

Summary
Background
Cerebrospinal fluid (CSF) biomarkers reflect changes in the brain, and contribute to early screening. Maternal inheritance is putatively stronger than paternal inheritance for late‐onset Alzheimer's disease (LOAD).
Methods
Clinical data of 162 cognitively normal subjects were reviewed. A standard questionnaire was used to identify LOAD family history. Mini‐mental state examination (MMSE) was used to evaluate cognition. CSF Aβ1‐40, Aβ1‐42, total and phosphorylated tau were measured using ELISA.
Aims
To compare biomarkers in cognitively normal elderly subjects with versus without LOAD family history.
Results
Among the 162 subjects, 38 and 60 had LOAD family history on paternal and maternal sides, respectively. The remaining 60 subjects had no family history. No difference was noted in age, gender, education level, MMSE score, and memory impairment complaint in the three groups. Aβ42 and the Aβ42/40 ratio were lower than in subjects with a maternal history than in subjects with a paternal history or without family history (P < 0.05 in both). Phosphorylated and total tau did not differ among the three groups.
Conclusion
Offspring with a family history of LOAD on the maternal side have lower Aβ42 and Aβ42/40 ratio in the CSF, and maybe at higher risk for developing AD.
Keywords: Biomarker, Cerebrospinal fluid, Family history, Late‐onset Alzheimer's disease, β‐Amyloid protein
Introduction
Alzheimer's disease (AD) is the most common type of senile dementia, and its incidence has been increasing over the years with the aging of the population 1. It has been noted that subclinical AD pathological changes such as amyloid plaque formation, gliosis, and neurofibrillary tangle formation start 10–15 years before the onset of dementia 2. Early detection of the pathological changes in subclinical AD could provide an opportunity to intervene at a stage before cognitive impairment develops. Currently, clinical examination cannot detect subclinical AD. There are also no reliable biomarkers that reveal early cellular and molecular changes of the brain.
Numerous studies have shown that the cerebrospinal fluid (CSF) represents an appealing source of biomarkers of AD pathological changes 3, 4, 5. The CSF is in direct contact with the brain and reflects biochemical changes in the brain. Currently, CSF biomarkers for AD primarily include β‐amyloid proteins (Aβ) and tau proteins. Total tau (T‐tau) is a neuron degeneration marker; hyperphosphorylated tau protein (P‐tau) is a marker of neurofibrillary tangles. Tau protein increased and Aβ1‐42 level decreased in the CSF of AD patients 6, 7, and these CSF markers help to discriminate AD patients from the healthy population and patients with other forms of senile dementia. These markers could also contribute to early screening in the preclinical population that has not manifested symptoms of dementia. Biomarkers most needed at this moment perhaps lie in the identification of individuals who are cognitively normal but are prone to the development of AD. Such individuals could represent the population most likely to benefit from future disease modifying therapies.
A family history of late‐onset AD (LOAD) in first‐degree relatives is a significant risk factor for AD. The genetic mechanism of this risk remains unclear 8. Findings suggested both maternal and paternal inheritance, but the proportion of maternal inheritance is higher than that of paternal inheritance. The offspring of females with LOAD are at a greater risk to develop LOAD than the offspring of males with LOAD. In this study, we measured the concentration of Aβ1‐42 and tau proteins in the CSF using enzyme‐linked immunosorbent assay (ELISA) in cognitively normal subjects without a family history of LOAD versus those with a maternal or paternal history of LOAD and investigated whether inheritance pattern influenced the risk of LOAD in their offspring.
Patients and Methods
Patients
A total of 300 cognitively normal subjects seeking medical treatment at the Department of Geriatrics of the Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China between September 2006 and August 2011 were enrolled in this study. A detailed medical history and interview with the patient and informant, neurological and physical examinations, a neuroimaging scan, neuropsychological testing, and screening laboratory studies were performed at the initial visit. Patients underwent lumbar puncture and were given an opportunity to provide CSF specimens for storage in a tissue bank for biomarker analyses. Subjects who were cognitively normal, defined as having a clinical dementia rating (CDR) of 0, were eligible for the study. A subject was excluded if he or she had concurrent or a history of diseases that could affect brain structure or function, such as stroke, diabetes, traumatic brain injury, neurodegenerative disorders, and depression, or if he or she was taking or had previously taken drugs that could affect the nervous system. A subject whose parents died before the age of 65 years was also excluded from this study.
