

Summary
Aims
To investigate effects of DNA methyltransferase (DNMT) inhibitors on dopaminergic neurons and its underlied mechanism.
Methods
The DNMT inhibitor 5‐aza‐2′‐deoxycytidine (5‐aza‐dC) was tested in cultured dopaminergic cells. Cell viability and apoptosis were assayed with 5‐aza‐dC alone. Neurotoxicity of 1‐methyl‐4‐phenylpyridinium (MPP +), 6‐hydroxydopamine or rotenone was also assayed with 5‐aza‐dC pretreatment. And mRNA levels of several key PD‐related genes were examined by semiquantitative RT‐PCR. Furthermore, CpG methylation of α‐synuclein promoter was examined by bisulfite sequencing.
Results
5‐aza‐dC resulted in decreased cell viability and increased apoptosis in dopaminergic neuronal cells. Pretreatment with 5‐aza‐dC exacerbated neurotoxic damage to dopaminergic neurons induced by MPP +, 6‐hydroxydopamine or rotenone. 5‐aza‐dC also induced transcriptional upregulation of the key PD‐related genes tyrosine hydroxylase and α‐synuclein. And demethylation of CpG in α‐synuclein promoter was also induced by 5‐aza‐dC and MPP +.
Conclusions
This DNMT inhibitor might influence pathogenesis of PD. And demethylation induced by DNMT inhibitor might contribute to dopaminergic neuron death, by increasing vulnerability of dopaminergic neurons to neurotoxins and by misregulating transcription of key PD‐related genes. Our data also suggested DNMT inhibitors may cause multiple effects on dopaminergic neurons.
Keywords: Demethylation, DNA methyltransferase inhibitor, Gene transcription, Neurotoxicity, Parkinson's disease
Introduction
DNA methylation, in which DNA methyltransferase (DNMT) transfers a methyl group from S‐adenosyl methionine (SAM) to a cytosine within a CpG dinucleotide, is one of the major epigenetic processes controlling gene transcription 1, 2. Hypomethylation (or demethylation) always reactivates gene transcription in cells, whereas hypermethylation represses gene transcription. Genes regulated by DNA methylation may play a critical role in the control of apoptosis, cell survival, and the response to internal and external environmental factors in neurons 2, 3, 4. Misregulated DNA methylation has been linked to several neurological disorders 1, 5, 6, 7. DNA methylation can be controlled by DNMT inhibitors 8, which induce demethylation of genes in adult neurons 7. DNA methylation and DNMT inhibitors have been implicated in gene regulation in the adult brain in normal and pathological conditions 1, 7.
Parkinson's disease (PD) is a neurodegenerative disease caused by progressive and selective loss of dopaminergic neurons in the substantia nigra pars compacta (SNc) in the midbrain 9. Both intracellular and extracellular factors in the SNc are believed to be pathogenic. Neurotoxic factors, PD‐related genes, and apoptotic genes 10, 11 might all contribute to the death of dopaminergic neurons in PD. However, the roles of DNA methylation and DNMT inhibitor in PD have not been extensively investigated.
DNA methyltransferase inhibitors are useful in identifying the role of DNA methylation in neurological disorders 5, 7. And several DNMT inhibitors have been tested in clinical trials of human disease 8, including 5‐aza‐2′‐deoxycytidine (5‐aza‐dC), one of the most commonly used DNMT inhibitors 8. Pharmacological modulation of DNA methylation in the brain can regulate neuronal function 2, 5, 6. And as such, the pharmacological effects of DNMT inhibitors need to be clearly demonstrated in neurons. Importantly, an understanding of epigenetic regulation (i.e., DNA methylation) within neurons will allow us an appreciation of the action mechanisms of environmental neurotoxic factors.
In PD, dopaminergic neurons are the most important targets for neurotoxic factors. Therefore, it is necessary to investigate the effects of DNMT inhibitors on dopaminergic neurons. Particular attention should be paid to the roles of DNMT inhibitors in the interaction between environmental neurotoxic factors and dopaminergic neuron survival. Answering these questions may lead to a better understanding of PD pathogenesis. In our studies, we treated cultured dopaminergic neuronal cells with DNMT inhibitor 5‐aza‐dC and investigated neuronal survival, apoptosis, neurotoxicity, gene transcription levels, and methylation status.
