

Summary
Background
Previous studies have demonstrated that endoplasmic reticulum (ER) stress is activated in Alzheimer's disease (AD) brains. ER stress–triggered unfolded protein response (UPR) leads to tau phosphorylation and neuronal death.
Aims
In this study, we tested the hypothesis that hypoxia‐induced m‐calpain activation is involved in ER stress‐mediated AD pathogenesis.
Method
We employed a hypoxic exposure in APP/PS1 transgenic mice and SH‐SY5Y cells overexpressing human Swedish mutation APP (APPswe).
Results
We observed that hypoxia impaired spatial learning and memory in the APP/PS1 mouse. In the transgenic mouse brain, hypoxia increased the UPR, upregulated apoptotic signaling, enhanced the activation of calpain and glycogen synthase kinase‐3β (GSK3β), and increased tau hyperphosphorylation and β‐amyloid deposition. In APPswe cells, m‐calpain silencing reduced hypoxia‐induced cellular dysfunction and resulted in suppression of GSK3β activation, ER stress and tau hyperphosphorylation reduction as well as caspase pathway suppression.
Conclusion
These findings demonstrate that hypoxia‐induced abnormal calpain activation may increase ER stress‐induced apoptosis in AD pathogenesis. In contrast, a reduction in the expression of the m‐calpain isoform reduces ER stress‐linked apoptosis that is triggered by hypoxia. These findings suggest that hypoxia‐triggered m‐calpain activation is involved in ER stress‐mediated AD pathogenesis. m‐calpain is a potential target for AD therapeutics.
Keywords: Alzheimer's disease, APP/PS1 transgenic mouse, Calpain, Endoplasmic reticulum stress, Glycogen synthase kinase 3, Hypoxia, RNA interference
Introduction
Alzheimer's disease (AD) is a chronic progressive neurodegenerative disorder that is clinically characterized by memory loss and cognitive impairment. Histopathologically, senile plaque (SP) deposition, neurofibrillary tangle (NFT) formation, and synapse loss represent the hallmarks of AD. The extracellular deposition of proteolytic fragments of the amyloid precursor protein (APP), termed β‐amyloid (Aβ), is the core component of SP 1. NFTs are composed of abnormal hyperphosphorylated tau, which forms polymeric filaments 2, 3. It has been reported that not only Aβ, but also hyperphosphorylated tau, can interfere with cellular processes such as calcium homeostasis and protein trafficking 4, 5. Importantly, hypoxia is a major vascular risk factor for AD 6, 7, 8 and can disturb calcium homeostasis 9. Calcium influx provokes the cleavage of functional proteins and enhances calpain activity in the brain 10. However, the precise contributions of hypoxia in facilitating AD pathogenesis remain unclear.
Previous studies have shown that hypoxia facilitates the abnormal cleavage of APP 11. An increase in the level of misfolded proteins in the endoplasmic reticulum (ER) can activate the unfolded protein response (UPR), which restores proteostasis in the ER 12, 13. The ER is a membranous organelle that provides an environment for the synthesis, initial posttranslational modification, proper folding, and maturation of secreted and transmembrane proteins. ER signaling interweaves the nucleus, mitochondria, and plasma membrane. The UPR includes three major pathways: inositol‐requiring enzyme 1 (IRE1), protein kinase RNA‐like ER kinase (PERK), and activating transcription factor 6 (ATF6). The ER provides quality control of proteins to ensure correct handling of the final product 14, 15. However, if a protein folding defect is not resolved, chronic activation of UPR signaling eventually induces apoptotic responses, resulting in ER stress 16, 17. Interestingly, ER stress is an important factor in the neuropathogenesis of a wide variety of neurological disorders 18, 19. It has been observed that the ER stress response is activated in AD 20, 21. ER stress‐induced APP undergoes amyloidogenesis cleavage 22. Furthermore, the UPR is activated in neurons in which pathological tau is present 21, 23, suggesting that the disruption of UPR signaling may potentially modulate AD pathogenesis. However, the links between hypoxia‐induced prolonged UPR activation and AD pathogenesis are still unclear.
Calpain, which is a member of the calcium‐activated intracellular cysteine protease family, is involved in neurotoxic insults in AD 24, 25. The two classical brain calpain isoforms are μ‐calpain and m‐calpain, which regulate Ca2+ levels at micromolar and millimolar concentrations, respectively 26. Ubiquitous μ‐calpain and m‐calpain have been hypothesized to be involved in apoptosis 27, 28. The μ‐calpain isoform is a key soluble neuron injury factor that drives reactive microgliosis 29. m‐calpain may play an important role in the apoptotic pathway that is triggered by ER stress 30. ER stress causes calcium efflux, which activates calpain 31, 32. Conversely, calpain activation leads to functional disturbance of the ER 33. In addition, sustained overactivation of calpain may trigger signaling cascades, such as the glycogen synthase kinase 3 (GSK‐3) pathway 34. It has been speculated that increased GSK‐3 signaling might contribute to tau hyperphosphorylation and Aβ generation in the AD brain 35. GSK‐3 is a key intermediate in apoptosis‐related signaling pathways that activate caspase‐3 36. Furthermore, calpain‐mediated proteolysis of recombinant GSK3β was shown to significantly increase GSK‐3 activity 37. Meanwhile, it has been reported that GSK‐3 plays an important role in ER stress‐induced apoptosis 38 and is a target of ER stress transducers, such as PERK 39. Importantly, Liang et al. 40 found that overexpression of endogenous calpain inhibitor calpastatin (CAST) in APP/PS1 mice caused remarkable decrease in Aβ plaque burdens and prevented tau phosphorylation and the loss of synapses. In APP23/CAST double transgenic mice, Aβ pathology is reduced compared with APP23 mice 41. Moreover, calpain inhibition can prevent hypoxia‐induced proteolysis 42. Hypoxia induced calpain activity in retinal endothelial cells and severely disrupted the actin cytoskeleton, whereas calpain inhibitors preserved actin cables under hypoxic conditions 43. Therefore, it is reasonable to speculate that calpains may be a common link between ER stress and AD pathogenesis following exposure to hypoxic conditions.
In the present study, we investigated the role of calpain in hypoxia‐induced ER stress using AD mouse and cell models. We found that hypoxia significantly increased UPR signaling, induced calpain activation, and increased the GSK3β activity. In addition, hypoxia increased tau hyperphosphorylation and Aβ deposition in the brains of APPswe/PS1dE9 (APP/PS1) transgenic mice. Mechanistically, siRNA‐mediated knockdown of m‐calpain significantly downregulated hypoxia‐triggered UPR signaling, resulted in the decrease in apoptotic cells, reduced GSK3β activity, Aβ generation and tau hyperphosphorylation in human neuroblastoma SH‐SY5Y cells overexpressing the Swedish mutation APP (APPswe) in vitro. These findings demonstrate that the hypoxia‐induced activation of m‐calpain is involved in ER stress‐mediated AD pathogenesis.
Materials and Methods
Experimental Animals and Treatments
Six‐month‐old female APP/PS1 mice (Jackson Laboratory, West Grove, PA, USA) and age‐matched C57BL/6 mice were randomly assigned to either the hypoxia or control group. The animals were group housed in a controlled environment (22–25°C, 50% humidity, 12 h light/dark cycle). A standard diet and distilled water were available ad libitum. The mice in the hypoxia group were exposed to hypoxic conditions once daily for 2 months. The procedures of the hypoxic treatment were performed exactly as previously described 44. Briefly, each mouse was placed in a 125‐mL jar with fresh room air, and then the jar was sealed tightly with a rubber plug. The mouse was immediately removed from the jar once the first gasping breath was observed. The time at which the first gasp was observed for each mouse was regarded as the tolerance limit in each experiment. Body weight of mice was monitored per week. When the mice reached 8 months of age, learning and memory evaluation, morphological assessment, and biochemical analyses were performed.
Morris Water Maze
Morris water maze tests were performed as previously described with minor modifications 45. The apparatus used was a circular tank equipped with a digital pick‐up camera for monitoring the animal behavior and a computer program for analyzing data (ZH0065, Zhenghua Bioequipments, Anhui, China). Briefly, mice were pretrained for 2 days by exposing them to the water maze apparatus with the platform placed in the center of the northwest quadrant in the tank. From the 3rd to 7th day, the platform was submerged 1 cm below the water surface (hidden platform) for the place navigation test. Each mouse was subjected to three trials with 1‐min intertrial interval per day. The escape latency to the hidden platform and the path length of each trial were recorded. On the 8th day, for the probe trial, the platform was removed. The times that the mouse crossed the center of the northwest quadrant at an interval of 1 min were recorded. The data of the escape latency, the path length, and the passing times among groups were analyzed.
Tissue Preparation
After water maze test, mice were anesthetized with sodium pentobarbital (50 mg/kg) by intraperitoneal (i.p.) injection and sacrificed by decapitation. The brains were immediately removed and divided into halves. The right hemisphere was placed in 4% paraformaldehyde and embedded in paraffin for morphological analyses, and the left hemisphere was kept at −80°C for biochemical assays.