The study protocol was approved by the local Institutional Review Board at the authors' affiliated institution. Signed informed consent was obtained from all participants or their legal surrogates. All aspects of this study were conducted according to the principles expressed in the Declaration of Helsinki.
Cognitive and Global Function Measures
The New York University Brain Aging Family History Questionnaire 9 was used to screen subjects with a family history of LOAD. A family history was defined as having at least one‐first‐degree relative with AD with an onset between 60 and 80 years of age. The subjects were asked to provide the name, age, and cause of death of all AD patients in their family within three generations and, in as much detail as possible, the clinical data of their parents, including the clinic/hospital of treatment, their physicians' name, medical records, imaging results, medication, and autopsy reports. The symptoms and disease progression of AD patients were also discussed with other family members of the subjects. Whenever possible, the diagnosis was confirmed by following up with the physician who made diagnosis.
Mini‐mental state examination (MMSE) was used to evaluate cognitive function and has contained 11 questions, with the highest possible score of 30 10. A score of 19–23 represents mild dementia, 10–18 moderate dementia, 1–9 severe dementia, and 0 extremely severe dementia 11. The Chinese version of MMSE was used for the evaluation 12. The cutoff score is 15–16 for participants with primary school education, and 23–24 for participants with secondary and higher education 12, 13. The Hachinski ischemic score 14 was obtained from the medical records of patients. In addition, all subjects were asked to complete the 15‐item geriatric depression scale (GDS‐15), and those with a total score of 7 or more were considered to have clinically significant depressive symptoms 15. The CDR scale examines functioning in six domains: memory, orientation, judgment/problem solving, community affairs, home/hobbies, and personal care and was conducted using a semi‐structured questionnaire with 0 indicating no impairment and 18 indicating maximum impairment 16.
Detection of CSF Biomarkers
In the morning after overnight fasting, CSF was obtained by lumbar puncture. CSF was centrifuged at 1500 g for 10 min, placed in polypropylene test tubes, and stored at −80°C. CSF Aβ1‐40, Aβ1‐42 and T‐tau and P‐tau concentration was measured in duplicate using well‐established double antibody sandwich ELISA after a single freeze‐thaw cycle (InnoTest, Ghent, Belgium). Absorbance (A) was read using a microplate reader (TECAN, Salzburg, Austria). Concentration was calculated using regression curves obtained with standard tau and Aβ protein in the CSF. The Aβ42/40 ratio was also calculated.
Statistical Analysis
Data are expressed as mean ± SD and analyzed using the SPSS 12.0 software (SPSS Inc., Chicago, IL, USA). The chi‐squared test (categorical variables) and t‐test (normal continuous variables) were used to compare data between the groups. Single‐factor analysis of variance was used to analyze the results of CSF biomarkers between the groups, and the least significant difference method was used for pair‐wise comparison. Linear regression analysis was used to analyze the relationship between CSF markers. P < 0.05 was considered statistically significant.
Results
Demographic and Disease Characteristics of Study Participants
Among the 300 cognitively normal subjects, there were 94 cases of upper respiratory tract infection, 82 cases of pneumonia, 37 cases of gallstone, 35 cases of peptic ulcers, 31 cases of benign prostatic hyperplasia, 15 cases of urinary tract infection, and 6 cases of bone fracture. The age of the subjects ranged between 55 and 70 years of age, with a mean of 61.9 ± 8.4 years. All subjects had received at least 9 years of education. The MMSE score was 28–30, CDR was 0, GDS score was 1 or 2, and the Hachinski ischemic score was <4. Eighty‐five patients were excluded from this study due to incomplete family history. Fifty‐three patients were excluded because both parents had AD (13 cases), or only their siblings had AD (12 cases), or their father and/or mother died before the age of 65 years (28 cases). A total of 162 subjects were included in the data analysis, including 60 (60/162, 37.0%) subjects without a family history of AD, 38 (38/162, 23.5%) subjects with a paternal history of LOAD and 64 (64/162, 39.5%) subjects with a maternal history of LOAD (Figure 1). The diagnosis of AD in the parents of seven subjects (three with a maternal history of AD and four with a paternal history of AD) was confirmed by autopsy reports. The diagnosis of AD in the parents of the remaining subjects was based on clinical appraisal. There were no significant differences in age, gender, education level, and complaints of memory impairment or MMSE score between the groups (Table 1).