Materials and Methods
Cell Culture and Drug Treatment
The dopaminergic neuronal cell lines human SH‐SY5Y, rat N27 and mouse MN9D were cultured in RPMI 1640 medium (Invitrogen, San Diego, CA, USA) supplemented with 10% FBS (Hiclone, Logan, UT, USA) and incubated at 37°C in a humidified chamber with 5% CO2. 5‐aza‐dC (Sigma, St Louis, MO, USA) and rotenone (Sigma) were dissolved in DMSO. MPP+ (Sigma), 6‐OHDA (Sigma), and SAM (Sigma) were dissolved in PBS. Cells were treated in medium containing MPP+, 6‐OHDA, or rotenone, with or without pretreatment with 5‐aza‐dC.
Cell Viability Assay
Cells were harvested and immediately stained with 0.02% trypan blue (Life Technologies, Inc., Gaithersburg, MD, USA). Viable cells were quantified using a hemocytometer under a microscope. Cell viability quantitatively was assessed using a Vi‐Cell XR Cell Viability Analyzer (Beckman Coulter, Fullerton, CA, USA). Experiments were performed in triplicate.
Assessment of Apoptosis
An annexin V‐FITC apoptosis detection kit (BD Pharmingen, San Diego, CA, USA) was used to detect apoptotic activity according to the manufacturer's instructions. Cells were stained with 0.5 mg/mL FITC‐conjugated annexin V and 1 mg/mL PI. Apoptotic cells (annexin V‐FITC positive) were microscopically quantified in at least five random fields. Experiments were performed in triplicate.
Apoptosis was also judged by DNA fragmentation analysis. Genomic DNA was extracted using a DNA isolation kit according to the manufacturer's instructions (BioDev, Beijing, China). The DNA laddering pattern was identified on 1.5% agarose gels.
Total RNA Extraction and Semiquantitative RT‐PCR
Total RNA was isolated using TRIzol (Invitrogen) and RT‐PCR was performed using an RNA PCR kit (TaKaRa Biotechnology, Dalian, China) according to the manufacturer's instructions. RNA was reversed transcribed with the AMV reverse transcriptase in the presence of an oligo(dT)15 primer. The cDNA was PCR amplified, using the primer sets indicated in Table S1, for 20, 25, 30, or 35 cycles. PCR products were visualized by electrophoresis on 1.2% agarose gels.
Protein Extraction and Western Blotting
Methods for protein extraction and Western blotting were modified from reference 12. Total protein (50 μg) was separated by SDS‐PAGE, electrotransferred onto nitrocellulose membranes, and probed with primary antibodies to p53 (1:2000; Cat# 2524, CST, Danvers, MA, USA) or actin (1:2000; Cat# A4700, Sigma). Signal was detected using anti‐mouse IDY800 (1:5000; Cat# 610‐132‐121, Rockland Immunochemicals, Gilbertsville, PA, USA) and scanned with an Odyssey® Imaging System (LI‐COR Bioscience Inc., Lincoln, NE, USA).
DNA Extraction and Bisulfite Sequencing
Methods for DNA isolation and bisulfite sequencing were modified from reference 13. Genomic DNA was extracted using a QIAamp DNA mini kits (Cat# 51304; QIAGEN, Hilden, Germany). Bisulfite conversion and clean‐up of DNA for methylation analysis were performed with the EpiTect Bisulfite kit (Cat# 59104, QIAGEN). Bisulfite‐treated DNA was amplified using primer pairs synF‐GGGAGGTTAAGTTAATAGGTGGTAA/synR‐CCCTCAACTATCTACCCTAAACAAAC with TaKaR Taq Hot Start version (Cat# DR007A; TaKaRa Biotechnology). PCR products were purified with Wizard® SV Gel and PCR Clean‐Up System (Cat# A9281, Promega, Madison, WI, USA) and then cloned into pGEM®‐T Vectors (Cat A1360, Promega). At least 10 clones from each experiment were sequenced by M13 primers. Quality control for DNA methylation data was performed using BiQ (software tool for DNA methylation analysis; http://biq-analyzer.bioinf.mpi-inf.mpg.de/).
Statistical Analysis
Values were represented as means ± SEM. Data were analyzed using one‐way ANOVA followed by a Student–Newman–Keuls test as a post hoc test. A value of P < 0.05 was considered statistically significant.
