C). *P < 0.05, **P < 0.01 Compared with control.
As impaired GS may induce a decrease in the production of glutamine, the glutamine levels in cortex and hippocampus were determined by HPLC (Figure 5A,B). Glutamine levels significantly decreased 10 days after withdrawal from a 2‐week diphenhydramine treatment. Supplementation with 2 g/kg glutamine once by gavage at that time reversed the increased seizure susceptibility induced by previous chronic diphenhydramine treatment as tested by PTZ‐induced seizure behavior (Figure 5C). Meanwhile, administration of glutamine to the control group did not affect the seizure behavior.
Figure 5.

Effect of chronic diphenhydramine treatment on glutamine after withdrawal in rats. Ten days after withdrawal from a 2‐week diphenhydramine (diphen) treatment, the total glutamine (Gln) content in the cortex (A) and hippocampus (B) was determined by high‐performance liquid chromatography. The pentylenetetrazol‐induced seizure behavioral stage was determined after supplementation with 2 g/kg Gln at 10 days after drug withdrawal from a 2‐week 20 mg/kg diphen treatment (C). Values are expressed as mean ± SEM. n = 7–9 (A, B); n = 7–11 (C). *P < 0.05, **P < 0.01 Compared with control; ## P < 0.01 compared with saline‐treated rats in control group; $ P < 0.05 compared with saline‐treated rats in diphen group.
To further study the effect of impaired GS on neurotransmitters in the brain, HPLC was used to determine the glutamate and GABA content 10 days after withdrawal from a 2‐week diphenhydramine treatment (Figure 6). The total GABA content was markedly decreased in cortex and hippocampus by both 10 and 20 mg/kg previous treatment of diphenhydramine; however, the total glutamate content was unaffected. In the thalamus, no significant change was found in neurotransmitter levels (Figure 6C,F).
Figure 6.

Effects of chronic diphenhydramine treatment on glutamate and gamma aminobutyric acid (GABA) content after drug withdrawal in rats. Ten days after drug withdrawal from a 2‐week diphenhydramine (diphen) treatment, the total glutamate (Glu, A–C) and GABA (D–F) contents in the cortex (A, D), hippocampus (B, E), and thalamus (C, F) were determined by high‐performance liquid chromatography. Values are expressed as mean ± SEM. n = 5–7. *P < 0.05, **P < 0.01 Compared with control.
Discussion and Conclusions
In the present study, we found for the first time that chronic H1‐antihistamine treatment enhanced seizure susceptibility in nonepileptic rodents after drug withdrawal. The effect of H1‐antihistamines on seizure susceptibility was reflected by decreased kindling threshold and accelerated progression of seizure stage in the amygdaloid kindling process, decreased MES threshold, and increased behavioral seizure stages induced by PTZ. More importantly, this effect can last at least for 10 days after H1‐antihistamine withdrawal in adults rats. These findings argue that nonepileptic people may also be cautious of taking H1 antagonists chronically, especially those who are at high risk of epileptogenesis such as patients suffering from traumatic brain injury, stroke, or infection.
