

SUMMARY
Aims: Pioglitazone, known as a peroxisome proliferator‐activated receptor γ (PPARγ) agonist, is used to treat type 2 diabetes mellitus (T2DM). T2DM has been associated with reduced performance on numerous domains of cognitive function. Here, we investigated the effects of pioglitazone on memory impairment in a mouse model with defects in insulin sensitivity and secretion, namely high‐fat diet (HFD) streptozotocin (STZ)‐induced diabetic mice. Methods: ICR mice were fed with HFD for 4 weeks and then injected with a single low dose of STZ followed by continued HFD feeding for an additional 4 weeks. Pioglitazone (18 mg/kg, 9 mg/kg body weight) was orally administered for 6 weeks once daily. Y‐maze test and Morris water maze test (MWM) were employed for testing learning and memory. Serum glucose, serum insulin, serum triglyceride, brain β‐amyloid peptide (Aβ), brain β‐site amyloid precursor protein cleaving enzyme (BACE1), brain nuclear factor κB (NF‐κB), and brain receptor for advanced glycation end products (RAGE) were also tested. Results: The STZ/HFD diabetic mice, characterized by hyperglycemia, hyperlipemia and hypoinsulinemia, performed poorly on Y‐maze and MWM hence reflecting impairment of learning and memory behavior with increases of Aβ40/Aβ42, BACE1, NF‐κB, and RAGE in brain. Treatment of PPARγ agonist, pioglitazone (18 or 9 mg/kg body weight), significantly reversed diabetes‐induced impairment of learning and memory behavior, which is involved in decreases of Aβ40/Aβ42 via inhibition of NF‐κB, BACE1 and RAGE in brain as well as attenuation of hyperglycemia, hyperlipemia, and hypoinsulinemia. Conclusions: It is concluded that PPARγ agonist pioglitazone may be considered as potential pharmacological agents for the management of cognitive dysfunction in T2DM.
Keywords: BACE1, β‐amyloid peptide, Cognition, Diabetes mellitus, NF‐κB, Pioglitazone
Introduction
Cognitive dysfunction and dementia have been proven to be common complications of diabetes mellitus [DM; Ref. 1]. Both type 1 and type 2 DM have been associated with reduced performance on numerous domains of cognitive function [2]. The exact pathophysiology of cognitive dysfunction in diabetes is not completely understood, though it was recognized as early as the 1920s that diabetes may affect cognition [3]. Very recently, a new term—“diabetes‐associated cognitive decline” (DACD) was proposed to facilitate research into this area and to increase recognition of the disorder [4]. This term is not suggestive of a particular pathogenesis, but merely describes a state of mild to moderate cognitive impairment, in particular, psychomotor slowing and reduced mental flexibility, not attributable to other causes.
Strong epidemiologic evidence supports an association between T2DM and Alzheimer's disease (AD) characterized by accumulation of β‐amyloid peptide (Aβ), elevation of hyperphosphorylated τ, neurite degeneration, and neuronal loss [5, 6]. There is a growing body of evidence that decreased insulin in T2DM may participate in amyloid precursor protein (APP) dysregulation and Aβ accumulation. Some reports showed that the increased Aβ level was present in the central nervous system of the diabetic patients [7, 8, 9]. Aβ accumulation also occurred in brain of the diabetic animals spontaneously [10] or induced by streptozotocin [a diabetogenic agent, toxic to β cell of pancreatic island; Refs. 11, 12], accompanied by cognitive impairment [13, 14]. Recent studies showed that insulin deficiency or insulin resistance exacerbated cerebral amyloidosis and behavioral deficits in an Alzheimer transgenic mouse model [15, 16, 17]. Furthermore, diabetes‐accelerated memory dysfunction was due to cerebrovascular inflammation and Aβ deposition in an Alzheimer mouse model with diabetes [18]. These findings suggest that Aβ accumulation in brain in diabetic condition may be one of the reasons for diabetes‐associated impairment of cognition. Peroxisome proliferator‐activated receptors (PPARs) are ligand‐dependent transcription factors. Activation of the PPARγ subtype is known to increase insulin sensitization, modulate glucose and lipid metabolism. Pioglitazone is a thiazoledinedione (TZD) and a highly selective PPARγ agonist. It is currently approved as an oral monotherapy and adjunctive therapy for patients with T2DM [19]. In more recent years, the focus on PPARγ agonists has intensified, as their novel biological roles have emerged, particularly for their therapeutic potential in neurodegenerative disorders, such as AD [20]. However, so far, less attention has been given to the effect of PPARγ agonists on cognitive impairment in T2DM. This study was undertaken to investigate the effects of pioglitazone on memory impairment and brain Aβ levels and other biochemical parameters in a mouse model with defects in insulin sensitivity and secretion, namely high‐fat diet (HFD) streptozotocin (STZ)‐induced diabetic mice.
Materials and Methods
Materials
Pioglitazone was purchased from Jiangsu Hengrui Medicine Co. Ltd (Nanjing, China). STZ was purchased from Sigma (St. Louis, MO, USA). Antibodies were purchased from different companies: anti‐Aβ42 from Bioworld Technology Co., Ltd (Minneapolis, MN, USA), anti‐receptor for advanced glycation end products (RAGE) and secondary antibody from Abcam Ltd. (Hongkong, China), anti‐β‐site APP cleaving enzyme 1(BACE1) from Cell Signaling Technology (USA). Mouse Insulin (INS) ELISA Kit, mouse Aβ40 ELISA Kit, mouse Aβ42 ELISA Kit and nuclear factor κB (NF‐κB) p65 ELISA Kit were purchased from R&D Systems (Shanghai, China), Coomassie (Bradford) protain assay kit, Glucose Oxidase Kit and Triglyceride (TG) Kit were purchased from Nanjing Jiancheng Biotech Institute (Nanjing, China). HFD was purchased from Medical Center of Yangzhou University (Yangzhou, China), consisted of 60% fat, 20% carbohydrate, and 20% protein, and all other chemicals were of analytical grade and commercially available.