Results
5‐aza‐dC Caused Decreased Survival of Dopaminergic Neuronal Cells
Misregulated DNA methylation can be induced by DNMT inhibitors (hypomethylation) or overabundance of the methyl donor SAM (hypermethylation) 1, 2, 8. To investigate the effects of a DNMT inhibitor on dopaminergic neurons, cultured human SH‐SY5Y, rat N27, and mouse MN9D cells were treated with 5‐aza‐dC. Treatment with 5‐aza‐dC (1, 5, or 10 μM) for 60 h in N27 cells (Figure 1A) or 72 h in MN9D cells (Figure 1B) resulted in a marked decrease in cell viability, compared with control cells. At doses of 0.25–1.0 μM or 5.0 ~50.0 μM for 12–96 h, 5‐aza‐dC resulted in a time‐ and dose‐dependent reduction in the number of viable cells, in both N27 and SH‐SY5Y cells (Figure 1D,E), compared with controls. Within 12–24 h, 5‐aza‐dC treatment of N27 (0.25–1.0 μM) and SH‐SY5Y (5.0–50.0 μM) cells resulted in low or no cytotoxic effects (Figure 1D,E). We also investigated the effects of excess SAM. Treatment with SAM (75–250 μg/mL) for 96 h resulted in a decreased number of viable N27 cells, compared with controls (Figure 1C).
Figure 1.
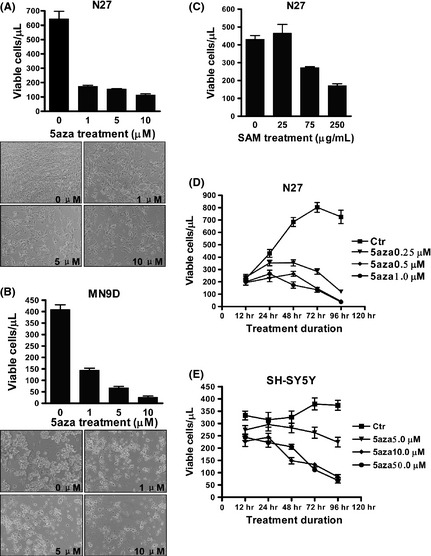
Decreased survival of dopaminergic neuronal cells following 5‐aza‐dC treatment. N27 cells (A) or MN9D cells (B) were treated with 1, 5, or 10 μM 5‐aza‐dC for 60 h or 72 h, respectively. N27 cells were treated with S‐adenosyl methionine (SAM) (25, 75, or 250 μg/mL) for 96 h (C). Cell growth was measured of N27 cells treated with 0.25, 0.5, or 1 μM 5‐aza‐dC (D) and SH‐SY5Y cells treated with 5, 10, or 50 μM 5‐aza‐dC (E) for 12–96 h. The values represent the means ± SEM of three experiments (n = 3). 5aza, 5‐aza‐2′‐deoxycytidine.
5‐aza‐dC Induced Apoptosis in Dopaminergic Neuronal Cells
To investigate whether the 5‐aza‐dC‐ or SAM‐induced decreased cell survival was mediated by apoptosis, the phosphatidylserine exposure of cell membranes and DNA strand breakages were analyzed. In N27 cells, 5‐aza‐dC (5 μM) for 24–48 h clearly resulted in apoptosis, as detected by annexin V‐FITC staining (Figure 2A). In SH‐SY5Y cells, 5‐aza‐dC (5 μM) for 24 h induced typical apoptosis observed under a fluorescence microscope (Figure 2D). DNA laddering assays showed that in N27 cells, 5‐aza‐dC (5 μM) for 24 h resulted in typical apoptosis, as indicated by DNA strand breaks (Figure 2C). In SH‐SY5Y cells, 5‐aza‐dC (10 μM) for 24 h did not result in apoptosis as assessed by DNA laddering assays (Figure 2C). SH‐SY5Y cells appeared to be more resistant than N27 cells to 5‐aza‐dC‐induced apoptosis. Annexin V‐FITC staining showed that SAM supplementation (up to 250 μg/mL) for 92 h did not cause significantly increased apoptosis in N27 cells (Figure 2B), suggesting that the SAM‐induced decreased survival of N27 cells (Figure 1C) might not be due to apoptosis.
Figure 2.