Previous clinical investigations have suggested that diphenhydramine at very high doses produces brief, self‐limited seizures, and patients may not even need medical attention after such attacks 29. The seizure‐promotive effect of H1‐antihistamines also appears a short time after drug administration in epileptic rats, within seconds or minutes 2, 30. In the present study, however, 10 days after H1‐antihistamine withdrawal, we still found increased seizure susceptibility in nonepileptic rats, as well as abnormal GS expression, activity, and GABA levels. Such high seizure susceptibility may be persistent after H1‐antihistamine withdrawal, because elevated behavioral seizure stages in PTZ‐induced seizure were also observed 1 and 5 days after withdrawal. Meanwhile, we found the GS activity decreased clearly at the same time after withdrawal (Figure S2). Interestingly, rats at 3 weeks of age were given 20 mg/kg diphenhydramine once daily for 2 weeks; then, 4 weeks after drug withdrawal, the seizure susceptibility was remarkably enhanced in the lithium–pilocarpine seizure model (W.W.H.; Q.F.; Z.H.X.; K.Z.; Z.C.). These data indicated that there exists the long‐lasting effect of H1‐antihistamines after withdrawal. The effects of H1‐antihistamines on seizure susceptibility and astrocytes were not attributed to the direct cytotoxic effects. On histological examination, loss of astrocytes was not evident after chronic diphenhydramine treatment. In addition, the dose of 20 mg/kg/day diphenhydramine for rats, which did not induce chemical kindling in our experiment, corresponds to about 200 mg/day for a standard 60 kg human by dose conversion according to body surface area 28. Such a dose may be not above the recommended therapeutic dose of diphenhydramine, which is 25–50 mg 3 or 4 times daily, but not exceeding 400 mg/day for >12‐year‐olds 31, 32. Moreover, it has been reported that diphenhydramine at 300 mg/day is given to treat refractory cancer pain 33. Consequently, the present findings suggest that attention needs to be paid to the long‐lasting effect of H1‐antihistamines, even at a routine dose. And because the terminal exponential half‐lives of diphenhydramine and pyrilamine are approximately 9 and 2 h, respectively 34, 35, the mechanism of the long‐lasting seizure‐promotive effect after H1‐antihistamine withdrawal demonstrated here may be different from the transitory effect reported in previous studies, which might be more likely related to direct alteration of neuronal excitability.
Our and other previous studies suggested that the epileptogenic activity of H1‐antihistamines is induced by blocking the action of histamine through the H1 receptor 6, 36. Therefore, we first demonstrated that the increased seizure susceptibility induced by H1‐antihistamines after withdrawal was also attributed to antagonism of the action of histamine, because the MES threshold decreased and the seizure stages induced by PTZ increased after diphenhydramine withdrawal in WT mice but not in HDC‐KO mice, which have a long‐term deficiency of histamine. Furthermore, we were interested to determine whether chronic H1‐antihistamine treatment induced a pronounced inhibition of GS, which is the critical enzyme for glutamate metabolism to glutamine and then GABA synthesis. We found that the expression or activity of GS significantly decreased after diphenhydramine or pyrilamine withdrawal (Figure 3; Figure S2). However, the expression of GLT‐1, another key component in glutamate metabolism, did not change after withdrawal from chronic H1‐antihistamine treatment in rats (data not shown). It has been reported that pharmacological inhibition of GS reduces the threshold for evoking epileptiform activity and induces recurrent seizures 13, 37. In our experiment, we confirmed that even a slight decline of GS activity induced by MSO, which was similar to the reduction after withdrawal from chronic H1‐antihistamine treatment, increased the susceptibility to lithium–pilocarpine‐induced seizures. The impaired GS also significantly decreased the glutamine level 10 days after drug withdrawal. As exogenous administration of glutamine can elevate glutamine and GABA levels 38, 39, we confirmed the involvement of impaired GS and the subsequent decrease in glutamine in H1‐antihistamine‐induced high seizure susceptibility by the reduction in PTZ‐induced behavioral seizure stage after supplementation with glutamine. Therefore, the long‐lasting effect of H1‐antihistamines on seizure susceptibility after drug withdrawal is possibly due to GS abnormality in astrocytes after blocking the action of histamine. And this further supports the importance of GS in astrocytes in epileptogenesis.