ICR male mice (8–10 weeks old, weighing 20–25 g) were purchased from Medical Center of Yangzhou University (Yangzhou, China). All animal procedures were performed in accordance with the guidelines of the institutional animal care and use committee of China. Mice were housed eight per cage and allowed access to diet and autoclaved water. Animal housing rooms were maintained at a constant room temperature (25°C) in a 12‐h light (7:00 A.M.) / dark (7:00 P.M.) cycle.
Generation of Diabetic Model and Treatment with Pioglitazone
The Mice were fed with HFD for 4 weeks and then injected once with low dose of STZ (in the tail vein at 100 mg/kg body weight) to induce partial insulin deficiency, followed by continued HFD feeding for an additional 4 weeks. The majority of STZ/HFD‐treated mice displayed hyperglycemia, insulin resistance, and glucose intolerance as previously reported [21]. Animals with similar degrees of hyperglycemia and body weight were randomly divided into three groups, STZ/HDF plus vehicle group (STZ/HDF+Veh), STZ/HDF plus pioglitazone (18 mg/kg body weight) group (STZ/HDF+Pio 18 mg/kg), and STZ/HDF plus pioglitazone (9 mg/kg body weight) group (STZ/HDF+Pio 9 mg/kg). The normal diet‐fed mice were divided into three groups, vehicle plus vehicle group (Veh+Veh), vehicle plus pioglitazone (18 mg/kg body weight) group (Veh+Pio 18 mg/kg) and vehicle plus pioglitazone (9 mg/kg body weight) group (Veh+Pio 9 mg/kg). Pioglitazone was dissolved in 0.5% sodium carboxymethyl cellulose (CMC‐Na) and the duration for oral administration was 6 weeks. Body weight was monitored every other day. After 5 weeks of pioglitazone treatment, Y‐maze test and subsequent Morris water maze (MWM) test were carried out for the evaluation of cognitive function. Blood glucose, serum insulin, and serum triglyceride levels were measured using assay kits following behavior tests. The mice were sacrificed under ether anesthesia for assays of Aβ, BACE1, NF‐κB, and RAGE in the brain.
Y‐Maze Test
This was performed as described previously [22] with minor modification. The Y‐maze was constructed of black plastic walls (10 cm high), consisting of three compartments (10 × 10 cm) connected with passages (4 × 5 cm), with the floor of 3.175 mm stainless steel rods (8 mm apart). The test was conducted for two consecutive days. On day 1 (learning trial), each mouse was placed in one of the compartments and allowed to move freely for 5 min (habituation) before moving to the next session with electric power on. During the training, electric shocks (2 HZ, 125 ms, 10 V) were available through the stainless steel grid floor in two of the compartments and the light was on in the shock‐free compartment. Each mouse was trained 10 times. The training was stopped once the mouse entered the shock‐free compartment and stayed for 30 seconds, which was recorded as a correct choice. If the mouse did not enter this compartment, it was gently navigated to the compartment and allowed to stay for 30 seconds. On day 2 (testing trial), each mouse was also tested for 10 times following the same procedures as on day 1. The number of correct choices in 10 times was recorded manually.
Morris Water Maze
Spatial memory was assessed by the MWM test, which consisted of 5‐day training (visible and invisible platform training sessions) and a probe trial on day 6. This was carried out as described previously [23] with some modifications. Mice were individually trained in a circular pool (120 cm diameter, 50 cm height) filled to a depth of 30 cm with water maintained at 25°C. The maze was located in a lit room with visual cues. A platform (9 cm diameter) was placed in the center of one quadrant of the pool. The platform's position was fixed throughout the training sessions; the starting points were pseudo‐randomized for each trial, with the animals facing toward the wall. Each mouse was individually trained in both visible‐platform (days 1–2) and hidden‐platform (days 3–5) versions. Visible platform training was performed for baseline differences in vision and motivation; the platform was placed 1 cm below the surface of the water and was indicated by a small flag (5 cm in height). The hidden‐platform version evaluates spatial learning and was used to determine the retention of memory to find the platform. During the training, the platform was placed 1 cm below the surface of the water and the flag was removed. The platform was always in the same place. On each day, the animal was subjected to four trials with a 1‐h interval between trials. Each trial lasted for 90 seconds unless the animal reached the platform first. If an animal failed to find the platform within 90 seconds, the test was ended and the animal was gently navigated to the platform by hand for 30 seconds. On day 6, the platform was removed and the probe trial started, during which animals had 90 seconds to search for the platform. The time spent and the number of entries in the target quadrant (i.e., the quadrant where the platform was previously located) and the number of times for the animals entering the quadrant was recorded. Data of the escape latency, the percentage of time and the number of entries in the target quadrant were collected by the video tracking equipment and processed by a computer equipped with an analysis‐management system (Viewer 2 Tracking Software, Ji Liang Instruments, China).