Nissl Staining
Coronal paraffin sections (6 μm) were dewaxed, rehydrated, and soaked in 0.1% cresyl violet at 37°C for 20 min. After dehydrated with a graded series of ethanol solutions, sections were placed under cover slips. Hippocampus was analyzed with a microscope. Cells with round and palely stained nuclei were considered as surviving cells. Five brain sections of each animal were selected and processed for counting. Surviving neurons/field was expressed as the percentage of WT group.
Cell Culture and Treatment
The APPswe and control cell lines were generated by Lipofectamine 2000 (Invitrogen, New York, NY, USA) transfection with human Swedish mutation APP or empty vectors (neo) pCLNCXv.2 in neuroblastoma SH‐SY5Y cells as we previously described 46. The APPswe cells were grown in DMEM/F12 (Gibco, New York, NY, USA) supplemented with 10% fetal bovine serum (Gibco) and 200 μg/mL G418 at 37°C in a humidified 5% CO2 incubator. APPswe cells were transfected with μ‐calpain siRNA, m‐calpain siRNA, or control siRNA using Lipofectamine 2000, according to the manufacturer's protocol. The μ‐calpain siRNA sequence was 5′‐GUGAAGGAGUUGCGGACAA‐3′, and the m‐calpain siRNA sequence was 5′‐ GGCAUUAGAAGAAGCAGGUTT‐3′ (GenBank). Scrambled siRNA from Qiagen (Valencia, CA, USA) was used as the control. The cells were harvested at 24, 48, 72, and 96 h post‐transfection to determine the optimal transfection time. The cell lysates were subjected to Western blot analysis. Based on the calpain expression levels after the indicated siRNA transfection times, 48 h was selected as the optimal treatment time. APPswe cells were then transfected with μ‐calpain, m‐calpain, or control siRNA for 48 h and then treated with 1 mM NiCl2 for 4 h, as previously described 47, to induce chemical hypoxia. The viability of APPswe cells was assessed by the MTT assays. At least three independent cell culture experiments were conducted. The cells and conditioned medium were harvested for analyses.
Western Blot
The APP/PS1 mouse cortex was minced and homogenized by ultrasonic treatment as previously described in chilled lysis buffer containing a protease inhibitor cocktail overnight at 4°C 48. The conditioned medium from APPswe cell cultures was concentrated. Cells pellets were lysed with lysis buffer for 2 h at 4°C, and the samples were then centrifuged. The supernatants were then collected, and total protein concentrations were determined using a UV 1700 PharmaSpec ultraviolet spectrophotometer (Shimadzu, Kyoto, Japan). Proteins (50 μg) were separated on 10% SDS–PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Temecula, MA, USA). After blocked, the membranes were then incubated with a primary antibody overnight at 4°C. The antibodies used in the study were as follows: rabbit anti‐PERK (1:800; Santa Cruz, Santa Cruz, CA, USA), rabbit anti‐p‐PERK (1:800; Santa Cruz), rabbit anti‐eIF2α (1:1000; Cell Signaling Technology, Danvers, MA, USA), rabbit anti‐p‐eIF2α (1:1000; Cell Signaling Technology), rabbit anti‐IRE1α (1:1000; Cell Signaling Technology), rabbit anti‐p‐IRE1α (1:1000; Abcam, Cambridge, UK), rabbit anti‐XBP‐1 (1:800; Santa Cruz), rabbit anti‐ATF6 (1:500, Abcam), goat anti‐Grp78/BiP (1:500; Santa Cruz), mouse anti‐CHOP/GADD153 (1:1000; Cell Signaling Technology), rabbit anticaspase‐12 (1:1000; Abcam), rabbit anti‐caspase‐3 (1:500; Santa Cruz), rabbit anticleaved caspase‐3 (1:1000; Cell Signaling Technology), mouse anti‐p‐tau Thr231 (1:1000; Invitrogen), rabbit anti‐p‐tau‐Thr205 (1:800; Abcam), rabbit anti‐p‐tau Thr404 (1:500; Santa Cruz), rabbit anti‐p‐tau‐Ser396 (1:500; Abcam), mouse anti‐tau‐5 (1:1000; Invitrogen), mouse anti‐tau C3 (1:1000; Millipore), rabbit anticalpain (1:3000; Abcam), rabbit antiphospho‐GSK3α/β (1:1000; Cell Signaling Technology), rabbit anti‐GSK3α/β (1:1000; Cell Signaling), and mouse anti‐GAPDH (1:10000; Kang Chen, Shanghai, China). Bound secondary antibodies were visualized using an enhanced chemiluminescence kit (Pierce, Rockford, IL, USA) using Chem Doc XRS with Quantity One software (Bio‐Rad, Kennedy BlvdUnion, CA, USA).
RT‐PCR
Total RNA was isolated from APP/PS1 mouse cortical tissue homogenates using the Trizol reagent (Invitrogen), according to the manufacturer's protocol. Total RNA (1 μg) was reverse‐transcribed with the reverse transcription system (Promega, Shanghai, China). The cDNAs (2 μL) were amplified with the following primers: Grp78/BiP: 5′‐CCACTAATGGAGATACTCACCTGGG‐3′ (forward), 5′‐GTAAGGGGACACACATCAAGCAG‐3′ (reverse); XBP‐1 gene segment containing the IRE1‐splicing region during UPR activation: 5′‐TTACGGGAGAAAACTCACGGA‐3′ (forward), 5′‐GGGTCCAACTTGTCCAGAATGC‐3′ (reverse); CHOP/GADD153: 5′‐GCCGGAACCTGAGGAGAGAGTGT‐3′ (forward), 5′‐GTGCGTGTGACCTCTGTTGGCC‐3′ (reverse); caspase‐3: 5′‐GAGCACTGGAATGTCATCTCGC‐3′ (forward), 5′‐AAGCATACAGGAAGTCAGCCTCC‐3′ (reverse); caspase‐12: 5′‐GCTGGCCACATTGCCAATTCCC‐3′ (forward), 5′‐GCCAGACGTGTTCGTCCCTCC‐3′ (reverse); GAPDH: 5′‐ACGGATTTGGTCGTATTGGG‐3′ (forward), 5′‐CGCTCCTGGAAGATGGTGAT‐3′ (reverse). The PCR products were normalized in relation to the GAPDH mRNA standard.
Calpain Enzymatic Activity Assay
The calpain enzymatic activity assay was performed as previously described 49. Briefly, brain tissue from APP/PS1 mice was homogenized in lysis buffer (25 mM HEPES, 5 mM MgCl2, 5 mM DTT, 5 mM EDTA, 2 mM PMSF, and 10 μg/mL pepstatin, pH 7.4) for three freeze–thaw cycles. The samples were then centrifuged, and the supernatants were collected. The supernatant (90 μL) was then incubated with 10 μL of fluorogenic substrate (Suc‐Leu‐Tyr‐AMC, 500 μM; Calbiochem, Darmstadt, MA, USA) for 1 h at 30°C, which allows measurement of activity through enzyme‐mediated cleavage of the substrate into the fluorescent product amino‐methylcoumarin. An equal volume of the assay buffer (100 mM HEPES containing 10 mM DTT, pH 7.4) was then added, and the fluorescence was measured with a fluorescence spectrophotometer (Hitachi, Tokyo, Japan) (λex 380 nm, λem 480 nm).
Confocal Laser Scanning Microscopy
For double immunofluorescence staining, serial 6‐μm coronal paraffin sections or cultured cells were prepared. Briefly, paraffin sections were dewaxed, rehydrated, and treated with 0.1 M Tris‐HCl buffer (pH 7.4) containing 3% hydrogen peroxide for 10 min to block endogenous peroxidase activity. APPswe cells were fixed with 4% paraformaldehyde for 20 min after the indicated treatment periods. After rinsing, the cells were preincubated with normal donkey serum (Jackson ImmunoResearch Laboratory, West Grove, PA, USA) for 1 h at room temperature. For Aβ immunofluorescence labeling, sections were incubated in primary antibody, mouse anti‐Aβ (1:200; Sigma, St. Louis, MO, USA). For confocal laser scanning microscopy analysis, brain sections and cells were incubated in a mixture of primary antibodies, mouse anti‐Aβ, rabbit anti‐p‐tau‐Thr205 (1:100; Abcam), rabbit anti‐p‐tau‐Ser404, or rabbit anti‐p‐tau‐Ser396 (1:200; Abcam) antibodies. The sections and cells incubated with normal donkey serum instead of primary antibodies were used as negative controls. After rinsing, the samples were incubated for 2 h with a mixture of secondary antibodies, including FITC‐conjugated donkey anti‐mouse IgG (1:50) and Texas red‐conjugated donkey anti‐rabbit IgG (1:50). The sections were examined with a confocal laser scanning microscope (SP2; Leica, Wetzlar, Germany). The p‐tau expression, number, and area of Aβ‐positive plaques were measured using Image‐Pro Plus 6.0 software (Media Cybernetics, Warrendale, MD, USA).
Quantification of Apoptosis
The apoptotic APPswe cells were quantified after the indicated treatment periods according to the protocol provided with the annexin V‐FITC/PI apoptosis detection kit (Biovision, Milpitas, CA, USA). Briefly, APPswe cells were trypsinized, rinsed with serum‐containing media, and resuspended in 500 μL of 1× Binding Buffer. The cells were then stained with 5 μL of annexin V‐FITC and 5 μL of propidium iodide (PI). Samples were incubated in the dark at room temperature for 5 min and then analyzed by flow cytometry using the FITC (FL1) and PI (FL2) signal detectors.