In addition, the low level of glutamine synthesis caused by abnormal function of GS in astrocytes may result in the loss of inhibition of neurons because of impaired vesicular release of GABA or accumulation of glutamate in the synaptic cleft 8, 40, 41. In this study, a marked decrease in GABA was found in the cortex and hippocampus, important areas implicated in seizure generation and propagation, 10 days after drug withdrawal, when GS activity and expression and the glutamine level also declined. Consequently, the decline of GABA here may be attributed to the deficiency of GS followed by reduced synthesis of the precursor glutamine, and this decline may cause disinhibition of the neuronal circuitry, generation of abnormal firing patterns and facilitation of their spread. The glutamate content in the brain was not affected after H1‐antihistamine withdrawal, presumably due to interference with both glutamate metabolism and synthesis caused by the deficiency of GS. Moreover, no evident change in neuron amount was found after chronic H1‐antihistamine treatment (Figure S3). Therefore, the neurotransmitter imbalance caused by the abnormal function of GS in astrocytes after drug withdrawal may be the specific mechanism underlying the high seizure susceptibility induced by H1‐antihistamines, although other direct effects of H1‐antihistamines on intrinsic neuronal properties cannot be ruled out.
In conclusion, this study showed that chronic treatment with H1‐antihistamines increased seizure susceptibility in nonepileptic rodents, and this effect lasted for a long time after drug withdrawal. The mechanism involved impairment of astrocytic GS expression or activity through blocking the action of histamine. It suggests that caution may be exercised by those who have chronically taken H1‐antihistamines, especially who are at high risk of epileptogenesis.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Figure S1 Representative examples of EEGs after diphenhydramine treatment. Figure S2 Chronic H1‐antihistamine treatment decreases GS activity 1 or 5 days after withdrawal in rats. Figure S3 Toluidine blue staining of hippocampus after chronic diphenhydramine treatment in rats.
Acknowledgments
This work was funded by the National Basic Research of China 973 Program (2011CB504403), the National Natural Science Foundation of China (81030061, 81173040, 81102429), and partly by the Youth Foundation of the Innovative Scientific Research of Zhejiang University (2009QNA7007). We are very grateful to Dr. Iain C. Bruce for reading the manuscript.
The first three authors contributed equally to this work.
References
- 1. Churchill JA, Gammon GD. The effect of antihistaminic drugs on convulsive seizures. J Am Med Assoc 1949;141:18–21. [DOI] [PubMed] [Google Scholar]
- 2. Fujii Y, Tanaka T, Harada C, Hirai T, Kamei C. Epileptogenic activity induced by histamine H(1) antagonists in amygdala‐kindled rats. Brain Res 2003;991:258–261. [DOI] [PubMed] [Google Scholar]
- 3. Yawata I, Tanaka K, Nakagawa Y, Watanabe Y, Murashima YL, Nakano K. Role of histaminergic neurons in development of epileptic seizures in EL mice. Brain Res Mol Brain Res 2004;132:13–17. [DOI] [PubMed] [Google Scholar]
- 4. Kamei C, Ohuchi M, Sugimoto Y, Okuma C. Mechanism responsible for epileptogenic activity by first‐generation H1‐antagonists in rats. Brain Res 2000;887:183–186. [DOI] [PubMed] [Google Scholar]
- 5. Thundiyil JG, Kearney TE, Olson KR. Evolving epidemiology of drug‐induced seizures reported to a Poison Control Center System. J Med Toxicol 2007;3:15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen Z, Li WD, Zhu LJ, Shen YJ, Wei EQ. Effects of histidine, a precursor of histamine, on pentylenetetrazole‐induced seizures in rats. Acta Pharmacol Sin 2002;23:361–366. [PubMed] [Google Scholar]