Figure 1.
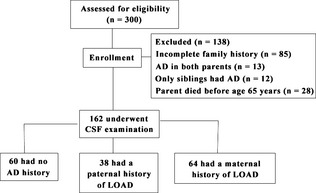
The study flow chart. AD, Alzheimer's disease; LOAD, late‐onset AD.
Table 1.
Demographic data and MMSE score of study participants (n = 162)
| NH (n = 60) | PH (n = 38) | MH (n = 64) | |
|---|---|---|---|
| Age (years) | 62.0 ± 8.1 | 59.8 ± 6.1 | 60.3 ± 5.9 |
| Gender (male/female) (n) | 28/32 | 18/20 | 31/33 |
| Education (years) | 10.5 ± 2.4 | 12.0 ± 2.8 | 11.2 ± 2.9 |
| Complaint of memory impairment (yes/no) | 6/54 | 3/35 | 7/57 |
| MMSE score | 29.2 ± 1.1 | 29.6 ± 0.8 | 29.6 ± 0.7 |
NH, subjects with a negative family history of Alzheimer's disease; PH, subjects with a paternal history of late‐onset Alzheimer's disease; MH, subjects with a maternal history of late‐onset Alzheimer's disease; MMSE, mini‐mental state examination.
Levels of CSF Biomarkers in Cognitively Normal Subjects
The Aβ40 levels were 7306 ± 2171 pg/mL for the study participants, 7318 ± 2454 pg/mL for subjects without a family history of AD, 6980 ± 1593 pg/mL for subjects with a paternal history of LOAD, and 7498 ± 2247 pg/mL for subjects with a maternal history of LOAD (Table 2). The Aβ42 levels were 753 ± 369 pg/mL for the study participants, 871 ± 518 pg/mL for subjects without a family history of AD, 790 ± 309 pg/mL for subjects with a paternal history of LOAD, and 622 ± 267 pg/mL for subjects with a maternal history of LOAD. The Aβ42/40 ratio was 0.12 ± 0.03 for subjects without a family history of AD, 0.11 ± 0.03 for subjects with a paternal history of LOAD, and 0.08 ± 0.03 for subjects with a maternal history of LOAD. The Aβ42/40 ratio was significantly lower in subjects with a maternal history of LOAD compared with those without a family history of AD or with a paternal history of LOAD (P < 0.05 for both).
Table 2.
Levels of cerebrospinal fluid biomarkers of study participants (n = 162) (pg/mL)
| NH (n = 60) | PH (n = 38) | MH (n = 64) | |
|---|---|---|---|
| Aβ40 | 7318 ± 2454 | 6980 ± 1593 | 7498 ± 2247 |
| Aβ42 | 871 ± 518 | 790 ± 309 | 622 ± 267 |
| Aβ42/40 | 0.119 ± 0.031 | 0.113 ± 0.032 | 0.083 ± 0.030* |
| P‐tau | 10.9 ± 12.8 | 9.8 ± 13.5 | 12.7 ± 20.0 |
| T‐tau | 322 ± 280 | 292 ± 124 | 315 ± 192 |
NH, subjects with a negative family history of Alzheimer's disease; PH, subjects with a paternal history of late‐onset Alzheimer's disease; MH, subjects with a maternal history of late‐onset Alzheimer's disease. *P < 0.05 versus the NH and PH group.
CSF T‐tau were 315 ± 200 pg/mL for the study participants, 322 ± 280 pg/mL for subjects without a family history of AD, 292 ± 124 pg/mL for subjects with a paternal history of LOAD, and 315 ± 192 pg/mL for subjects with a maternal history of LOAD. P‐tau levels were 11.4 ± 15.9 pg/mL for the study participants, 10.9 ± 12.8 pg/mL for subjects without a family history of AD, 9.8 ± 13.5 pg/mL for subjects with a paternal history of LOAD, and 12.7 ± 20.0 pg/mL for subjects with a maternal history of LOAD. There was no significant difference in P‐tau, T‐tau or Aβ40 levels among subjects without a family history of AD, those with a paternal history of LOAD and those with a maternal history of LOAD (P > 0.05). There was no significant difference in the levels of any of the biomarkers between subjects with a paternal history of LOAD and those with a maternal history of LOAD.