Statistical Analysis
All values were expressed as mean ± standard error of the mean (SEM). Comparisons were analyzed using SPSS 16.0 software (IBM, Chicago, NY, USA). Statistical significance between the groups was determined using a Student's t‐test as well as one‐way analysis of variance (ANOVA) and Post hoc Fisher's protected least significant difference (PLSD) for cultured cells. For water maze analysis of latency and path length, repeated measures ANOVA were performed; P < 0.05 was considered statistically significant.
Results
Hypoxia Treatment Impaired Spatial Learning and Memory, and Increased Hippocampal Neuronal Apoptosis in the CA1 and CA3 of APP/PS1 Mouse
Six‐month‐old female APP/PS1 double transgenic (Tg) mice and age‐matched wild‐type (WT) C57BL/6 mice were treated under repeated hypoxic conditions (hypo) once daily for 2 months. During the treatment, there was no significant difference in the body weight between these groups (Figure 1A). Morris water maze tests were performed to evaluate the effect of hypoxia treatment on learning and memory of the mice. As shown in Figure 1B–D, the procedure included visible platform training (2 days), hidden platform tests (5 days), and a probe trial (1 day) after the last hidden platform test. There was no statistical difference in the escape latency and path length of visible platform tests (P > 0.05; Figure 1B,C), which indicated that hypoxia treatment did not markedly alter motility or vision in the mice. In hidden platform tests, the mice of Tg + Hypo group showed a longer escape latency and a longer path length compared with the WT and Tg mice (P < 0.01; Figure 1B,C). In addition, the probe trial indicated that the passing times of Tg + Hypo mice crossed the center of the northwest quadrant, where the hidden platform was previously placed, were significantly less compared with WT and Tg group (P < 0.01; Figure 1D), whereas, hypoxia treatment did not induce statistical alterations in the spatial learning and memory in WT mice. And at the age of 8 month old, there was no significant difference in spatial learning and memory between WT and Tg mice.
Figure 1.
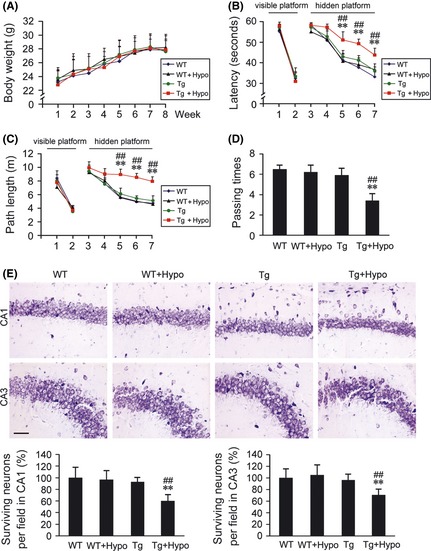
Effects of hypoxia treatment on spatial learning and memory, and the hippocampal neuronal apoptosis. (A) During 2 months hypoxia intervention, the body weight evaluation of hypoxia‐treated transgenic APP/PS1 mice (Tg + Hypo) and age‐matched wild‐type C57BL/6 mice (WT + Hypo) or the groups under control (Tg or WT), respectively. (B–D) Morris water maze tests showed the spatial learning and memory capabilities of these four groups. Mice exhibited the similar escape latency and path length in the visible platform training from day 1st to 2nd, whereas, from day 5th to 7th, mice in Tg + Hypo group performed the longest latency and escape length in hidden platform tests. In the probe trial on the 8th day, the mice in Tg + Hypo group exhibited the less passing times into the northwest quadrant, where the platform was previously located; All values are mean ± SEM (n = 6); **P < 0.01 versus WT group, ## P < 0.01 versus Tg group (repeated measures ANOVA). (E) Nissl staining showed the surviving neurons (round and palely stained nuclei) and the apoptosis neurons (shrunken neurons with pyknotic nuclei) in the CA1 and CA3 of the hippocampus. Scale bar = 20 μm. All values are mean ± SEM (n = 6); **P < 0.01 versus WT group, ## P < 0.01 versus Tg group (two‐way ANOVA was adopted for factorial designs).
We then assessed the hypoxia‐triggered effects on hippocampal neuronal apoptosis by Nissl. As shown in Figure 1E, more nuclear breakdown, less nuclear integrity, and intact Nissl substance in the neurons were observed in hippocampal subareas of CA1 and CA3 in the hypoxia‐treated APP/PS1 mouse brain. While, there were no statistical differences in the neuronal apoptosis between WT, WT + Hypo, and Tg groups.
Hypoxia Activates the UPR and Increases ER Stress‐Induced Apoptosis in the APP/PS1 Mouse Brain
To investigate the molecular pathway involved in hypoxia‐mediated AD pathogenesis‐linked ER stress in vivo, we examined UPR signaling by evaluating the phosphorylated form of PERK (p‐PERK), eukaryotic translation initiation factor 2 α subunit (p‐eIF2α), IRE1, and ATF6. Hypoxia treatment increased the ratios of p‐PERK/PERK and p‐eIF2α/eIF2α to 163.27 ± 19.24% (P < 0.01; Figure 2A) and 133.81 ± 10.78% (P < 0.05; Figure 2A), respectively, compared with the control group. Analysis of the ER stress signaling molecules ATF6, IRE1, and X‐box binding protein 1 (XBP‐1) was also performed. The cleavage form of ATF6 was increased to 146.34 ± 20.04% (P < 0.01; Figure 2A), and the ratio of p‐IRE1/IRE1 was markedly increased to 222.60 ± 42.48% (P < 0.01; Figure 2A), compared with the controls. Western blot assays showed that hypoxia treatment increased IRE1‐activated XBP‐1 to 134.91 ± 21.65%, compared with the controls (P < 0.01; Figure 2B). Unconventional splicing of the small intron of XBP‐1 by the ER stress sensor IRE1 is another consequence of ER stress 50. To investigate the effect of hypoxia on the spliced forms of XBP‐1, the expression levels of unspliced (U) and spliced (S) XBP‐1 in the APP/PS1 mouse brain were measured by RT‐PCR. In agreement with the hypoxia‐mediated increase in XBP‐1 expression, RT‐PCR analysis indicated that hypoxia treatment induced an increase in XBP‐1 splicing to 149.43 ± 25.45% of control (P < 0.05; Figure 2C).
Figure 2.
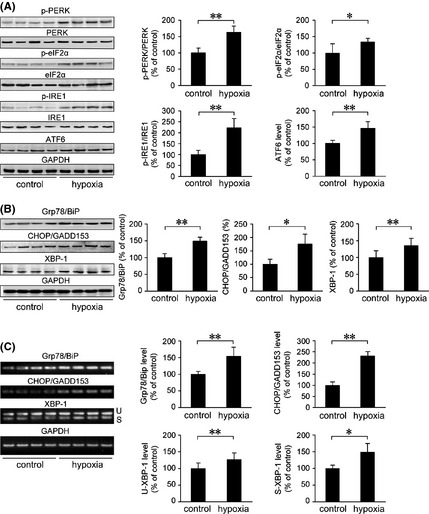
Hypoxia induces the activation of UPR and increases the expression of ER‐resident chaperones in the APP/PS1 mouse brain. Six‐month‐old female APP/PS1 transgenic mice were exposed to hypoxic conditions once daily for 2 months. UPR signaling and ER stress–response proteins were analyzed by Western blot and RT‐PCR. GAPDH was used as an internal control. (A) The increase in p‐PERK/PERK, p‐eIF2α/eIF2α, and p‐IRE1/IRE1 in the APP/PS1 mouse brain exposed to hypoxic conditions indicated the activation of PERK, eIF2α, and IRE1, respectively. In addition, Western blot analysis of ATF6 (active form of ATF6, 50 kDa) showed a significant increase in the hypoxia group. (B) The protein level of ER‐resident chaperones Grp78/BiP was markedly increased, and CHOP/GADD153 and XBP‐1 expression levels were significantly elevated by hypoxia treatment, compared with the control. The changes in mRNA levels of Grp78/BiP and CHOP/GADD153 detected by RT‐PCR were in agreement with the protein expression analysis. RT‐PCR showed the levels of unspliced (U) and spliced (S) XBP‐1. (C) Analysis of XBP‐1 mRNA splicing indicated that hypoxia treatment significantly increased the presence of spliced XBP‐1 mRNA in the APP/PS1 mouse brain. All values are mean ± SEM (n = 6); *P < 0.05, **P < 0.01 versus control group (Student's t‐test).