- 7. Chen Z, Li Z, Sakurai E, et al. Chemical kindling induced by pentylenetetrazol in histamine H(1) receptor gene knockout mice (H(1)KO), histidine decarboxylase‐deficient mice (HDC(−/−)) and mast cell‐deficient W/W(v) mice. Brain Res 2003;968:162–166. [DOI] [PubMed] [Google Scholar]
- 8. Eid T, Thomas MJ, Spencer DD, et al. Loss of glutamine synthetase in the human epileptogenic hippocampus: Possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 2004;363:28–37. [DOI] [PubMed] [Google Scholar]
- 9. Eid T, Williamson A, Lee TS, Petroff OA, de Lanerolle NC. Glutamate and astrocytes–key players in human mesial temporal lobe epilepsy? Epilepsia 2008;49(Suppl 2):42–52. [DOI] [PubMed] [Google Scholar]
- 10. van Gassen KL, van der Hel WS, Hakvoort TB, Lamers WH, de Graan PN. Haploinsufficiency of glutamine synthetase increases susceptibility to experimental febrile seizures. Genes Brain Behav 2009;8:290–295. [DOI] [PubMed] [Google Scholar]
- 11. Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA‐glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem 2006;98:641–653. [DOI] [PubMed] [Google Scholar]
- 12. Battaglioli G, Martin DL. GABA synthesis in brain slices is dependent on glutamine produced in astrocytes. Neurochem Res 1991;16:151–156. [DOI] [PubMed] [Google Scholar]
- 13. Eid T, Ghosh A, Wang Y, et al. Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain 2008;131:2061–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rodriguez J, Moran J, Blanco I, Patel AJ. Effect of histamine on the development of astroglial cells in culture. Neurochem Res 1989;14:693–700. [DOI] [PubMed] [Google Scholar]
- 15. Jin CL, Yang LX, Wu XH, et al. Effects of carnosine on amygdaloid‐kindled seizures in Sprague–Dawley rats. Neuroscience 2005;135:939–947. [DOI] [PubMed] [Google Scholar]
- 16. Hosli L, Hosli E, Schneider U, Wiget W. Evidence for the existence of histamine H1‐ and H2‐receptors on astrocytes of cultured rat central nervous system. Neurosci Lett 1984;48:287–291. [DOI] [PubMed] [Google Scholar]
- 17. Paxinos G, Watson C, editors. The rat brain stereotaxic coordinates. Sydney: Academic Press, 1986. [Google Scholar]
- 18. Wang S, Wu DC, Ding MP, Li Q, Zhu‐Ge ZB, Zhang SH, Chen Z. Low‐frequency stimulation of cerebellar fastigial nucleus inhibits amygdaloid kindling acquisition in Sprague–Dawley rats. Neurobiol Dis 2008;29:52–58. [DOI] [PubMed] [Google Scholar]
- 19. Wu DC, Xu ZH, Wang S, et al. Time‐dependent effect of low‐frequency stimulation on amygdaloid‐kindling seizures in rats. Neurobiol Dis 2008;31:74–79. [DOI] [PubMed] [Google Scholar]
- 20. Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 1972;32:281–294. [DOI] [PubMed] [Google Scholar]
- 21. Nishida N, Huang ZL, Mikuni N, Miura Y, Urade Y, Hashimoto N. Deep brain stimulation of the posterior hypothalamus activates the histaminergic system to exert antiepileptic effect in rat pentylenetetrazol model. Exp Neurol 2007;205:132–144. [DOI] [PubMed] [Google Scholar]
- 22. Galic MA, Riazi K, Heida JG, et al. Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci 2008;28:6904–6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lian XY, Khan FA, Stringer JL. Fructose‐1,6‐bisphosphate has anticonvulsant activity in models of acute seizures in adult rats. J Neurosci 2007;27:12007–12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fischer W, Kittner H, Regenthal R, Malinowska B, Schlicker E. Anticonvulsant and sodium channel blocking activity of higher doses of clenbuterol. Naunyn Schmiedebergs Arch Pharmacol 2001;363:182–192. [DOI] [PubMed] [Google Scholar]