ELISA Assays
For ELISA assay of the brain tissues, mice (n = 4) were sacrificed by cervical dislocation, and brains were snap‐frozen in dry ice for dissection of the hippocampus and cortex, which were then homogenated, 20% tissue homogenates were prepared in 0.1M PBS, pH 7.4, and stored at −70°C before use. All the procedures were performed following the manufacturer's instructions. In brief, the sample (50 μL) was added into the precoated plate and incubated for 30 min at 37°C in the dark. After washing each well of the precoated plate with washing buffer, 50 μL labeled antibody solution was added and the mixture was incubated at 37°C in the dark for 30 min. Chromogen was then added after washing and the mixture was incubated at 37°C in the dark for 15 min. After the addition of the stop solution, the resulting color was recorded at 450 nm using a microplate absorbance reader. The protein content was measured using Coomassie blue‐based assay reagent and bovine serum albumin as standard. Results were expressed by μg of Aβ40 and Aβ42 per gram of protein, ng of NF‐κB p65 per gram of protein.
Immunohistochemical Staining
Mice were euthanized with diethyl ether and perfused with 0.1 M PBS followed by 4% paraformaldehyde. The brains were collected and immediately fixed in 4% paraformaldehyde for 24 h before transferred successively to 10%, 20% and 30% sucrose solutions, after which the brains were frozen on a cold stage and sectioned in a cryostate (40 μm‐thick). Sections were treated with endogenous peroxidase (3% H2O2 in PBS), for 10 min, followed by 0.01 M PBS blocking buffer containing 10% bovine serum albumin in PBS for 40 min. The sections were then incubated with rabbit polyclonal antibody against Aβ42 (1:100) overnight. After which sections were washed in PBS and incubated with the biotinylated secondary antibodies (1:500) for 30 min. The sections were washed with PBS, incubated with the avidin–biotin complex (Vector Laboratories, Burlingame, CA, USA) for 30 min, and visualized by chromogen DAB (Vector Laboratories) reaction. The sections were dehydrated in ethanol, cleared in xylene, and mounted with permaunt (Fisher Scientific, PA, USA).
Western Blotting
Mice hippocampus and cerebral cortex (n= 3) were chopped into small pieces, homogenized in 0.5 mL of RIPA buffer. The dissolved proteins were collected from the supernatant after centrifugation at 12,000 g for 15 min, Protein concentrations were determined using Coomassie blue‐based assay reagent and then assessed for expression of BACE1 and RAGE proteins. Protein extracts were separated by a SDS‐polyacrylamide gel electrophoresis and then transferred onto a PVDF membrane. The membrane was blocked with 5% skim milk in Tris buffer saline and then incubated at 4°C overnight with respective primary antibodies for rabbit anti‐BACE1 antibody (1:1000), anti‐RAGE antibody (1:500) or β‐actin (inner control, 1:500). After washing with TBST, the membranes were incubated with a horseradish peroxidase conjugated secondary antibody (1:5000) for 2 h at room temperature. The antibody‐reactive bands were visualized by using the enhanced chemiluminescence detection reagents by a gel imaging system (ChemiScope 2850, Clinx Science Instruments Co., Ltd, Shanghai, China).
Statistical Analysis
Data shown are expressed as means ± standard error (SEM). All the data, unless specified, were analyzed using one‐way ANOVA followed by Newman–Keuls tests for post hoc comparisons between groups. When two factors were assessed, the significance of differences was determined using two‐way ANOVAs. Statistical significance was considered when P < 0.05.
Results
Effects of Pioglitazone on Cognitive Impairment in the Diabetic Mice
We first assessed the performance of mice in the Y‐maze. At the learning trial (day 1), mice of all groups entered the compartment randomly, and there were no significant differences among groups (data not shown). In the testing trial (day 2), mice in the STZ/HFD+Veh group showed significant decreases in the number of correct compared to those of Veh+Veh group, and mice in STZ/HDF+Pio (18 or 9 mg/kg) group showed significant increases in the number of correct compared to those of STZ/HFD+Veh group (F[5,59]= 13.296, P < 0.001, Figure 1A), indicating that pioglitazone at the dosage of 18 or 9 mg/kg produced significant improvement on learning deficits in STZ/HFD diabetic mice.
Figure 1.
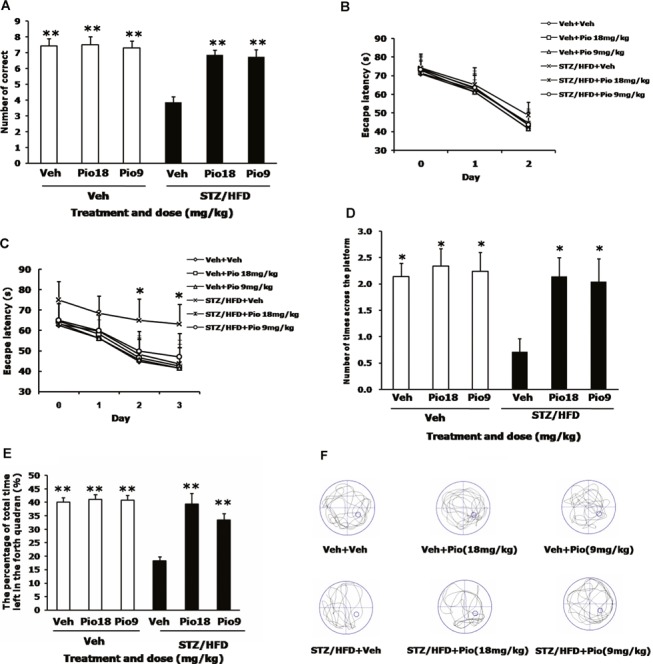
Effects of pioglitazone on memory impairment in STZ/HFD diabetic mice. The number of correct choices in the Y‐maze test was shown (A). In MWM test, day 0 indicates performance on the first trial, and subsequent points represent average of all daily trials. (B) No differences were found in the escape latency among all groups during the 2‐day visible platform test. (C) Changes in escape latency to reach the hidden platform during the 3‐day acquisition trials. The number of times of crossing the platform (D) and the percentage of time spent in the target quadrant (E) during the probe trial test. Representative swim paths of individual mice in each group (F). Values are expressed as the mean ± SEM (n= 10). *P < 0.05, **P < 0.01, versus STZ/HFD+Veh group.