The expression of ER chaperone glucose‐regulated protein 78/immunoglobulin‐heavy‐chain binding protein (Grp78/BiP) was increased in the cortex and hippocampus of AD postmortem brain, which correlated with an increase in UPR signaling 20, 21. Therefore, we analyzed the expression level of Grp78/BiP in the APP/PS1 mouse brain. As shown in Figure 2B, the protein expression of Grp78/BiP was increased to 149.25 ± 11.83% (P < 0.01), compared with the control group. C/EBP homologous transcription factor/growth arrest and DNA damage‐inducible gene 153 (CHOP/GADD153) is a pro‐apoptotic translational‐dependent mediator of ER stress and is involved in cellular redox and cell death signaling 51, 52. Under ER stress conditions, an increased expression of CHOP/GADD153 has been shown to be a marker of cells undergoing apoptosis 53. To evaluate hypoxia‐induced ER stress‐related apoptosis, the expression levels of CHOP/GADD153 were examined. Our data showed that the levels of both CHOP/GADD153 protein (P < 0.05; Figure 2B) and mRNA (P < 0.01; Figure 2C) were increased in the hypoxia‐treated group compared with the controls. Furthermore, Grp78/BiP mRNA expression was significantly upregulated under hypoxia treatment (P < 0.01; Figure 2C), which was consistent with the protein level alterations.
The activation of caspase‐12 has been shown to mediate ER‐specific apoptosis 54. Therefore, we assessed the expression levels of caspase‐12 and a downstream effector, caspase‐3. As shown in Figure 3, the levels of caspase‐12 protein and mRNA in the hypoxia‐treated mouse brain were increased to 178.02 ± 35.02% and 144.81 ± 12.74%, respectively, compared with the controls (P < 0.01). Moreover, the levels of cleaved caspase‐12 increased to 165.97 ± 25.49% (P < 0.01) under hypoxia treatment compared with the controls. No statistically significant difference in caspase‐3 levels was observed between the two groups (P > 0.05). In contrast, the levels of cleaved caspase‐3 were significantly increased to 153.79 ± 22.87% under hypoxia treatment compared with the controls (P < 0.01).
Figure 3.
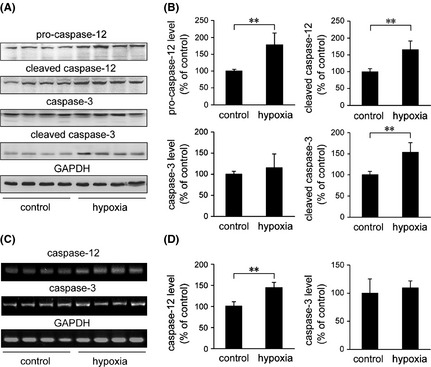
Hypoxia increases susceptibility to ER stress‐induced cell apoptosis in the APP/PS1 mouse brain. (A) The Western blot shows the expression levels of caspase‐12, cleaved caspase‐12, caspase‐3, and cleaved caspase‐3 in the APP/PS1 mouse brain. A GAPDH immunoblot was used as a loading control. (B) The levels of caspase‐12, cleaved caspase‐12, and cleaved caspase‐3 were elevated in the APP/PS1 mouse brain exposed to hypoxic conditions. Hypoxia and control APP/PS1 mice exhibited a similar level of caspase‐3. (C) The levels of caspase‐12 and caspase‐3 mRNA were detected by semiquantitative RT‐PCR. Hypoxia induced an increase in caspase‐12 mRNA, whereas there was no statistical difference in the mRNA level of caspase‐3 in the brain of APP/PS1 transgenic mice between the hypoxia‐treated and control groups (D). All values are mean ± SEM (n = 6); **P < 0.01 versus control group (Student's t‐test)
Hypoxia Enhances Calpain Activation and Elevates GSK‐3 Activity in the APP/PS1 Mouse Brain
To investigate whether ER‐specific apoptosis is related to calpain‐mediated GSK‐3 activation, calpain activity was measured by assessing the fluorescence emitted upon cleavage of a fluorogenic substrate. As shown in Figure 4A, the calpain activity in the APP/PS1 mouse brain increased to 199.25 ± 22.53% of control (P < 0.01) in the hypoxia group.
Figure 4.
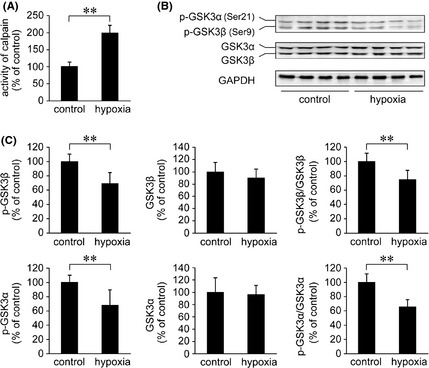
Effects of hypoxia on calpain activity and GSK3α/β in the APP/PS1 mouse brain. (A) Calpain activity was measured by a fluorometric enzyme assay. The results show that hypoxia induces an increase in calpain activity compared with the controls. (B) The activity of GSK3α/β was measured by activity‐dependent phosphorylation antibodies against p‐GSK3α (Ser21) and p‐GSK3β (Ser9). (C) The Western blot shows that the levels of p‐GSK3β and p‐GSK3α were markedly reduced under hypoxia treatment. In addition, the ratios of p‐GSK3β/GSK3β and p‐GSK3α/GSK3α were significantly downregulated in the hypoxia‐treated mouse brain. However, hypoxia did not affect the total protein levels of GSK3β and GSK3α. All values are mean ± SEM (n = 6); **P < 0.01 versus control group (Student's t‐test).
The activation of GSK3β is regulated by phosphorylation at serine 9 (Ser9), and GSK3α activity is inhibited by phosphorylation at serine 21 (Ser21) 55. To assess the activity of GSK3α/β, phosphorylation at these residues was measured using antiphospho‐GSK3β‐Ser9 (p‐GSK3β) and anti‐phospho‐GSK3α‐Ser21 (p‐GSK3α) antibodies, respectively. Western blot analysis showed that the levels of p‐GSK3β and p‐GSK3α were significantly reduced in the APP/PS1 mouse brain under hypoxia treatment compared with the controls (P < 0.01; Figure 4B,C). The levels of GSK3β and GSK3α were not altered.
Hypoxia Increases Tau Phosphorylation and Aβ Deposition in the APP/PS1 Mouse Brain
In AD, tau undergoes abnormal hyperphosphorylation of several residues, including threonine 181 and 231 as well as serine 199, 235, 396, and 404 56. To determine whether repeated hypoxia elevates tau phosphorylation in the APP/PS1 mouse brain, different protein phosphorylation sites were examined, including threonine‐205 (p‐tau‐Thr205), threonine‐231 (p‐tau‐Thr231), serine‐404 (p‐tau‐Ser404), and serine‐396 (p‐tau‐Ser396) as well as total protein level by Western blot (Figure 5). The p‐tau‐Ser396, p‐tau‐Thr205, p‐tau‐Ser404, and p‐tau‐Thr231 levels were significantly increased to 142.65 ± 11.60% (P < 0.01) of control, 139.45 ± 29.17% of control (P < 0.05), 211.75 ± 35.29% of control (P < 0.01), and 178.64 ± 16.23% of control (P < 0.01), respectively, in the hypoxia‐treated APP/PS1 mouse brain (Figure 5A,B). Hypoxia did not affect total tau protein levels (data not shown). In addition, we detected different forms of cleaved tau using anti‐tau‐5 and anti‐tau‐C3 antibodies, which can detect the calpain‐mediated 17 kDa cleaved tau fragment and caspase‐3‐mediated tau truncation at Asp421, respectively. As shown in Figure 5A,C, a significant increase in the 17 kDa tau fragment (P < 0.01) and ~50 kDa caspase‐3‐mediated truncated tau fragment (P < 0.05) were observed in the hypoxia‐treated APP/PS1 mouse brain.
Figure 5.
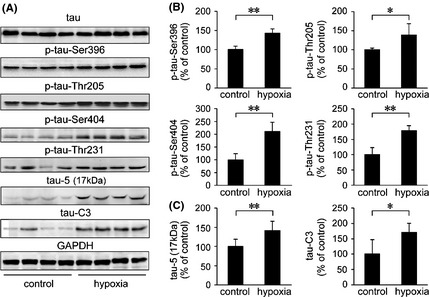
The effects of hypoxia on the phosphorylation of tau in the APP/PS1 mouse brain. (A) Phosphorylation levels of tau in the APP/PS1 mouse brain were measured by p‐tau‐Ser396, p‐tau‐Thr205, p‐tau‐Ser404, and p‐tau‐Thr231 antibodies and normalized against tau total protein. In addition, anti‐tau‐5 and anti‐tau‐C3 antibodies were used to identify calpain‐dependant tau cleavage fragments and truncated tau at residue Asp421, respectively. (B) Quantification of tau phosphorylation shows that p‐tau‐Ser396, p‐tau‐Thr205, p‐tau‐Ser404, and p‐tau‐Thr231 were markedly increased in the hypoxia‐treated APP/PS1 mouse brain, compared with the control group. (C) The protein expression of the 17 kDa tau‐5 and tau‐C3 was increased in the hypoxia‐treated mouse brain, compared with the controls.