- 25. Zhu YY, Zhu‐Ge ZB, Wu DC, Wang S, Liu LY, Ohtsu H, Chen Z. Carnosine inhibits pentylenetetrazol‐induced seizures by histaminergic mechanisms in histidine decarboxylase knock‐out mice. Neurosci Lett 2007;416:211–216. [DOI] [PubMed] [Google Scholar]
- 26. Gilby KL, Sydserff S, Patey AM, Thorne V, St‐Onge V, Jans J, McIntyre DC. Postnatal epigenetic influences on seizure susceptibility in seizure‐prone versus seizure‐resistant rat strains. Behav Neurosci 2009;123:337–346. [DOI] [PubMed] [Google Scholar]
- 27. Ohtsu H, Tanaka S, Terui T, et al. Mice lacking histidine decarboxylase exhibit abnormal mast cells. FEBS Lett 2001;502:53–56. [DOI] [PubMed] [Google Scholar]
- 28. Huang JH, Huang XH, Chen ZY, Zheng QS, Sun RY. Dose conversion among different animals and healthy volunteers in pharmacological study. Chin J Clin Pharmacol Ther 2004;9:1069–1072. [Google Scholar]
- 29. Olson KR, Kearney TE, Dyer JE, Benowitz NL, Blanc PD. Seizures associated with poisoning and drug overdose. Am J Emerg Med 1994;12:392–395. [DOI] [PubMed] [Google Scholar]
- 30. Yokoyama H, Sato M, Iinuma K, Onodera K, Watanabe T. Centrally acting histamine H1 antagonists promote the development of amygdala kindling in rats. Neurosci Lett 1996;217:194–196. [PubMed] [Google Scholar]
- 31. Simons FE. Advances in H1‐antihistamines. N Engl J Med 2004;351:2203–2217. [DOI] [PubMed] [Google Scholar]
- 32. Nobles ST. Delmar's drug reference for health care professionals. Australia: Cengage Learning, 2001. [Google Scholar]
- 33. Santiago‐Palma J, Fischberg D, Kornick C, Khjainova N, Gonzales G. Diphenhydramine as an analgesic adjuvant in refractory cancer pain. J Pain Symptom Manage 2001;22:699–703. [DOI] [PubMed] [Google Scholar]
- 34. Kelly DW, Slikker W Jr. The metabolism and elimination of pyrilamine maleate in the rat. Drug Metab Dispos 1987;15:460–465. [PubMed] [Google Scholar]
- 35. Toothaker RD, Barker SH, Gillen MV, Helsinger SA, Kindberg CG, Hunt TL, Powell JH. Absence of pharmacokinetic interaction between orally co‐administered naproxen sodium and diphenhydramine hydrochloride. Biopharm Drug Dispos 2000;21:229–233. [DOI] [PubMed] [Google Scholar]
- 36. Yokoyama H. The role of central histaminergic neuron system as an anticonvulsive mechanism in developing brain. Brain Dev 2001;23:542–547. [DOI] [PubMed] [Google Scholar]
- 37. Wang Y, Zaveri HP, Lee TS, Eid T. The development of recurrent seizures after continuous intrahippocampal infusion of methionine sulfoximine in rats: A video‐intracranial electroencephalographic study. Exp Neurol 2009;220:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liebschutz J, Airoldi L, Brownstein MJ, Chinn NG, Wurtman RJ. Regional distribution of endogenous and parenteral glutamate, aspartate and glutamine in rat brain. Biochem Pharmacol 1977;26:443–446. [DOI] [PubMed] [Google Scholar]
- 39. Wang L, Maher TJ, Wurtman RJ. Oral L‐glutamine increases GABA levels in striatal tissue and extracellular fluid. FASEB J 2007;21:1227–1232. [DOI] [PubMed] [Google Scholar]
- 40. Liang SL, Carlson GC, Coulter DA. Dynamic regulation of synaptic GABA release by the glutamate–glutamine cycle in hippocampal area CA1. J Neurosci 2006;26:8537–8548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ortinski PI, Dong J, Mungenast A, et al. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci 2010;13:584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Representative examples of EEGs after diphenhydramine treatment. Figure S2 Chronic H1‐antihistamine treatment decreases GS activity 1 or 5 days after withdrawal in rats. Figure S3 Toluidine blue staining of hippocampus after chronic diphenhydramine treatment in rats.