To confirm the results observed in the Y‐maze test, we also carried out the MWM test. Mice in each group exhibited similar escape latency in the 2‐day visible‐platform test, suggesting no differences in vision or basal motivation among all groups (F[5,239]= 1.018, P= 0.418, Figure 1B). We then tested the mice in the 3‐day spatial hidden‐platform variant; the results indicated that mice in the STZ/HFD+Veh group showed increase in escape latencies compared to those of the corresponding controls (Veh+Veh group) (F[5,719]= 5.611, P < 0.001, Figure 1C), and these were reversed by pioglitazone (4 trials/day for 3 days, P < 0.05). In the probe trial, a putative measure of spatial learning and memory retention, swim path (Figure 1F) showed that all the mice showed preference for the target quadrant, with the exception of the mice in STZ/HFD+Veh group which displayed significant decrease in the number of times of crossing the platform (F[5,59]= 6.665, P < 0.001, Figure 1D) and the percentage of time (F[5,59]= 28.179, P < 0.001, Figure 1E) in the target quadrant compared to those of the Veh+Veh group. In contrast, STZ/HFD diabetic mice treated with pioglitazone (18 or 9 mg/kg) showed significant increases in both indices compared to the STZ/HFD diabetic mice with vehicle (P < 0.05 for the number of entries and P < 0.01 for the percentage of time). No differences can be observed in behavior tests between Veh+Veh group and Veh+Pio (18 or 9 mg/kg) group.
Effects of Pioglitazone On Body Weight, Serum Glucose, Triglyceride, and Insulin Levels in Diabetic Mice
No significant differences can be observed in the mean body weight among all groups (data not shown), whereas the combination of HFD and STZ treatment led to frank hyperglycemia, elevation of the serum triglyceride levels and a reduction of serum insulin levels. Pioglitazone at the dose of 18 and 9 mg/kg can reverse these hyperglycemia (F[5,59]= 30.508, P < 0.001, Table 1), hypertriglyceride (F[5,59]= 88.364, P < 0.001, Table 1), and hypoinsulinemia [F[5,59]= 3.814, P= 0.005, Table 1) situation. Effects of pioglitazone on serum glucose, triglyceride and insulin were not found in age‐matched normal mice.
Table 1.
Effects of pioglitazone on serum glucose, serum insulin and serum triglyceride levels in the STZ/HFD diabetic mice
| Groups | Serum glucose (mmol/L) | Serum insulin (mIU/L) | Serum trigly‐ceride (mmol/L) |
|---|---|---|---|
| Veh+Veh | 5.31 ± 0.22** | 22.61 ± 2.00* | 0.48 ± 0.12*** |
| Veh+Pio 18 mg/kg | 5.51 ± 0.37** | 23.61 ± 1.84* | 0.44 ± 0.15*** |
| Veh+Pio 9 mg/kg | 5.41 ± 0.30** | 21.61 ± 1.49* | 0.49 ± 0.15*** |
| STZ/HFD+Veh | 10.90 ± 0.73 | 15.92 ± 1.52 | 1.92 ± 0.38 |
| STZ/HFD+Pio 18 mg/kg | 6.24 ± 0.49** | 21.74 ± 2.08* | 0.80 ± 0.20*** |
| STZ/HFD+Pio 9 mg/kg | 6.61 ± 0.76** | 22.37 ± 2.22* | 0.78 ± 0.19*** |
Values are expressed as the mean ± SEM (n= 10). *P < 0.05, **P < 0.01, ***P < 0.001 vs. STZ/HFD+Veh group.
Effects of Pioglitazone on the Aβ40 and Aβ42 in the Brain of Diabetic Mice
To examine whether pioglitazone could affect the Aβ generation, we measured the Aβ levels in the brain using ELISA assays. Compared with age‐matched Veh+Veh group, STZ/HFD diabetic mice showed a significant increase in both Aβ40 and Aβ42 levels, by 15.6% (F[3,15]= 6.129, P= 0.009) and 27.8% (F[3,15]= 12.085, P= 0.001) in hippocampus or by 20.7% (F[3,15]= 5.538, P= 0.013) and 19.3% (F[3,15]= 5.057, P= 0.017) in cerebral cortex. As expected, treatment of pioglitazone at 18 or 9 mg/kg blocked the increased levels of Aβ40 and Aβ42 in both brain regions of STZ/HFD diabetic mice (Figure 2A and B). To further confirm this, we use the immunohistochemical staining, this result bolstered our ELISA findings that the immunoreactivity of Aβ42 was significantly increased in the brain of STZ/HFD diabetic mice, while the level of Aβ42 immunoreactivity returned to the control level after treatment of diabetic mice with pioglitazone at the dose of 18 and 9 mg/kg (Figure 2C).