To investigate whether hypoxia‐induced ER stress response and caspase activation were accompanied by histopathological alterations in the APP/PS1 mouse brain, Aβ plaque depositions in the APP/PS1 mouse brain were determined using an anti‐Aβ antibody. Quantification indicated that the number and area of Aβ plaques were increased to 129.66 ± 16.38% of control and 127.57 ± 14.55% of control, respectively, in the cortex (P < 0.01; Figure 6A,B), and were increased to 137.54 ± 18.63% of control and 189.88 ± 21.36% of control in the hippocampus of hypoxic transgenic mouse brain (P < 0.01; Figure 6A,B). The expression and distribution of tau phosphorylation and Aβ deposition were determined using immunofluorescence labeling. As shown in Figure 7, confocal laser scanning microscopy of p‐tau and Aβ indicated that p‐tau‐Thr205, p‐tau‐Ser‐396, and p‐tau‐Ser404 were extensively distributed surrounding the Aβ plaques and significantly increased after hypoxia treatment, which were in agreement with the Western blot results.
Figure 6.
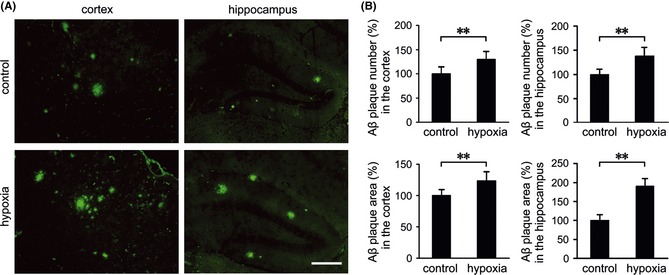
The effects of hypoxia on Aβ plaque deposition in the APP/PS1 mouse brain. (A) Brain sections of APP/PS1 mice from the control and hypoxia groups were labeled with anti‐Aβ to detect Aβ plaques by immunofluoresence. (B) Quantification shows that more Aβ plaques were stained in the cortex and hippocampus of the hypoxic mouse brain. Scale Bar = 100 μm. Values are mean ± SEM (n = 6); **P < 0.01 versus control group (Student's t‐test).
Figure 7.
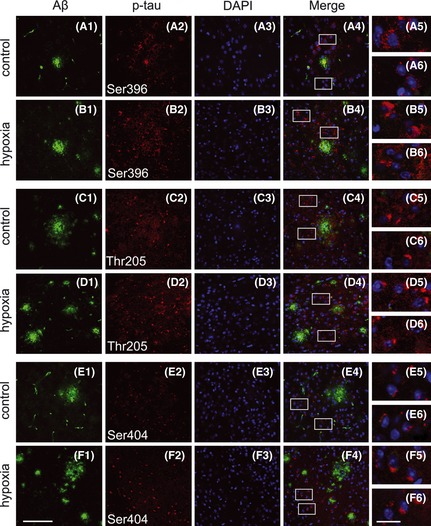
Confocal laser scanning microscopy analysis of Aβ plaques and p‐tau in the APP/PS1 mouse brain. Sections were costained with anti‐Aβ (A1–F1) and antiphosphorylated tau (A2–F2) antibodies to label Aβ plaques and p‐tau, respectively. The nuclei were stained with DAPI (A3–F3). Positive expression of p‐tau‐Ser396 (A4 and B4), p‐tau‐Thr205 (C4 and D4), and p‐tau‐Ser404 (E4 and F4) was extensively distributed surrounding the Aβ plaques in the cortex. Insets show a higher‐magnification view of p‐tau (A5, A6 to F5 and F6). Scale Bar = 50 μm (F1) and 10 μm (F6).
Silencing of m‐Calpain Protects APPswe Cells from Hypoxia‐Mediated ER Stress‐Induced Apoptosis
We next determined whether silencing of calpain could reduce the levels of μ‐calpain and m‐calpain isoforms. APPswe cells were transfected with μ‐calpain siRNA, m‐calpain siRNA, and scrambled siRNA for 24, 48, 72, and 96 h, respectively. Western blot analysis showed that the levels of both calpain isoforms were normal at 24 h and were significantly reduced at 48 and 72 h after μ‐calpain and m‐calpain siRNA treatment, respectively (P < 0.01). After 72 h, the expression levels of calpain were restored to a level similar to the control (Figure 8A). The effects of μ‐calpain and m‐calpain siRNA‐mediated knockdown on ER stress‐induced apoptosis triggered by hypoxia in APPswe cells were then investigated. APPswe cells were treated with calpain siRNA for 48 h and then treated with 1 mM NiCl2 for 4 h to induce chemical hypoxia. Cell lysates were then examined for the expression of ER stress–response proteins Grp78/BiP, CHOP/GADD153, and XBP‐1 by Western blot. We found that hypoxia treatment increased the protein expression of XBP‐1, Grp78/BiP, and CHOP/GADD153 to 170.20 ± 12.07% of control, 130.37 ± 13.90% of control, and 187.23 ± 9.53% of control, respectively (P < 0.01; Figure 8B,C). RNAi silencing of m‐calpain significantly reduced the levels of Grp78/BiP, CHOP/GADD153, and XBP‐1 to 65.99 ± 6.02%, 80.34 ± 8.82%, and 65.81 ± 9.54%, respectively, compared with the hypoxic group without siRNA treatment (P < 0.01; Figure 8B,C). No change in the levels of these proteins was observed in cells treated with μ‐calpain siRNA (Figure 8B,C). It has been previously shown that the ER stress‐mediated apoptotic signaling pathway, which may contribute to AD pathogenesis, is involved in the activation of the ER‐resident protease caspase‐12 54. We found that hypoxia‐induced upregulation of caspase‐12 was suppressed by m‐calpain siRNA (P < 0.01; Figure 8B,D), suggesting that the silencing of m‐calpain inhibits hypoxia‐induced caspase‐12 activation. We then assessed apoptosis in these cells by analyzing caspase‐3 activation and annexin V/PI staining. As shown in Figure 8B,D, hypoxia treatment led to an increase in the amount of cleaved caspase‐3 compared with the control group (P < 0.01; Figure 8B,D). Pretreatment with m‐calpain siRNA partially suppressed the effects. The level of caspase‐3 cleavage was reduced compared with the hypoxic group without m‐calpain siRNA pretreatment (P < 0.01; Figure 8B, D). Furthermore, flow cytometric analysis indicated that the apoptotic rate increased after hypoxia treatment compared with the control (P < 0.01; Figure 8E, e2 vs. e1), whereas pretreatment with m‐calpain RNAi significantly reduced the percentage of apoptotic cells (P < 0.01; Figure 8E, e3 vs. e2).
Figure 8.

Pretreatment with m‐calpain siRNA increases resistance to hypoxia‐triggered ER stress‐induced apoptosis in APPswe cells (A) Overexpression of the Swedish mutation APP (APPswe) in human neuroblastoma SH‐SY5Y cells followed by transfection with μ‐calpain siRNA (μ‐cal RNAi) and m‐calpain siRNA (m‐cal RNAi) at 24, 48, 72, and 96 h, respectively. Scrambled siRNA treatment was used as a negative control. Western blot analysis indicates that the expression of calpain is significantly suppressed after transfection with μ‐cal RNAi or m‐cal RNAi for 48 and 72 h. GAPDH was used as an internal control. (B) APPswe cells were treated with 1 mM NiCl2 for 4 h with or without pretreatment with calpain siRNA for 48 h. The ER stress–response proteins Grp78/BiP, CHOP/GADD153, and XBP‐1 were detected. The Western blot shows the levels of caspase‐12, cleaved caspase‐12, and cleaved caspase‐3. (C) The expression levels of hypoxia‐induced Grp78/BiP, CHOP/GADD153, and XBP‐1 are significantly reduced by RNAi‐mediated knockdown of m‐calpain. μ‐cal RNAi had no affect on these proteins. (D) The m‐cal RNAi pretreatment suppresses the hypoxia‐triggered increase in caspase‐12 and cleaved caspase‐12. Hypoxia treatment causes an increase in cleaved caspase‐3. The siRNA‐mediated knockdown of m‐calpain suppresses the effects induced by hypoxia. (E) Annexin V/PI staining was performed to measure apoptotic cells by flow cytometry. The lower‐right quadrant represents apoptotic cells. The data indicate that hypoxia significantly increases the apoptosis of APPswe cells (e2) compared with the controls (e1). The siRNA‐mediated knockdown of m‐calpain prior to hypoxic exposure markedly reduces the percentage of apoptotic cells (e3) induced by hypoxia compared with hypoxic cells without m‐cal RNAi. All results are presented as the mean ± SEM of at least three independent experiments; *P < 0.05, **P < 0.01 versus control group; ## P < 0.01 versus hypoxia group without siRNA pretreatment (one‐way ANOVA).
Silencing of m‐Calpain Suppresses Hypoxia‐Induced GSK‐3 Activation in APPswe Cells
A previous study indicated that ER stress agents can induce apoptosis in SH‐SY5Y cells by activating GSK3β 38. To test whether siRNA‐mediated knockdown of m‐calpain can increase the resistance of cells to ER stress‐related apoptosis induced by hypoxia, the GSK‐3 activity was measured in APPswe cells. The expression levels of p‐GSK3α/β and total GSK3α/β were measured (Figure 9A). Our findings indicated that the hypoxia‐induced reduction in p‐GSK3α/β was suppressed by m‐calpain siRNA treatment (P < 0.01; Figure 9B,C).
Figure 9.