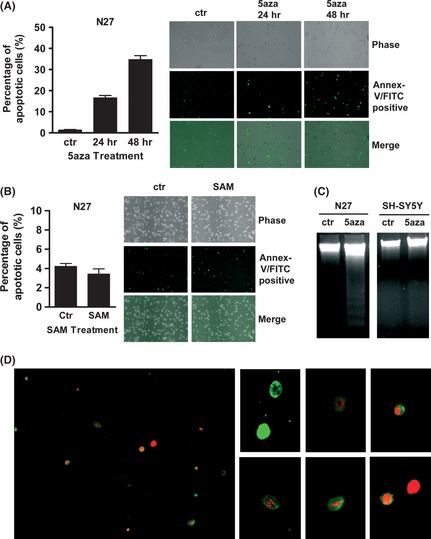
5‐aza‐dC–induced apoptosis in dopaminergic neuronal cells. N27 cells, treated with 5‐aza‐dC (5 μM) for 24–48 h (A) or S‐adenosyl methionine (SAM) (250 μg/mL) for 92 h (B), were stained with annexin V‐FITC. DNA laddering assay (C) in N27 cells treated with 5‐aza‐dC (5 μM) for 24 h and in SH‐SY5Y cells treated with 5‐aza‐dC (10 μM) for 24 h. SH‐SY5Y cells (D) treated with 5‐aza‐dC (5 μM) for 24 h were stained with annexin V‐FITC and propidium iodide (PI) and then viewed under a fluorescence microscope (green only, early apoptotic stage; green and red together, middle apoptotic stage; red only, late apoptotic stage). 5aza, 5‐aza‐2′‐deoxycytidine.
5‐aza‐dC Exacerbated the Neurotoxic Damage Induced by MPP+, 6‐OHDA, or Rotenone in Dopaminergic Neuronal Cells
We pretreated dopaminergic neuronal cells with low cytotoxic doses of 5‐aza‐dC to investigate its effects on the toxicity of neurotoxins commonly used in PD models: MPP+, 6‐OHDA, and rotenone. In N27 cells, pretreatment with 5‐aza‐dC (0.5 μM) for 24 h exacerbated the neurotoxic damage induced by 12‐h treatment with MPP+ (200 μM), 6‐OHDA (15 μM), or rotenone (0.5 μM) (Figure 3A). 5‐aza‐dC also exacerbated the neurotoxic damage accrued during longer (24 h) neurotoxin treatments (Figure 3B). Similarly, in SH‐SY5Y cells, 24 h pretreatment with 5‐aza‐dC (10 or 50 μM) exacerbated the neurotoxic damage induced by 12‐h (Figure 3C) or 24‐h (Figure 3D) treatment with 400 μM MPP+. To confirm our findings, we assessed cell viability quantitatively with a cell viability analyzer (Vi‐Cell XR Cell Viability Analyzer; Beckman Coulter). Consistently, pretreatment with 5‐aza‐dC exacerbated neurotoxic damage induced by MPP+ in dopaminergic neuronal cells (Figure 3E).
Figure 3.
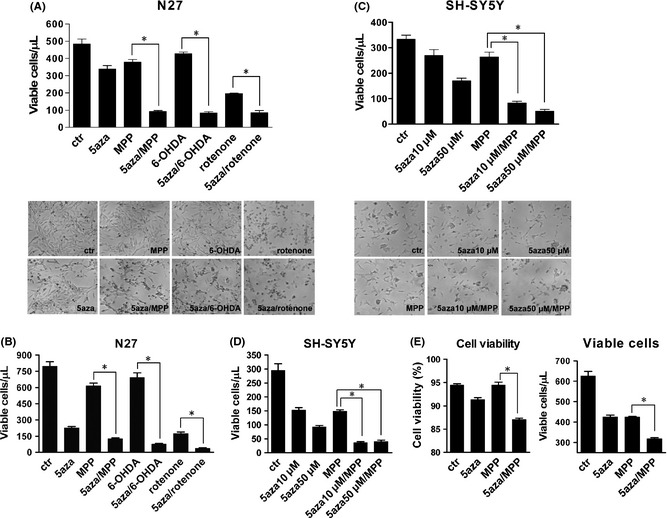
Pretreatment with 5‐aza‐dC increased the neurotoxic damages of MPP +, 6‐OHDA, and rotenone on dopaminergic neuronal cells. N27 cells were pretreated with 5‐aza‐dC (0.5 μM) for 24 h, followed by 12 h (A) or 24 h (B) of MPP + (200 μM), 6‐OHDA (15 μM) or rotenone (0.5 μM), then counted by trypan blue staining (upper panel) and visualized by morphological observation (lower panel). SH‐SY5Y cells were pretreated with 5‐aza‐dC (10 or 50 μM) for 24 h, followed by MPP + (400 μM) for 12 h (C) or for 24 h (D). N27 cells (E) were pretreated with 5‐aza‐dC (0.5 μM) for 24 h, followed by 12 h of MPP + (200 μM). Cell viability was assayed using a cell viability analyzer. The values represent the means ± SEM of three experiments (n = 3). *P < 0.05. 5aza, 5‐aza‐2′‐deoxycytidine; MPP, 1‐methyl‐4‐phenylpyridinium (MPP +); 6‐OHDA, 6‐hydroxydopamine.