Figure 2.
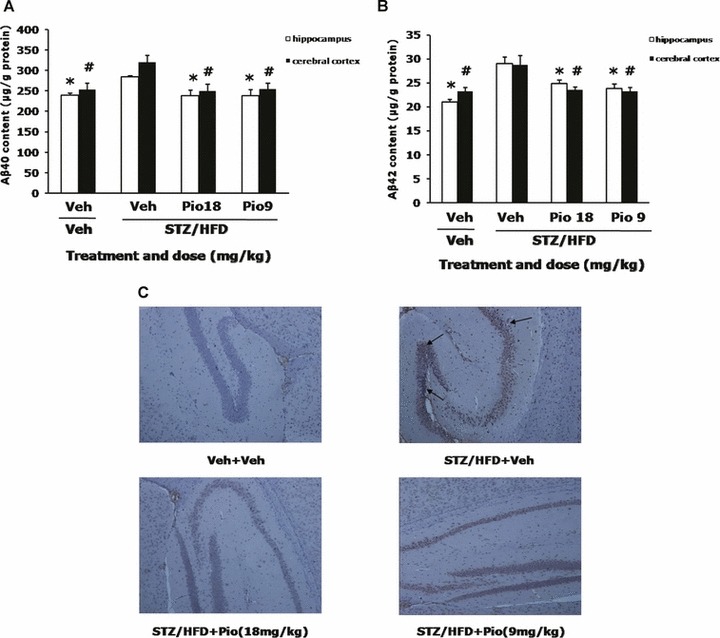
Effects of pioglitazone on the Aβ40 and Aβ42 levels in the brain of STZ/HFD diabetic mice. Aβ40 (A) and Aβ42 (B) levels in the hippocampus and cerebral cortex were measured by ELISA assays. Immunohistochemistry of Aβ42 was also conducted and representative immunohistochemical microphotographs of Aβ42 are shown (C). Values are expressed as the mean ± SEM (n= 4). *P < 0.05 versus hippocampus of STZ/HFD+Veh group, # P < 0.05 versus cerebral cortex of STZ/HFD+Veh group.
Effects of Pioglitazone on the BACE1 Levels in the Brain of Diabetic Mice
BACE1 is a transmembrane aspartic proteinase responsible for cleaving the APP to generate the soluble ectodomain sAPP and its C‐terminal fragment CTF and thus plays an important role in Aβ production [24]. To examine whether BACE1 contributed to the Aβ production in the STZ/HFD diabetic mice, the expression of BACE1 was detected by western blotting. As shown in Figure 3 that BACE1 levels significantly increased 1.7 times in the hippocampus and 3.0 times in the cerebral cortex in the STZ/HFD diabetic mice, compared with those of Veh+Veh group. Treatment of the STZ/HFD diabetic mice with pioglitazone at 18 or 9 mg/kg caused significant reductions in the protein levels of BACE1 both in hippocampus (F[3,11]= 72.793, P < 0.001) and cerebral cortex (F[3,11]= 345.218, P < 0.001, Figure 3A and B).
Figure 3.
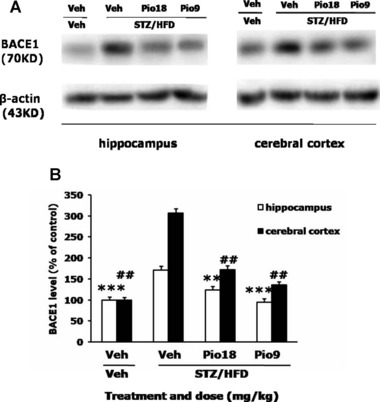
Effects of pioglitazone on BACE1 content in the brain of STZ/HFD diabetic mice. Left panel is representative immunoblots of BACE1 and β‐actin (inner control) measured by western bloting using respective antibodies in hippocampus, left panel is immunoblots in cerebral cortex (A). Quantification of BACE1 is expressed as the ratio (in percentage) of control (B). **P < 0.01, ***P < 0.001 versus hippocampus of STZ/HFD+Veh group, ## P < 0.01 versus cerebral cortex of STZ/HFD+Veh group.
Effects of Pioglitazone on the NF‐κB p65 Levels in the Brain of Diabetic Mice
To test whether BACE1 upregulation is involved in NF‐κB activation, we used the ELISA assay to measure NF‐κB p65 levels in the brain. Our data showed that NF‐κB p65 levels significantly increased in both hippocampus and cerebral cortex in STZ/HFD diabetic mice compared with those of Veh+Veh group (P < 0.05, P < 0.01). Pioglitazone at 18 or 9 mg /kg significantly decreased NF‐κB p65 levels in hippocampus (F[3,15]= 23.800, P < 0.001) and in the cerebral cortex (F[3,15]= 5.849, P= 0.011, Figure 4) compared with STZ/HFD+Veh group.
Figure 4.
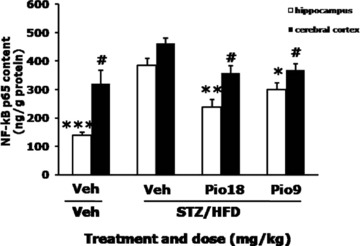
Effects of pioglitazone on NF‐κB content in the brain of STZ/HFD diabetic mice. NF‐κB p65 levels in the hippocampus and cerebral cortex were measured by ELISA assays. Values are expressed as the mean ± SEM (n= 4). *P < 0.05, **P < 0.01, ***P < 0.001 versus hippocampus of STZ/HFD+Veh group, # P < 0.05 versus cerebral cortex of STZ/HFD+Veh group.