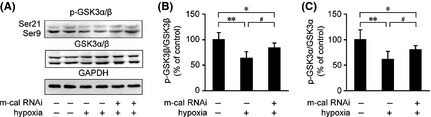
Silencing of m‐calpain in APPswe cells inhibits the hypoxia‐induced activation of GSK‐3. (A) A Western blot was performed to detect the levels of p‐GSK3α/β and GSK3α/β in APPswe cells under hypoxia treatment with or without m‐calpain siRNA pretreatment. GAPDH was used as the endogenous control. (B) The ratio of p‐GSK3β/GSK3β was significantly downregulated under hypoxia treatment. Silencing of m‐calpain increased the ratio of p‐GSK3β/GSK3β compared with the hypoxic group without m‐calpain siRNA pretreatment. (C) Hypoxia reduces the ratio of p‐GSK3α/GSK3α, whereas m‐calpain siRNA suppresses the reduction of p‐GSK3α/GSK3α in APPswe cells. All results are presented as the mean ± SEM of at least three independent experiments; *P < 0.05, **P < 0.01 versus control group; # P < 0.05 versus hypoxia group without m‐calpain siRNA pretreatment (one‐way ANOVA).
Silencing of m‐Calpain Reduces Hypoxia‐Induced Tau Phosphorylation and Aβ Secretion
To examine the effect of m‐calpain siRNA on the phosphorylation of tau under hypoxic conditions in vitro, we assessed the total tau expression levels and Ser396, Thr205, Ser404, and Thr231 phosphorylation by Western blot. As shown in Figure 10A,B, the expression levels of total tau were not altered. The hypoxia‐induced increase in tau phosphorylation at these sites in APPswe cells was suppressed by m‐calpain siRNA pretreatment (Figure 10A,B). In addition, m‐calpain siRNA treatment partially suppressed the increase in Aβ secretion induced by hypoxia (P < 0.01; Figure 10A,C).
Figure 10.
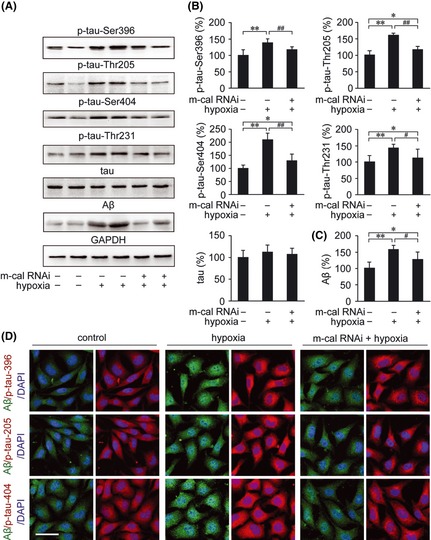
The effects of m‐calpain siRNA on Aβ generation and phosphorylation of tau in APPswe cells under hypoxia treatment. (A) The concentrations of conditioned medium and the cell lysates of APPswe cells were analyzed for the expression of Aβ and tau phosphorylation, respectively. (B) The Western blot shows that m‐calpain siRNA suppresses the hypoxia‐mediated increase in tau phosphorylation at Ser396, Thr205, Ser404, and Thr231. The Aβ secretion was also significantly increased in the medium of APPswe cells under hypoxia treatment. (C) Pretreatment with m‐calpain siRNA reduces the Aβ generation compared with hypoxic cells that were not treated with m‐calpain siRNA. (D) Immunofluorescence labeling using anti‐Aβ (green) and anti‐phosphorylated tau (red) antibodies show the distribution and expression of Aβ and p‐tau in APPswe cells. DAPI was used to detect the nucleus (blue). Scale bar = 30 μm. All results are presented as the mean ± SEM of at least three independent experiments; *P < 0.05, **P < 0.01 versus control group; # P < 0.05, ## P < 0.01 versus hypoxia group without m‐calpain siRNA pretreatment (one‐way ANOVA).
We next analyzed the distribution and expression of Aβ and phosphorylated tau in APPswe cells by immunofluorescence. Consistent with the Western blot results, hypoxia significantly increased the intensity of p‐tau‐Ser396, p‐tau‐Thr205, and p‐Ser404 as well as the Aβ in APPswe cells compared with the control group. Moreover, pretreatment with m‐calpain siRNA markedly suppressed these responses (Figure 10D).
Discussion
It has been previously reported that hypoxia can increase Aβ generation and neuritic plaque formation 47. Prenatal hypoxia aggravates the cognitive impairment and AD neuropathology in APPSwe/PS1A246E transgenic mice 57. In addition to alterations in Aβ, which is an AD hallmark, hypoxia facilitated chronic inflammatory processes in the APP/PS1 experimental model of AD 58. Most importantly, the ER stress response is also involved in AD pathogenesis 20. The intracellular calcium levels as well as the calcium released from the ER provide important contributions in the control of cell death and have a key role in ER–mitochondria interactions 59. Calpain, which is a Ca2+‐dependent protease, has also been implicated in the early stages of AD 24, 25. Calpain activation plays an important role in APP processing and plaque formation in APP/PS1 mice 40. Calpain system in neurons is more responsive to calpain inhibition under conditions of Aβ pathology 41. We postulated that calpain may play an important role in the hypoxia‐induced alterations of AD pathogenesis. In this study assessing hypoxia treatment in APP/PS1 transgenic mice, we observed that hypoxia impaired spatial learning and memory and increased hippocampal neuronal apoptosis. While, hypoxic treatment did not cause significant alterations on the spatial learning and memory, as well as the neuronal apoptosis in the hippocampus of age‐matched WT mice. It is proposed that chronic hypoxia‐induced significant impairment in spatial learning and memory might be related the hippocampal neuronal apoptosis in the AD mouse but not WT mouse. We confirmed the effects of hypoxia on specific markers of ER stress and found that hypoxia‐induced ER stress‐related apoptosis was partially suppressed by m‐calpain siRNA in APPswe cells.
Repeated Exposure to Hypoxic Conditions Induces Prolonged ER Stress and Activates the Terminal UPR, Leading to Apoptosis in the APP/PS1 Mouse Brain
The ER is an important perinuclear organelle that connects the nuclear envelope to the Golgi complex. The ER performs multiple vital functions involved in regulating cellular calcium homeostasis, folding and trafficking of secretory proteins, and modulating cellular survival by complex signal transduction. The ER is highly sensitive to alterations in cellular homeostasis. Disturbances in the balance between protein load and folding capacity of the ER trigger the UPR, which is a self‐protective signaling pathway 14, 60. Three classes of sensors, PERK, IRE1, and ATF6, recognize unfolded proteins. The ER chaperone BiP binds to unfolded proteins to support correct protein folding. Activation of the UPR protects cells from misfolded protein aggregation. Recently, Lee et al. 61 found that activation of the PERK/eIF2α pathway markedly increased the level of Grp78/BiP and attenuated apoptosis in Aβ‐treated neurons. On the other hand, prolonged activation of the UPR induces cell death and may therefore be related to the pathogenesis of protein folding diseases, such as AD. The protein level of Grp78/BiP was found to be increased in the temporal cortex and hippocampus of AD postmortem brain 20. Moreover, the ER stress sensor PERK and its target, eIF2α, are activated 21. PS1 mutations have been shown to increase translational regulation of CHOP/GADD153, which subsequently sensitizes cells to the negative effects of ER stress 62. Chronic hypoxia appears to be a key factor in the neurodegenerative damage involved in prolonged ER stress. In this study, UPR activation was accompanied by ER stress‐mediated elevation of the pro‐apoptotic transcription factors CHOP/GADD153, Grp78/BiP, and XBP1 in the APP/PS1 mouse brain after repeated hypoxia exposure. Furthermore, chronic hypoxia significantly increased the levels of caspase‐12 protein and mRNA in the APP/PS1 mouse brain. Cleaved forms of caspase‐3 were also significantly increased, suggesting caspase‐3 activation under hypoxia treatment. The results indicate that apoptosis caused by chronic exposure to hypoxic conditions in the APP/PS1 mouse brain may be mediated by prolonged ER stress, which could activate the terminal UPR.