Correlation Between CSF Biomarkers in Cognitively Normal Subjects
A significant correlation was noted between P‐tau and T‐tau levels among subjects without a family history of AD, those with a paternal history of LOAD, or those with a maternal history of LOAD (NH: R 2 = 0.25, P = 0.013; PH: R 2 = 0.31, P = 0.009; MH: R 2 = 0.36, P = 0.005 for all). There was also a significant correlation between Aβ40 and Aβ42 levels and the Aβ42/40 ratio (NH: R 2 = 0.20, P = 0.037; PH: R 2 = 0.28, P = 0.022; MH: R 2 = 0.32, P = 0.007 for all subgroups). No significant correlation was observed between tau and Aβ (P > 0.05).
Discussion
In recent years, the study of AD has mainly focused on quantitative investigations of CSF Aβ1‐42 and tau protein that may contribute to the typical pathological changes that include senile plaques and neurofibrillary tangles 6, 7. van Oijen et al. 17 reported an association of elevated Ab40 but not Ab42 in the baseline with increased risk of AD in a large study with extended follow‐up. At first look, our results (lower Aβ42 but not Aβ40 in the MH‐NL group) seem inconsistent. But at a deeper, our findings are consistent with the higher susceptibility in subjects with maternal transmission, and probably also reflects the fact that our study is limited in sample size. Consistent with our report, Graff‐Radford previously reported decreasing serum Aβ42 in subjects with impaired cognitive function (12% each year) and increasing Aβ42 in the cognitively intact control population 18. The study results suggested that changes in Aβ40 and Aβ42 could vary significantly across subjects depending upon the health/disease type and over the course of AD. Finally, together with the limited sample size of our study, the results presented in this article must be considered preliminary and need further studies. Significantly lower Aβ1‐42 but higher tau level has been noted in the CSF of AD patients versus that the normal elderly population 6, 7, 19. Identification of individuals who are cognitively normal, without evidence of AD pathology, but are prone to AD, could allow development of interventions at a stage prior to the onset of cognitive impairment. Detection of Aβ1‐42 and tau changes in the CSF may contribute to early diagnosis in populations suspected of having AD or having subclinical AD. The results of the current study confirmed the role of CSF markers in AD pathology. Briefly, we found more pronounced change in Aβ levels in subjects with a maternal history of LOAD compared with those without a family history and those with a paternal history of LOAD. These findings suggest that offspring with a maternal history of LOAD are at increased risk for the development of AD, possibly due to the maternally inherited defects in oxidative metabolism and the increase in extracellular Aβ deposits 20, 21.
Oxidative stress is extensively implicated in AD pathology 22. Aβ42 acts as a neurotoxin in AD pathogenesis and induces oxidative stress, thus compromising mitochondrial function and stimulating the neurotoxic cascade 23, 24, 25. Increased Aβ42 also leads to an excess of free radicals and neuronal membrane lipid peroxides. Oxidative stress also promotes Aβ deposition. It is believed that Aβ‐related oxidative damage could be an early manifestation of AD, or at least in maternally inherited subjects 26. Such changes may occur prior to the formation of neurofibrillary tangles and neuronal degeneration. Here, we showed that Aβ levels were decreased in subjects with a maternal history of LOAD, but found no significant differences in P‐tau or T‐tau levels. The findings are generally consistent with previous results 24, 25. P‐tau and T‐tau levels were correlated within each group, suggesting a progressive neural aging process that includes neural degeneration and formation of neurofibrillary tangles.
Reduced Aβ levels suggest impending cognitive function decline and development into AD in cognitively normal elderly subjects 27. Our findings further showed that those who have a maternal history of LOAD could display AD pathologies even although they are cognitively normal. We speculate that maternally inherited defects in oxidative metabolism could be the basis of such vulnerability for AD. These results are consistent with those of positron emission tomography imaging studies, which showed subjects with a maternal history of AD are prone to hypometabolism and increased pathological changes in Aβ fibers 28. Longitudinal study is needed to verify the dynamic changes of AD biomarkers in the temporal lobe and to further clarify whether CSF abnormalities could predict future cognitive function impairment.
Reduced Aβ42 level could reflect early Aβ deposition in the brain and could lead to the formation of amyloid precursor protein and the mutation of the presenilin gene 29, 30. An increased Aβ42/Aβ40 ratio in the brain tissue and a correspondingly decreased Aβ42/Aβ40 ratio in the CSF would follow, leading to early onset AD. The results of this study showed that Aβ42 level is significantly decreased in cognitively normal subjects with a maternal history of LOAD, but not in subjects with a parental history. CSF abnormalities in cognitively normal subjects with a maternal history of LOAD may be affected by other potential risk factors, including age, gender, education level, and the presence of the ApoEε4 gene.