Effects of Pioglitazone on the RAGE Levels in the Brain of Diabetic Mice
We also detected the expression of RAGE by western blot. Compared with those of Veh+Veh group, the RAGE levels significantly increased in both the hippocampus and in the cerebral cortex in the STZ/HFD+Veh group. Pioglitatone treatment (18 or 9 mg/kg) significantly reversed the elevated RAGE levels in both the hippocampus (F[3,11]= 197.401, P < 0.001) and the cerebral cortex (F[3,11]= 32.126, P < 0.001, Figure 5A and B).
Figure 5.
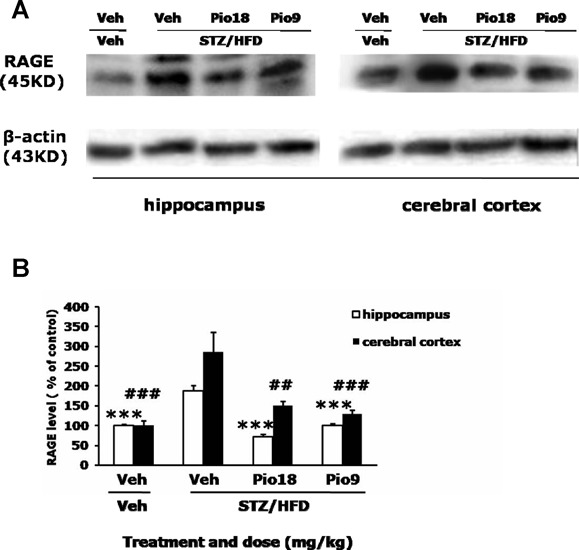
Effects of pioglitazone on RAGE content in the brain of STZ/HFD diabetic mice. Left‐panel is representative immunoblots of RAGE and β‐actin (inner control) measured by western bloting using respective antibodies in hippocampus, left‐panel is immunoblots in cerebral cortex (A). Quantification of RAGE is expressed as the ratio (in percentage) of control (B). ***P < 0.001 versus hippocampus of STZ/HFD+Veh group, ## P < 0.01, ### P < 0.001 versus cerebral cortex of STZ/HFD+Veh group.
Discussion
Clinical and experimental studies indeed show that altered glucose regulation impairs learning and memory [25]. The potential mechanisms for this not only include direct effects of hypo‐ or hyperglycaemia and hypo‐ or hyperinsulima, but also indirect effects via cerebrovascular alterations [26, 27]. In this study, STZ/HFD diabetic mice, characterized by hyperglycemia, hyperlipemia and hypoinsulinemia, produced marked impairment in cognitive function which was coupled with marked increases in Aβ40 and Aβ42 levels in brain. Chronic treatment with PPARγ agonist pioglitazone significantly ameliorated cognitive deficits, with significant decreases in Aβ40 and Aβ42 levels via inhibition of BACE1, RAGE, and NF‐κB in brain as well as attenuation of hyperglycemia, hyperlipemia, and hypoinsulinemia.
Animal models of type 1 and type 2 diabetes exhibit Alzheimer‐like changes, such as accumulation of APP and Aβ[10, 11, 12]. These changes were more marked in a T2DM animal model than in a T1DM model, and, thereby, Aβ accumulation in brain is one of the reasons for DM‐associated cognitive impairment. In this study, ICR mice were fed with HFD for 4 weeks and then injected with a single low dose of STZ followed by continued HFD feeding for an additional 4 weeks to generate a nongenetic rodent model mimicking human T2DM characterized by insulin resistance, insulin deficiency, and significant cognitive impairment evidenced by decrease in the number of correct in Y‐maze test as well as decreases in number of times of crossing the platform and percentage of time spent in the target quadrant in the MWM test, coupled with the elevated Aβ40 and Aβ42 levels in brain. It is well known that accumulation of Aβ in brain may initiate pathogenic cascades mediating neurovascular and neuronal dysfunctions associated with the development of cerebral β‐amyloidosis and cognitive decline in patients with diabetes [2, 28]. Aβ originates from proteolysis of the APP by the sequential enzymatic actions of BACE1, a β‐secretase, and γ‐secretase. BACE1 upregulation was recently reported in a diabetic animal model, which is one of the reasons for Aβ accumulation in brain [29]. BACE1 promoter contains NF‐κB binding site [30], which is a representative transcription factor activated by RAGE‐ligand interactions [31]. Hyperglycemia, a consequence of diabetes, enhances the formation of advanced glycation end products (AGEs), senescent protein derivatives that result from the auto‐oxidation of glucose and fructose [32]. Binding of AGEs to the receptor for AGEs (RAGE), activates intracellular signaling processes, such as NF‐κB pathway, thus mediating pro‐inflammatory effects. The formation and accumulation of AGEs have been known to progress at an accelerated rate in diabetes [33, 34]. Activation of the AGEs/RAGE axis, as a result of hyperglycemia, driving the upregulation of the key enzyme, BACE1, for Aβ production via NF‐κB activation, results in Aβ accumulation in diabetic animal [29]. In addition, the increasing AGEs or Aβ continued binding RAGE, in turn, caused NF‐κB activation, thereby triggering a positive feedback loop in which RAGE expression is upregulated and thus enhances the binding capacity of the AGEs and Aβ ligands [29]. As expected, activation of NF‐κB pathway characterized by the elevation of NF‐κB p65 protein level and its mediated‐upregulation of BACE1 and RAGE were observed in the STZ/HFD diabetic mice.