Calpain Activation Enhances GSK‐3 Activity and Increases Aβ Generation and Tau Phosphorylation
A number of neurological insults activate calpain, which causes synaptic dysfunction and neuronal degeneration 26. It has been suggested that calpain inhibition can prevent neuronal degeneration induced by multiple neurotoxic factors 63. GSK‐3 is a calpain substrate and can be cleaved by the enzyme. Calpain‐mediated proteolysis of recombinant GSK3β significantly increases GSK‐3 activity 37. Moreover, GSK3β has recently been shown to be an important intermediate factor in apoptotic signaling pathways that lead to caspase‐3 activation 36. In vitro evidence has indicated that tau is a GSK3β substrate 64, 65. In the brains of patients with AD and APP transgenic mice 65, 66, GSK3β activation is predominantly associated with tau phosphorylation and neurofibrillary tangle formation 67, 68. Several reports have demonstrated that Cdk5, one of the enzymes responsible for phosphorylation of tau, is hyperactivated by the calpain‐mediated cleavage of p35 to p25 69, 70, 71. Meanwhile, p25‐induced Cdk5 activation can indirectly mediate abnormal tau phosphorylation via deregulation of GSK‐3 72. Calpastatin, a calpain inhibitor protein, regulates the cleavage of the Cdk5 activator p35 to p25 73. In the present study, the increase in calpain‐dependent cleavage of the 17 kDa tau fragment suggests that calpain activity is elevated in the hypoxic APP/PS1 mouse brain. We also found that hypoxia treatment induced GSK3β activation and increased tau hyperphosphorylation at Ser396, Thr205, Ser404, and Thr231 in vivo in the APP/PS1 transgenic mouse brain. Previous studies have shown that increased GSK3α activity is mainly involved in APP processing and Aβ generation 74. In this study, the increase in Aβ deposition may be involved in the activation of GSK3α, and the downregulation of p‐GSK3α (Ser21) confirmed these results.
m‐Calpain is Involved in the Regulation of Hypoxia‐Triggered ER Stress in AD
In the present study, siRNA‐mediated knockdown of m‐calpain blocked the induction of the ER stress molecules CHOP/GADD153 and Grp78/BiP after exposure to hypoxic conditions in APPswe cells. Hypoxia‐induced XBP‐1 upregulation was also altered by siRNA‐mediated knockdown of m‐calpain. These results suggest that m‐calpain contributes to the observed ER stress under hypoxic conditions. Whereas siRNA‐mediated knockdown of μ‐calpain did not significantly alter hypoxia‐induced ER stress in APPswe cells, m‐calpain siRNA inhibited GSK3β and reduced tau phosphorylation at Ser396, Thr205, Ser404, and Thr231 in APPswe cells. These results indicate that the alteration of GSK3β activity regulated by ER stress might be mediated by m‐calpain. Furthermore, hypoxia increased Aβ generation in APPswe cells. Increased Aβ production can sensitize cells to the toxicity generated by ER stress 75. In our study, m‐calpain siRNA pretreatment reduced Aβ generation and suppressed apoptosis‐related signaling in hypoxic APPswe cells. We hypothesize that the siRNA‐mediated knockdown of m‐calpain may protect cells from ER stress‐induced dysfunction and may occur as a result of GSK‐3 inhibition, reduction in tau hyperphosphorylation, and suppression of caspase cleavage.
The results presented in this study have identified a pathway linking calpain and GSK‐3 to ER stress. In the present study, we showed that hypoxia caused calpain activation, led ER stress, and enhanced AD pathology in vivo in APP/PS1 transgenic mouse. Meanwhile, m‐calpain silencing rescued ER stress and APP pathology in SH‐SY5Y cells over expressing human Swedish mutation APP in vitro. These results may help to determine the molecular events of hypoxia‐induced alterations in AD, suggesting that the activation of m‐calpain under hypoxic conditions might be related to the regulation of the UPR signaling pathway in the AD brain. Furthermore, calpain activation may regulate GSK‐3 activity and affect tau hyperphosphorylation in the hypoxia‐treated APP/PS1 mouse brain.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by the China Postdoctoral Science Foundation (20100471481, 2012M510849), the Natural Science Foundation of China (81100808, 81071004, 81100810, 81200972), the Specialized Research Fund for the Doctoral Program of Higher Education of China (20112104120010).
The first two authors equally contributed to this work.
References
- 1. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science 2002;297:353–356. [DOI] [PubMed] [Google Scholar]
- 2. Trojanowski JQ, Lee VM. Phosphorylation of paired helical filament tau in Alzheimer's disease neurofibrillary lesions: Focusing on phosphatases. FASEB J 1995;9:1570–1576. [DOI] [PubMed] [Google Scholar]
- 3. Iqbal K, Liu F, Gong CX, Grundke‐Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res 2010;7:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Khachaturian ZS. Calcium, membranes, aging, and Alzheimer's disease. Introduction and overview. Ann N Y Acad Sci 1989;568:1–4. [DOI] [PubMed] [Google Scholar]
- 5. LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci 2002;3:862–872. [DOI] [PubMed] [Google Scholar]
- 6. Bazan NG, Palacios‐Pelaez R, Lukiw WJ. Hypoxia signaling to genes: Significance in Alzheimer's disease. Mol Neurobiol 2002;26:283–298. [DOI] [PubMed] [Google Scholar]
- 7. Skoog I, Gustafson D. Update on hypertension and Alzheimer's disease. Neurol Res 2006;28:605–611. [DOI] [PubMed] [Google Scholar]
- 8. Borenstein AR, Wu Y, Mortimer JA, et al. Developmental and vascular risk factors for Alzheimer's disease. Neurobiol Aging 2005;26:325–334. [DOI] [PubMed] [Google Scholar]
- 9. Szegezdi E, Fitzgerald U, Samali A. Caspase‐12 and ER‐stress‐mediated apoptosis: The story so far. Ann N Y Acad Sci 2003;1010:186–194. [DOI] [PubMed] [Google Scholar]
- 10. Wang KK. Calpain and caspase: Can you tell the difference? by kevin K.W. WangVol. 23, pp. 20–26. Trends Neurosci 2000;23:59. [DOI] [PubMed] [Google Scholar]
- 11. Sun X, He G, Qing H, et al. Hypoxia facilitates Alzheimer's disease pathogenesis by up‐regulating BACE1 gene expression. Proc Natl Acad Sci U S A 2006;103:18727–18732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER‐associated degradation. Cell 2000;101:249–258. [DOI] [PubMed] [Google Scholar]
- 13. Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000;101:451–454. [DOI] [PubMed] [Google Scholar]
- 14. Rutkowski DT, Kaufman RJ. A trip to the ER: Coping with stress. Trends Cell Biol 2004;14:20–28. [DOI] [PubMed] [Google Scholar]
- 15. Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 2003;22:8608–8618. [DOI] [PubMed] [Google Scholar]
- 16. Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol 2006;18:444–452. [DOI] [PubMed] [Google Scholar]
- 17. Fribley A, Zhang K, Kaufman RJ. Regulation of apoptosis by the unfolded protein response. Methods Mol Biol 2009;559:191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: A cell biologist thinks about neurodegenerative diseases. Neuron 2001;29:15–32. [DOI] [PubMed] [Google Scholar]
- 19. Zhang K, Kaufman RJ. The unfolded protein response: A stress signaling pathway critical for health and disease. Neurology 2006;66:S102–S109. [DOI] [PubMed] [Google Scholar]
- 20. Hoozemans JJ, Veerhuis R, Van Haastert ES, et al. The unfolded protein response is activated in Alzheimer's disease. Acta Neuropathol 2005;110:165–172. [DOI] [PubMed] [Google Scholar]
- 21. Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer's disease hippocampus. Am J Pathol 2009;174:1241–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takahashi K, Niidome T, Akaike A, Kihara T, Sugimoto H. Amyloid precursor protein promotes endoplasmic reticulum stress‐induced cell death via C/EBP homologous protein‐mediated pathway. J Neurochem 2009;109:1324–1337. [DOI] [PubMed] [Google Scholar]
- 23. Unterberger U, Hoftberger R, Gelpi E, Flicker H, Budka H, Voigtlander T. Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. J Neuropathol Exp Neurol 2006;65:348–357. [DOI] [PubMed] [Google Scholar]
- 24. Nixon RA. A “protease activation cascade” in the pathogenesis of Alzheimer's disease. Ann N Y Acad Sci 2000;924:117–131. [PubMed] [Google Scholar]
- 25. Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 2000;405:360–364. [DOI] [PubMed] [Google Scholar]
- 26. Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev 2003;83:731–801. [DOI] [PubMed] [Google Scholar]
- 27. Wood DE, Thomas A, Devi LA, et al. Bax cleavage is mediated by calpain during drug‐induced apoptosis. Oncogene 1998;17:1069–1078. [DOI] [PubMed] [Google Scholar]
- 28. Benetti R, Del Sal G, Monte M, Paroni G, Brancolini C, Schneider C. The death substrate Gas2 binds m‐calpain and increases susceptibility to p53‐dependent apoptosis. EMBO J 2001;20:2702–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Levesque S, Wilson B, Gregoria V, et al. Reactive microgliosis: Extracellular micro‐calpain and microglia‐mediated dopaminergic neurotoxicity. Brain 2010;133:808–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakagawa T, Yuan J. Cross‐talk between two cysteine protease families. Activation of caspase‐12 by calpain in apoptosis. J Cell Biol 2000;150:887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Siman R, Flood DG, Thinakaran G, Neumar RW. Endoplasmic reticulum stress‐induced cysteine protease activation in cortical neurons: Effect of an Alzheimer's disease‐linked presenilin‐1 knock‐in mutation. J Biol Chem 2001;276:44736–44743. [DOI] [PubMed] [Google Scholar]