Accurate AD diagnosis could only be made upon post‐mortem analysis. In the current study, only subjects whose parents were diagnosed by specialists using a standard set of diagnostic criteria were included. The data from the family history questionnaire are consistent with the clinical and neuropathological results, and thus, reduced the possibility of misclassification. Clinical diagnostic data for all 162 subjects in the final analysis were complete. As the diagnosis of AD was only confirmed by autopsy reports in the parents of 7 subjects, this study may have included subjects whose parents did have other forms of dementia. This may lead to data bias, and could have decreased the differences among the groups. However, only subjects with normal cognitive function, as reflected by using the current, universal clinical assessment criteria, and neuropsychological evaluation, were included. Aβ levels in the CSF could be abnormal in subjects who had a maternal history of AD but normal cognitive function, suggesting that biochemical abnormalities occur prior to clinical symptoms. Therefore, the inclusion of biomarkers in clinical evaluations is helpful for the diagnosis. The study demonstrates that the offspring of female patients with LOAD are more vulnerable to Aβ‐related oxidative stress, further supporting the theory that genetic variation of Aβ catabolism may increase the risk of LOAD.
In summary, cognitively normal subjects with a maternal history of AD exhibit an AD‐prone biological phenotype, and may increase the risk of LOAD. However, this study was cross‐sectional, and the sample size was small. As a result,the findings need to be further confirmed by large‐scale, longitudinal clinical studies.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This study was supported by Special Research Foundation of Chinese College Doctoral Program (#J20100047).
References
- 1. Jack CR Jr. Alzheimer disease: new concepts on its neurobiology and the clinical role imaging will play. Radiology 2012;263:344–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Braak H, Braak E. Frequency of stages of Alzheimer‐related lesions in different age categories. Neurobiol Aging 1997;18:351–357. [DOI] [PubMed] [Google Scholar]
- 3. Grønning H, Rahmani A, Gyllenborg J, Dessau RB, Høgh P. Does Alzheimer's disease with early onset progress faster than with late onset? A case–control study of clinical progression and cerebrospinal fluid biomarkers. Dement Geriatr Cogn Disord 2012;33:111–117. [DOI] [PubMed] [Google Scholar]
- 4. Ringman JM, Coppola G, Elashoff D, et al. Cerebrospinal fluid biomarkers and proximity to diagnosis in preclinical familial Alzheimer's disease. Dement Geriatr Cogn Disord 2012;33:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rosén C, Andreasson U, Mattsson N, et al. Cerebrospinal fluid profiles of amyloid β‐related biomarkers in Alzheimer's disease. Neuromolecular Med 2012;14:65–73. [DOI] [PubMed] [Google Scholar]
- 6. Parnetti L, Chiasserini D, Eusebi P, et al. Performance of aβ1‐40, aβ1‐42, total tau, and phosphorylated tau as predictors of dementia in a cohort of patients with mild cognitive impairment. J Alzheimers Dis 2012;29:229–238. [DOI] [PubMed] [Google Scholar]
- 7. Fagan AM, Shaw LM, Xiong C, et al. Comparison of analytical platforms for cerebrospinal fluid measures of β‐amyloid 1‐42, total tau, and p‐tau181 for identifying Alzheimer disease amyloid plaque pathology. Arch Neurol 2011;68:1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bakulski KM, Dolinoy DC, Sartor MA, et al. Genome‐wide DNA methylation differences between late‐onset Alzheimer's disease and cognitively normal controls in human frontal cortex. J Alzheimers Dis 2012;29:571–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mosconi L, Mistur R, Glodzik L, et al. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer's. Neurology 2009;72:513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 11. Tiraboschi P, Hansen LA, Alford M, et al. The decline in synapses and cholinergic activity is asynchronous in Alzheimer's disease. Neurology 2000;55:1278–1283. [DOI] [PubMed] [Google Scholar]