The synthetic TZDs, such as pioglitazone and rosiglitazone, which serve as PPARγ agonists, are widely prescribed for the treatment of T2DM, and have also been shown to be efficacious in a number of CNS disease models [35, 36]. Suppression of RAGE expression may be a molecular target of PPARγ agonists [37]. Marx et al. reported that the stimulation of human endothelial cell with PPARγ agonists such as rosiglitazone and pioglitazone decreased basal as well as tumor necrosis factor‐alpha‐induced RAGE expression via suppression of NF‐κB activation [38]. Our data suggested that pioglitazone treatment deceased either RAGE or BACE1 by interference with NF‐κB activation in hippocampus and cortex of STZ/HFD diabetic mice, and such mechanisms may explain its decrease of intracerebral Aβ accumulation that strongly correlated with cognitive decline in the STZ/HFD diabetic mice. We used two higher dosages of pioglitazone, 9 mg/kg body weight (equivalent to the maximum dosage for patients, 0.75 mg/kg body weight), and 18 mg/kg body weight in the studies owing to its poor BBB permeability, so that the level of intracerebral pioglitazone was elevated based on the characteristic of its pharmacokinetics [39]. The beneficial effects of pioglitazone at lower dosage such as 4.5 mg/kg body weight for the central nervous system in the STZ/HFD diabetic animals were not observed.
This study has important implications for diabetic patients. In T2DM, the development of cognitive dysfunction is substantially accelerated. Our data indicated that PPARγ agonist pioglitazone may provide a dual benefit for T2DM by ameliorating insulin resistance and its metabolic sequelae as well as the improvement of cognitive impairment.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
Research grants were received from the Graduate School of China Pharmaceutical University. We also acknowledge the editorial assistance of Dr. Zhang Yu, China Pharmaceutical University.
References
- 1. S Roriz‐Filho J, Sá‐Roriz TM, Rosset I, et al (Pre)diabetes, brain aging, and cognition. Biochim Biophys Acta 2009;1792:432–443. [DOI] [PubMed] [Google Scholar]
- 2. Kodl CT, Seaquist ER. Cognitive dysfunction and diabetes mellitus. Endocr Rev 2008;29:494–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miles WR, Root HF. Psychologic tests applied in diabetic patients. Arch Intern Med 1922;30:767–777. [Google Scholar]
- 4. Mijnhout GS, Scheltens P, Diamant M, et al Diabetic encephalopathy: A concept in need of a definition. Diabetologia 2006;49:1447–1448. [DOI] [PubMed] [Google Scholar]
- 5. Janson J, Laedtke T, Parisi JE, O'Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004;53:474–481. [DOI] [PubMed] [Google Scholar]
- 6. Han W, Li C. Linking type 2 diabetes and Alzheimer's disease. Proc Natl Acad Sci U S A 2010;107:6557–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Peila R, Rodriguez BL, Launer LJ. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu‐Asia Aging Study. Diabetes 2002;51:1256–1262. [DOI] [PubMed] [Google Scholar]
- 8. Yoneda S, Hara H, Hirata A, Fukushima M, Inomata Y, Tanihara H. Vitreous fluid levels of beta‐amyloid (1–42) and tau in patients with retinal diseases. Jpn J Ophthalmol 2005;49:106–108. [DOI] [PubMed] [Google Scholar]
- 9. Valente T, Gella A, Fernàndez‐Busquets X, Unzeta M, Durany N. Immunohistochemical analysis of human brain suggests pathological synergism of Alzheimer's disease and diabetes mellitus. Neurobiol Dis 2010;37:67–76. [DOI] [PubMed] [Google Scholar]
- 10. Li ZG, Zhang W, Sima AA. Alzheimer‐like changes in rat models of spontaneous diabetes. Diabetes 2007;56:1817–1824. [DOI] [PubMed] [Google Scholar]
- 11. Liu Y, Liu H, Yang J, et al Increased amyloid beta‐peptide (1–40) level in brain of streptozotocin‐induced diabetic rats. Neuroscience 2008;153:796–802. [DOI] [PubMed] [Google Scholar]
- 12. Hong H, Liu LP, Liao JM, et al Downregulation of LRP1 at the blood‐brain barrier in streptozotocin‐induced diabetic mice. Neuropharmacology 2009;56:1054–1059. [DOI] [PubMed] [Google Scholar]
- 13. Kamal A, Biessels GJ, Duis SE, Gispen WH. Learning and hippocampal synaptic plasticity in streptozotocin‐diabetic rats: interaction of diabetes and ageing. Diabetologia 2000;43:500–506. [DOI] [PubMed] [Google Scholar]
- 14. Sima AA, Li ZG. The effect of C‐peptide on cognitive dysfunction and hippocampal apoptosis in type 1 diabetic rats. Diabetes 2005;54:1497–1505. [DOI] [PubMed] [Google Scholar]
- 15. Jolivalt CG, Hurford R, Lee CA, Dumaop W, Rockenstein E, Masliah E. Type 1 diabetes exaggerates features of Alzheimer's disease in APP transgenic mice. Exp Neurol 2010;223:422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang X, Zheng W, Xie JW, et al Insulin deficiency exacerbates cerebral amyloidosis and behavioral deficits in an Alzheimer transgenic mouse model. Mol Neurodegener 2010;5:46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ho L, Qin W, Pompl PN, et al Diet‐induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. FASEB J 2004;18:902–904. [DOI] [PubMed] [Google Scholar]