5‐aza‐dC Resulted in Transcriptional Upregulation of the Key PD‐Related Genes TH and α‐Synuclein in Human Neuronal Cells
Gene transcription can be reactivated by DNMT inhibitor–induced hypomethylation of cytosines. To investigate transcriptional regulation of genes important in PD, we analyzed the mRNA levels of several PD‐related genes (TH, α‐synuclein, parkin, UCHL1, DJ‐1) and an apoptosis‐related gene (p53) after 5‐aza‐dC treatment in SH‐SY5Y cells. The mRNA levels of reelin were also measured as a control, because reelin has been reported to be upregulated by DNMT inhibitor–induced hypomethylation 14. Treatment with 5‐aza‐dC (50 μM) for 24 h resulted in upregulation of TH and α‐synuclein (SNCA) genes (Figure 4A). Similarly, lower doses of 5‐aza‐dC (2 μM or 10 μM) for 24 h also resulted in marked transcriptional upregulation of TH, though not of SNCA (Figure 4B). Transcription levels of the other PD‐related genes, parkin, UCHL1, and DJ‐1, were unaffected (Figure 4A). The expression levels of p53 protein were not upregulated by 5‐aza‐dC (Figure 4C).
Figure 4.
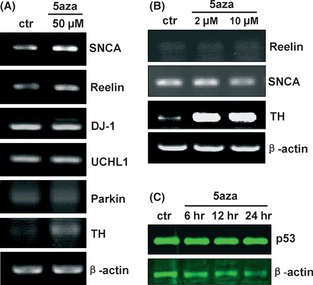
Regulation of gene transcription and expression in 5‐aza‐dC–treated SH‐SY5Y cells. Semiquantitative RT‐PCR showing the mRNA levels of tyrosine hydroxylase (TH), α‐synuclein (SNCA), parkin, ubiquitin carboxy‐terminal esterase hydrolase L1 (UCHL1), DJ‐1, and reelin in SH‐SY5Y cells treated with 50 μM (A), 2 μM, or 10 μM (B) of 5‐aza‐dC for 24 h. Expression levels of p53 protein (C) in N27 cells treated with 5‐aza‐dC (5 μM) for 6, 12, or 24 h, as assessed by immunoblotting. 5aza, 5‐aza‐2′‐deoxycytidine.
Demethylation Status of CpG Island at Promoter Region of α‐Synuclein Gene Was Induced by DNMT Inhibitor 5‐aza‐dC and Neurotoxin MPP+
Upregulation or reactivation of gene transcription in cells associates with demethylation of CpG island in promoter region. To identify whether demethylation was induced by 5‐aza‐dC in neuronal cells, we therefore analyzed methylation status of CpG island in α‐synuclein (SNCA) gene promoter region (Figure 5). A fragment (‐686/‐535) containing 13 CpG sites (Figure 5A) in this SNCA CpG island was investigated. Results of bisulfite sequencing showed that in SH‐SY5Y neuronal cells, DNMT inhibitor 5‐aza‐dC resulted in a reduced methylation levels (at 36.9–41.5% methylated), compared with control (at 49.2% methylated) (Figure 5B). Interestingly, neurotoxin MPP+ also induced a reduced methylation levels (at 43.8–47.7% methylated) (Figure 5B). The detailed analysis of different CpG site showed that CpG sites 2, 3, 4, 5, 6, 7, 8, 10, and 13 were demethylated by 5‐aza‐dC (Figure 5C). These DNMT inhibitor–demethylated CpG sites could be candidate targets for epigenetically pharmaceutical manipulation, associated with regulation of SNCA gene transcription. In addition, neurotoxin MPP+ also induced the demethylation at CpG sites 2, 3, 4, 6, 7, 10, and 13 (Figure 5C).
Figure 5.