- 12. Guo NW, Liu HC, Wong PF, et al. Chinese version and norms of the Mini‐Mental State Examination. J Rehabil Med Assoc 1988;16:52–59. [Google Scholar]
- 13. Guo NW, Liu HC, Wong PF, et al. Introduction of the Chinese version of the Mini‐Mental State Examination. Clin Med Res 1989;23:39–42. [Google Scholar]
- 14. Hachinski VC, Iliff LD, Zilhka E. Cerebral blood flow in dementia. Arch Neurol 1975;32:632–637. [DOI] [PubMed] [Google Scholar]
- 15. Almeida OP, Almeida SA. Short versions of the geriatric depression scale: a study of their validity for the diagnosis of a major depressive episode according to ICD‐10 and DSM‐IV . Int J Geriatr Psychiatry 1999;14:858–865. [DOI] [PubMed] [Google Scholar]
- 16. Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Dementia 1982;140:566–572. [DOI] [PubMed] [Google Scholar]
- 17. van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler MM. Plasma Abeta(1‐40) and Abeta(1‐42) and the risk of dementia: a prospective case‐cohort study. Lancet Neurol 2006;5:655–660. [DOI] [PubMed] [Google Scholar]
- 18. Graff‐Radford NR, Lucas JA, Petersen RC, Younkin LH, Younkin SG. P3‐011 Analysis of plasma Aβ42 ASA premorbid biomarker for late onset Alzheimer's disease. Neurobiol Aging 2004;25:S354. [Google Scholar]
- 19. Ganzer S, Arlt S, Schoder V, et al. CSF‐tau, CSF‐Abeta1‐42, ApoE‐genotype and clinical parameters in the diagnosis of Alzheimer's disease: combination of CSF‐tau and MMSE yields highest sensitivity and specificity. J Neural Transm 2003;110:1149–1160. [DOI] [PubMed] [Google Scholar]
- 20. Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta‐peptide. Nat Rev Mol Cell Biol 2007;8:101–112. [DOI] [PubMed] [Google Scholar]
- 21. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006;443:787–795. [DOI] [PubMed] [Google Scholar]
- 22. Castellani RJ, Moreira PI, Perry G, Zhu X. The role of iron as a mediator of oxidative stress in Alzheimer disease. BioFactors 2012;38:133–138. [DOI] [PubMed] [Google Scholar]
- 23. Butterfield DA. Amyloid beta‐peptide (1‐42)‐induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer's disease brain. A review. Free Radic Res 2002;36:1307–1313. [DOI] [PubMed] [Google Scholar]
- 24. Mohmmad Abdul H, Sultana R, Keller JN, St Clair DK, Markesbery WR, Butterfield DA. Mutations in amyloid precursor protein and presenilin‐1 genes increase the basal oxidative stress in murine neuronal cells and lead to increased sensitivity to oxidative stress mediated by amyloid beta‐peptide (1‐42), HO and kainic acid: implications for Alzheimer's disease. J Neurochem 2006;96:1322–1335. [DOI] [PubMed] [Google Scholar]
- 25. Pensalfini A, Zampagni M, Liguri G, et al. Membrane cholesterol enrichment prevents Aβ‐induced oxidative stress in Alzheimer's fibroblasts. Neurobiol Aging 2011;32:210–222. [DOI] [PubMed] [Google Scholar]
- 26. Jack CR Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010;8:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Skoog I, Davidsson P, Aevarsson O, Vanderstichele H, Vanmechelen E, Blennow K. Cerebrospinal fluid beta‐amyloid 42 is reduced before the onset of sporadic dementia: a population‐based study in 85‐year‐olds. Dement Geriatr Cogn Disord 2003;15:169–176. [DOI] [PubMed] [Google Scholar]
- 28. Yates PA, Sirisriro R, Villemagne VL, et al. Cerebral microhemorrhage and brain β‐amyloid in aging and Alzheimer disease. Neurology 2011;77:48–54. [DOI] [PubMed] [Google Scholar]
- 29. Kasuga K, Tokutake T, Ishikawa A, et al. Differential levels of alpha‐synuclein, beta‐amyloid42 and tau in CSF between patients with dementia with Lewy bodies and Alzheimer's disease. J Neurol Neurosurg Psychiatry 2010;81:608–610. [DOI] [PubMed] [Google Scholar]
- 30. Boche D, Denham N, Holmes C, Nicoll JA. Neuropathology after active Abeta42 immunotherapy: implications for Alzheimer's disease pathogenesis. Acta Neuropathol 2010;120:369–384. [DOI] [PubMed] [Google Scholar]