- 18. Takeda S, Sato N, Uchio‐Yamada K, et al Diabetes‐accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A 2010;107:7036–7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sood V, Colleran K, Burge MR. Thiazolidinediones: A comparative review of approved uses. Diabetes Technol Ther 2000;2:429–440. [DOI] [PubMed] [Google Scholar]
- 20. Nicolakakis N, Hamel E. The Nuclear receptor PPARgamma as a therapeutic target for cerebrovascular and brain dysfunction in Alzheimer's disease. Front Aging Neurosci 2010;2:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luo J, Quan J, Tsai J, et al Nongenetic mouse models of non‐insulin‐dependent diabetes mellitus. Metabolism 1998;47:663–668. [DOI] [PubMed] [Google Scholar]
- 22. Kimura R, Devi L, Ohno M. Partial reduction of BACE1 improves synaptic plasticity, recent and remote memories in Alzheimer's disease transgenic mice. J Neurochem 2011;113:248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morris R. Developments of a water‐maze procedure for studying spatial learning in the rat. J Neurosci Methods 1984;11:47–60. [DOI] [PubMed] [Google Scholar]
- 24. Joanna M. Exclusively targeting β‐secretase to lipid rafts by GPI‐anchor addition up‐regulates β‐site processing of the amyloid precursor protein. Proc Natl Acad Sci U S A 2003;100:11735–11740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Messier C, Gagnon M. Glucose regulation and cognitive functions: Relation to Alzheimer's disease and diabetes. Behav Brain Res 1996;75:1–11. [DOI] [PubMed] [Google Scholar]
- 26. Brands AM, Kessels RP, de Haan EH, Kappelle LJ, Biessels GJ. Cerebral dysfunction in type 1 diabetes: Effects of insulin, vascular risk factors and blood glucose levels. Eur J Pharmacol 2004;490:159–168. [DOI] [PubMed] [Google Scholar]
- 27. Lobnig BM, Kromeke O, Optenhostert‐Porst C, Wolf OT. Hippocampal volume and cognitive performance in long‐standing Type 1 diabetic patients without macrovascular complications. Diabet Med. 2005;23:32–39. [DOI] [PubMed] [Google Scholar]
- 28. Mackic JB, Bading J, Ghiso J, Walker L, Wisniewski T, Frangione B, Zlokovic BV. 2002. Circulating amyloid‐β peptide crosses the blood‐brain barrier in aged monkeys and contributes to Alzheimer's disease lesions. Vasc Pharmacol 38:303–313. [DOI] [PubMed] [Google Scholar]
- 29. Guglielmotto M, Aragno M, Tamagno E, et al AGEs/RAGE complex upregulates BACE1 via NF‐kappaB pathway activation. Neurobiol Aging 2012;33:196.e13–27. [DOI] [PubMed] [Google Scholar]
- 30. Liu LP, Hong, H , Liao JM, et al Up‐regulation of RAGE at the blood‐brain barrier in streptozotocin‐induced diabetic mice. Synapse 2009;63:636–642. [DOI] [PubMed] [Google Scholar]
- 31. Bourne KZ, Ferrari DC, Lange‐Dohna C, Rossner S, Wood TG, Perez‐Polo JR. Differential regulation of BACE1 promoter activity by nuclear factor‐B in neurons and glia upon exposure to beta‐amyloid peptides. J Neurosci Res 2007;85:1194–1204. [DOI] [PubMed] [Google Scholar]
- 32. Bucala R, Cerami A. Advanced glycosylation: Chemistry, biology, and implications for diabetes and aging. Adv Pharmacol 1992;23:1–34. [DOI] [PubMed] [Google Scholar]
- 33. Thorpe SR, Baynes JW. Maillard reaction products in tissue proteins: New products and new perspectives. Amino Acids 2003;25:275–281. [DOI] [PubMed] [Google Scholar]
- 34. Yamagishi S, Matsui T, Nakamura K. Kinetics, role and therapeutic implications of endogenous soluble form of receptor for advanced glycation end products (sRAGE) in diabetes. Curr Drug Targets 2007;8:1138–1143. [DOI] [PubMed] [Google Scholar]
- 35. Derosa G. Efficacy and tolerability of pioglitazone in patients with type 2 diabetes mellitus: Comparison with other oral antihyperglycaemic agents. Drugs 2010;70:1945–1961. [DOI] [PubMed] [Google Scholar]
- 36. Mandrekar‐Colucci S, Landreth GE. Nuclear receptors as therapeutic targets for Alzheimer's disease. Expert Opin Ther Targets. 2011;15:1085–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yamagishi S, Nakamura K, Matsui T. Regulation of advanced glycation end product (AGE)‐receptor (RAGE) system by PPAR‐gamma agonists and its implication in cardiovascular disease. Pharmacol Res 2009;60:174–178. [DOI] [PubMed] [Google Scholar]
- 38. Marx N, Walcher D, Ivanova N, et al Thiazolidinediones reduce endothelial expression of receptors for advanced glycation end products. Diabetes 2004;53:2662–2668. [DOI] [PubMed] [Google Scholar]
- 39. Maeshiba Y, Kiyota Y, Yamashita K, Yoshimura Y, Motohashi M, Tanayama S. Disposition of the new antidiabetic agent pioglitazone in rats, dogs, and monkeys. Arzneimittelforschung 1997;47:29–35. [PubMed] [Google Scholar]