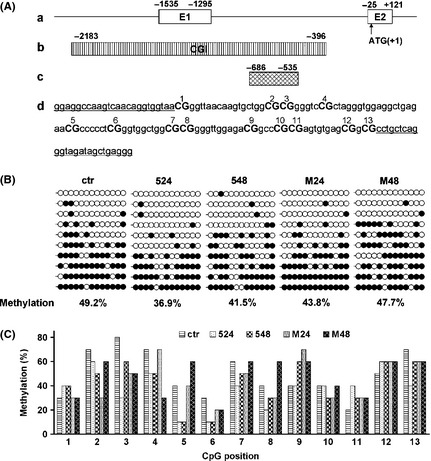
Methylation status of CpG island in SNCA promoter region. (A) The CpG island at 5′ end of the interested SNCA region. (a) Schematic drawing of the five region of SNCA with exons 1 and 2 (boxes) and the five UTR and intron 1 (line). Numbers are relative to the translation start site (ATG 1). (b) CpG island (CGI) is depicted by the stripped box. (c) Fragments in CpG island were used for bisulfite sequencing. (d) Location of CpG dinucleotides (CpGs) in this fragments of CpG island. There are 13 CpGs in the CpG region. Each CpG is labeled with number. Sequences with under bar show primer locations of the bisulfite PCR. (B) Bisulfite sequence analysis of the CpG region. DNA was purified from SH‐SY5Y cells, and bisulfite sequence analysis was performed. At least 10 clones from each drug‐treated group were analyzed. Open circles indicate unmethylated CpGs, whereas closed circles indicate methylated CpGs. The degree of methylation was calculated as methylated CpG/total CpG and shown below (% methylation). (C) Detailed comparison of methylation at individual CpG sites. The degree of methylation at each CpG site was calculated as methylated CpG/total CpG. Total 10 clones per group were analyzed by bisulfite sequencing. ctr, control; 524, 5‐aza‐dC (50 μM) for 24 h; 548, 5‐aza‐dC (50 μM) for 48 h; M24, MPP + (400 μM) for 24 h; M48, MPP + (400 μM) for 48 h.
Discussion
DNA methyltransferase inhibitors such as 5‐aza‐dC have been used in experimental studies and clinical trials to induce DNA hypomethylation and reactivate transcription 8, 13, 15. It is necessary to confirm its side effects on nervous system. We investigated its effects on dopaminergic neurons and its underlined mechanism that might involve in demethylation. Our result suggested that 5‐aza‐dC and 5‐aza‐dC‐induced demethylation might associate with PD. Based on these findings, we suggested there might be adverse effects on dopaminergic neuron, and use of 5‐aza‐dC in epigenetic therapy needs to be prudent.
The DNA methylation status of a given neuronal genome may play a critical role in its response to challenges or injury 5. Epigenetic effects in the mammalian brain may represent a major molecular mechanism mediating dynamic gene–environment interactions 2. Aberrant DNA methylation status might lead to vulnerability to environmental toxins or other neurotoxic factors 4. Here, we demonstrated for the first time that a DNMT inhibitor reduced survival and increased apoptosis in dopaminergic neuronal cells, and most importantly, that it exacerbated the neurotoxic damage induced by MPP+, 6‐OHDA, or rotenone in dopaminergic neuronal cells. Demethylation and transcriptional upregulation of genes induced by 5‐aza‐dC might contribute to enhancement of vulnerability of dopaminergic neurons to these neurotoxic damages. In our study, 5‐aza‐dC induced CpG demethylation in promoter and upregulated transcriptional levels of α‐synuclein gene. Upregulation of α‐synuclein gene transcription might contribute to vulnerability of dopaminergic neuron loss in PD 9, 16. MPP+/6‐OHDA‐caused neurotoxicity is mainly through mitochondria dysfunction and ROS, with which demethylation‐induced upregulation of gene transcription might cooperate leading to an exacerbated neurotoxicity. Our data suggested that DNMT inhibitor and demethylation induced by it may influence the susceptibility of dopaminergic neurons to environmental neurotoxic factors.