- 32. Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: The calcium‐apoptosis link. Nat Rev Mol Cell Biol 2003;4:552–565. [DOI] [PubMed] [Google Scholar]
- 33. Muruganandan S, Cribb AE. Calpain‐induced endoplasmic reticulum stress and cell death following cytotoxic damage to renal cells. Toxicol Sci 2006;94:118–128. [DOI] [PubMed] [Google Scholar]
- 34. Shiurba RA, Ishiguro K, Takahashi M, et al. Immunocytochemistry of tau phosphoserine 413 and tau protein kinase I in Alzheimer pathology. Brain Res 1996;737:119–132. [DOI] [PubMed] [Google Scholar]
- 35. Giese KP. GSK‐3: A key player in neurodegeneration and memory. IUBMB Life 2009;61:516–521. [DOI] [PubMed] [Google Scholar]
- 36. Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol 2001;65:391–426. [DOI] [PubMed] [Google Scholar]
- 37. Goni‐Oliver P, Lucas JJ, Avila J, Hernandez F. N‐terminal cleavage of GSK‐3 by calpain: A new form of GSK‐3 regulation. J Biol Chem 2007;282:22406–22413. [DOI] [PubMed] [Google Scholar]
- 38. Song L, De Sarno P, Jope RS. Central role of glycogen synthase kinase‐3beta in endoplasmic reticulum stress‐induced caspase‐3 activation. J Biol Chem 2002;277:44701–44708. [DOI] [PubMed] [Google Scholar]
- 39. Baltzis D, Pluquet O, Papadakis AI, Kazemi S, Qu LK, Koromilas AE. The eIF2alpha kinases PERK and PKR activate glycogen synthase kinase 3 to promote the proteasomal degradation of p53. J Biol Chem 2007;282:31675–31687. [DOI] [PubMed] [Google Scholar]
- 40. Liang B, Duan BY, Zhou XP, Gong JX, Luo ZG. Calpain activation promotes BACE1 expression, amyloid precursor protein processing, and amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Biol Chem 2010;285:27737–27744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morales‐Corraliza J, Berger JD, Mazzella MJ, et al. Calpastatin modulates APP processing in the brains of beta‐amyloid depositing but not wild‐type mice. Neurobiol Aging 2012; 33: 1125 e1129‐1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim MJ, Oh SJ, Park SH, et al. Hypoxia‐induced cell death of HepG2 cells involves a necrotic cell death mediated by calpain. Apoptosis 2007;12:707–718. [DOI] [PubMed] [Google Scholar]
- 43. Hoang MV, Smith LE, Senger DR. Calpain inhibitors reduce retinal hypoxia in ischemic retinopathy by improving neovascular architecture and functional perfusion. Biochim Biophys Acta 2011;1812:549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shao G, Gao CY, Lu GW. Alterations of hypoxia‐inducible factor‐1 alpha in the hippocampus of mice acutely and repeatedly exposed to hypoxia. Neurosignals 2005;14:255–261. [DOI] [PubMed] [Google Scholar]
- 45. Qing H, He G, Ly PT, et al. Valproic acid inhibits Abeta production, neuritic plaque formation, and behavioral deficits in Alzheimer's disease mouse models. J Exp Med 2008;205:2781–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang CY, Zheng W, Wang T, et al. Huperzine A activates Wnt/beta‐catenin signaling and enhances the nonamyloidogenic pathway in an Alzheimer transgenic mouse model. Neuropsychopharmacology 2011;36:1073–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li L, Zhang X, Yang D, Luo G, Chen S, Le W. Hypoxia increases Abeta generation by altering beta‐ and gamma‐cleavage of APP. Neurobiol Aging 2009;30:1091–1098. [DOI] [PubMed] [Google Scholar]
- 48. Wang CY, Wang T, Zheng W, et al. Zinc overload enhances APP cleavage and Abeta deposition in the Alzheimer mouse brain. PLoS ONE 2010;5:e15349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boland B, Campbell V. beta‐Amyloid (1‐40)‐induced apoptosis of cultured cortical neurones involves calpain‐mediated cleavage of poly‐ADP‐ribose polymerase. Neurobiol Aging 2003;24:179–186. [DOI] [PubMed] [Google Scholar]
- 50. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007;8:519–529. [DOI] [PubMed] [Google Scholar]
- 51. Paz Gavilan M, Vela J, Castano A, et al. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol Aging 2006; 27: 973–982. [DOI] [PubMed] [Google Scholar]
- 52. Spear E, Ng DT. The unfolded protein response: No longer just a special teams player. Traffic 2001;2:515–523. [DOI] [PubMed] [Google Scholar]
- 53. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 2004;11:381–389. [DOI] [PubMed] [Google Scholar]
- 54. Nakagawa T, Zhu H, Morishima N, et al. Caspase‐12 mediates endoplasmic‐reticulum‐specific apoptosis and cytotoxicity by amyloid‐beta. Nature 2000;403:98–103. [DOI] [PubMed] [Google Scholar]
- 55. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase‐3 by insulin mediated by protein kinase B. Nature 1995;378:785–789. [DOI] [PubMed] [Google Scholar]
- 56. Blennow K, Hampel H. CSF markers for incipient Alzheimer's disease. Lancet Neurol 2003;2:605–613. [DOI] [PubMed] [Google Scholar]
- 57. Zhang X, Li L, Xie W, et al. Prenatal hypoxia may aggravate the cognitive impairment and Alzheimer's disease neuropathology in APPSwe/PS1A246E transgenic mice. Neurobiol Aging 2013;34:663–678. [DOI] [PubMed] [Google Scholar]
- 58. de Lemos ML, de la Torre AV, Petrov D, Brox S, Folch J, et al. Evaluation of hypoxia inducible factor expression in inflammatory and neurodegenerative brain models. Int J Biochem Cell Biol 2013;45:1377–1388. [DOI] [PubMed] [Google Scholar]
- 59. Verkhratsky A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev 2005;85:201–279. [DOI] [PubMed] [Google Scholar]
- 60. Forman MS, Lee VM, Trojanowski JQ. ‘Unfolding’ pathways in neurodegenerative disease. Trends Neurosci 2003;26:407–410. [DOI] [PubMed] [Google Scholar]
- 61. Lee do Y, Lee KS, Lee HJ, et al. Activation of PERK signaling attenuates Abeta‐mediated ER stress. PLoS ONE 2010; 5: e10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Milhavet O, Martindale JL, Camandola S, et al. Involvement of Gadd153 in the pathogenic action of presenilin‐1 mutations. J Neurochem 2002;83:673–681. [DOI] [PubMed] [Google Scholar]
- 63. Rao MV, Mohan PS, Peterhoff CM, et al. Marked calpastatin (CAST) depletion in Alzheimer's disease accelerates cytoskeleton disruption and neurodegeneration: Neuroprotection by CAST overexpression. J Neurosci 2008;28:12241–12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Johnson GV, Hartigan JA. Tau protein in normal and Alzheimer's disease brain: An update. J Alzheimers Dis 1999;1:329–351. [DOI] [PubMed] [Google Scholar]
- 65. Lucas JJ, Hernandez F, Gomez‐Ramos P, Moran MA, Hen R, Avila J. Decreased nuclear beta‐catenin, tau hyperphosphorylation and neurodegeneration in GSK‐3beta conditional transgenic mice. EMBO J 2011;20:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ishizawa T, Sahara N, Ishiguro K, et al. Co‐localization of glycogen synthase kinase‐3 with neurofibrillary tangles and granulovacuolar degeneration in transgenic mice. Am J Pathol 2003;163:1057–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Baum L, Hansen L, Masliah E, Saitoh T. Glycogen synthase kinase 3 alteration in Alzheimer disease is related to neurofibrillary tangle formation. Mol Chem Neuropathol 1996;29:253–261. [DOI] [PubMed] [Google Scholar]
- 68. Ma QL, Lim GP, Harris‐White ME, et al. Antibodies against beta‐amyloid reduce Abeta oligomers, glycogen synthase kinase‐3beta activation and tau phosphorylation in vivo and in vitro. J Neurosci Res 2006;83:374–384. [DOI] [PubMed] [Google Scholar]
- 69. Gao L, Tian S, Gao H, Xu Y. Hypoxia increases abeta‐induced tau phosphorylation by calpain and promotes behavioral consequences in AD transgenic mice. J Mol Neurosci 2013;51:138–147. [DOI] [PubMed] [Google Scholar]
- 70. Rosales JL, Nodwell MJ, Johnston RN, Lee KY. Cdk5/p25(nck5a) interaction with synaptic proteins in bovine brain. J Cell Biochem 2000;78:151–159. [DOI] [PubMed] [Google Scholar]
- 71. Cruz JC, Tsai LH. Cdk5 deregulation in the pathogenesis of Alzheimer's disease. Trends Mol Med 2004;10:452–458. [DOI] [PubMed] [Google Scholar]
- 72. Plattner F, Angelo M, Giese KP. The roles of cyclin‐dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J Biol Chem 2006;281:25457–25465. [DOI] [PubMed] [Google Scholar]
- 73. Sato K, Minegishi S, Takano J, et al. Calpastatin, an endogenous calpain‐inhibitor protein, regulates the cleavage of the Cdk5 activator p35 to p25. J Neurochem 2011;117:504–515. [DOI] [PubMed] [Google Scholar]
- 74. Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK‐3alpha regulates production of Alzheimer's disease amyloid‐beta peptides. Nature 2003;423:435–439. [DOI] [PubMed] [Google Scholar]
- 75. Chafekar SM, Zwart R, Veerhuis R, Vanderstichele H, Baas F, Scheper W. Increased Abeta1‐42 production sensitizes neuroblastoma cells for ER stress toxicity. Curr Alzheimer Res 2008;5:469–474. [DOI] [PubMed] [Google Scholar]