Both TH and α‐synuclein belong to the key PD‐related genes. Besides, the important PD‐related genes include parkin, UCHL1, and DJ‐1 genes detected in the present study and others. TH is a key rate‐limited enzyme in synthesis of dopamine, which is one of the most important factors in physiological function of dopaminergic neurons. In PD, loss of TH‐positive neurons (dopaminergic neurons) in SNc leads to depletion of dopamine in its projected striatum 9. Upregulation of TH expression and activity is neuroprotective to patients with PD. Genes α‐synuclein, parkin, UCHL1, and DJ‐1 may contribute to PD neurodegeneration, through cellular machinery involved in protein misfolding, protein aggregation, mitochondrial dysfunction, and oxidative stress 17. Overexpression or mutation of α‐synuclein, especially, may associate with family and sporadic PD. And it might relate to the selective neurodegeneration of PD 18.
Epigenetic modifications, including DNA methylation, control the transcription potential of genes 1, 5. DNA methylation misregulation could lead to abnormality of gene transcription, through the silencing of normally active genes or reactivation of normally silent genes 3. And DNA methylation misregulation usually associates with human disorders 3. In the present study, DNMT inhibitor upregulated transcription of key PD‐related genes α‐synuclein and TH in dopaminergic neuronal cells. It suggests a mechanism that transcription of α‐synuclein and TH genes could be misregulated by aberrant DNA methylation status. Up‐ or downregulation of these key PD‐related gene transcriptions depends on DNA methylation modification. Given that α‐synuclein could be upregulated by demethylation or TH could be downregulated by hypermethylation in aging through unknown mechanism, it mostly likely would develop PD. In clinic, loss of TH activity, degeneration of TH‐positive dopaminergic neurons, and aggregation of α‐synuclein in SNc are primary characteristics of PD. And it has been reported that α‐synuclein might relate to the selective neurodegeneration of PD 18. Our results suggest that DNA methylation misregulation, for example, demethylation induced by DNMT inhibitor, might lead to abnormal transcription of key PD‐related genes such as α‐synuclein and eventually might associate with the selective neurodegeneration in PD.
Overexpression of α‐synuclein may play important roles in pathogenesis of PD 19, 20, whereas upregulation of TH transcription may be neuroprotective in PD therapy 21, 22. In the present study, transcriptions of both α‐synuclein and TH were upregulated by DNMT inhibitor. These data are consistent with other reports that DNMT inhibitors upregulate these key PD‐related genes 13, 23. Both upregulation of α‐synuclein and TH at the same time are opposite or conflict effects in PD, which normally would not happen. Although these opposite or conflict effects caused by DNMT inhibitor in our experiments could not happen in real case of PD, our data suggest that DNMT inhibitors could cause multiple, complicated, and even conflicting effects on dopaminergic neurons. Thus, it is important to exercise caution in the use of DNMT inhibitors in epigenetic therapy for human disease.
The most commonly studied epigenetic modifications include DNA methylation and histone modifications 1, 5. Degenerative disorders of the CNS have been demonstrated may depend, at least in part, on epigenetic mechanisms 2, 5, 6, 24. In PD, recent report suggested that epigenetic mechanisms including DNA methylation might be involved 13, 15. Our findings suggest that DNMT inhibitor and DNMT inhibitor–misregulated DNA methylation might associate with PD. This supports the notion that epigenetic misregulation might involve in PD. And aberrant DNA methylation might play a role in PD pathogenesis.
In summary, DNMT inhibitor and its induced demethylation might influence PD pathogenesis through two ways (Figure 6): (1) by increasing vulnerability of dopaminergic neurons to neurotoxins and (2) by misregulating transcription of key PD‐related genes. This suggests that DNA methylation misregulation might involve in PD. In addition, our data suggest that DNMT inhibitors may cause multiple effects on dopaminergic neurons. And it is important to exercise caution in the use of DNMT inhibitors in epigenetic therapy for human disease. Further understanding involved in the molecular mechanism of epigenetic process of PD is needed.
Figure 6.
A speculated model for the roles of DNA methylation modification in Parkinson's disease. There might two ways that DNA methylation misregulation may play roles in PD. One is that at cellular level, it may influence the susceptibility of dopaminergic neurons to environmental neurotoxic factors, associating with the progressive loss of dopaminergic neurons in PD. Another is that at molecular level, it may misregulate transcription of key PD‐related genes such as α‐synuclein or TH, associating with the selective neurodegeneration of PD.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Table S1. Primer sets for semi‐quantitative RT‐PCR.
Acknowledgments
This work was supported by grants from the Chinese National Basic Research Program (2011CB504100) and the Beijing Natural Science Foundation (7082008).
References
- 1. Jiang Y, Langley B, Lubin FD, et al. Epigenetics in the nervous system. J Neurosci 2008;28:11753–11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mehler MF. Epigenetic principles and mechanisms underlying nervous system functions in health and disease. Prog Neurobiol 2008;86:305–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature 2007;447:433–440. [DOI] [PubMed] [Google Scholar]
- 4. Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet 2007;8:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. MacDonald JL, Roskams AJ. Epigenetic regulation of nervous system development by DNA methylation and histone deacetylation. Prog Neurobiol 2009;88:170–183. [DOI] [PubMed] [Google Scholar]
- 6. Gräff J, Mansuy IM. Epigenetic dysregulation in cognitive disorders. Eur J Neurosci 2009;30:1–8. [DOI] [PubMed] [Google Scholar]
- 7. Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 2007;8:355–367. [DOI] [PubMed] [Google Scholar]
- 8. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004;429:457–463. [DOI] [PubMed] [Google Scholar]
- 9. Dauer W, Przedborski S. Parkinson's disease: Mechanisms and models. Neuron 2003;39:889–909. [DOI] [PubMed] [Google Scholar]
- 10. Mattson MP, Magnus T. Ageing and neuronal vulnerability. Nat Rev Neurosci 2006;7:278–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tatton WG, Chalmers‐Redman R, Brown D, Tatton N. Apoptosis in Parkinson's disease: Signals for neuronal degradation. Ann Neurol 2003;53:S61–S72. [DOI] [PubMed] [Google Scholar]
- 12. Wang Y, Wang X, Liu L, Wang X. HDAC inhibitor trichostatin A‐inhibited survival of dopaminergic neuronal cells. Neurosci Lett 2009;467:212–216. [DOI] [PubMed] [Google Scholar]
- 13. Jowaed A, Schmitt I, Kaut O, Wullner U. Methylation regulates alpha‐synuclein expression and is decreased in Parkinson's disease patients' brains. J Neurosci 2010;30:6355–6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grayson DR, Jia X, Chen Y, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci USA 2005;102:9341–9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matsumoto L, Takuma H, Tamaoka A, et al. CpG demethylation enhances alpha‐synuclein expression and affects the pathogenesis of Parkinson's disease. PLoS ONE 2010;5:e15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee VMY, Trojanowski JQ. Mechanisms of Parkinson's disease linked to pathological α‐synuclein: New targets for drug discovery. Neuron 2006;52:33–38. [DOI] [PubMed] [Google Scholar]
- 17. Vila M, Przedborski S. Genetic clues to the pathogenesis of Parkinson's disease. Nat Med 2004;10(Suppl):S58–S62. [DOI] [PubMed] [Google Scholar]
- 18. Xu J, Kao SY, Lee FJ, et al. Dopamine‐dependent neurotoxicity of alpha‐synuclein: A mechanism for selective neurodegeneration in Parkinson disease. Nat Med 2002;8:600–606. [DOI] [PubMed] [Google Scholar]
- 19. Maries E, Dass B, Collier TJ, Kordower JH, Steece‐Collier K. The role of [alpha]‐synuclein in Parkinson's disease: Insights from animal models. Nat Rev Neurosci 2003;4:727–738. [DOI] [PubMed] [Google Scholar]
- 20. Sulzer D. Clues to how alpha‐synuclein damages neurons in Parkinson's disease. Mov Disord 2010;25:S27–S31. [DOI] [PubMed] [Google Scholar]
- 21. Falk T, Zhang S, Sherman SJ. Pigment epithelium derived factor (PEDF) is neuroprotective in two in vitro models of Parkinson's disease. Neurosci Lett 2009;458:49–52. [DOI] [PubMed] [Google Scholar]
- 22. Biju KC, Zhou Q, Li G, et al. Macrophage‐mediated GDNF delivery protects against dopaminergic neurodegeneration: A therapeutic strategy for Parkinson's disease. Mol Ther 2010;18:1536–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aranyi T, Faucheux BA, Khalfallah O, et al. The tissue‐specific methylation of the human Tyrosine Hydroxylase gene reveals new regulatory elements in the first exon. J Neurochem 2005;94:129–139. [DOI] [PubMed] [Google Scholar]
- 24. Scarpa S, Cavallaro RA, D'Anselmi F, Fuso A. Gene silencing through methylation: An epigenetic intervention on Alzheimer disease. J Alzheimers Dis 2006;9:407–414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sets for semi‐quantitative RT‐PCR.