

SUMMARY
Background: Previous studies have suggested that manic states and sleep deprivation could contribute to the pathophysiology of bipolar disorder (BD) through protein kinase C (PKC) signaling abnormalities. Moreover, adjunctive therapy has become a standard strategy in the management of BD patients who respond poorly to current pharmacological treatments. Aim: Thus, the aim of this study was to investigate the possible involvement of PKC inhibition by tamoxifen both separately or in combination with lithium, in paradoxical sleep deprivation (PSD)‐induced hyperactivity, one facet of mania‐like behavior. Materials & Methods: Adult male C57BL/6J mice were randomly distributed (n = 7/group) in 24‐h PSD or control groups and injected intraperitoneally (i.p.) with vehicle, lithium (50, 100, or 150 mg/kg) or tamoxifen (0.5, 1.0, or 2.0 mg/kg – experiment 1). In a second experiment, mice were injected i.p. with vehicle or a combination of subeffective doses of lithium and tamoxifen. Animals were subjected to a protocol based on repetitive PSD conditions, followed by assessment of locomotion activity in the open‐field task. Results: PSD significantly increased locomotor activity in both experiments. These behavioral changes were prevented by a treatment with lithium or tamoxifen, or a combined treatment with both lithium and tamoxifen. Discussion: Therefore, our findings suggest that lithium and tamoxifen exert reversal effects against PSD‐induced hyperactivity in mice. Conclusion: Furthermore, tamoxifen as an adjunct to lithium therapy provides support for an alternative treatment of individuals who either do not respond adequately or cannot tolerate the adverse effects associated with therapeutic doses of lithium.
Keywords: Adjunctive therapy, Lithium, Mania, Mice, Paradoxical sleep deprivation, Protein kinase C, Tamoxifen
Introduction
Bipolar disorder (BD) is a common psychiatric disorder that is characterized by recurrent manic, mixed, and depressive episodes. Life prevalence is estimated at 0.8–1.6%, but may be higher depending on the diagnostic criteria [1]. It is also frequently associated with a wide range of physiological perturbations and medical problems that result in cognitive, motor, autonomic, endocrine, and sleep‐wake impairments, which culminate in decreased quality of life [2, 3].
Although lithium remains the drug of choice for treating acute states and preventing new episodes of this disabling disease, many patients do not respond to it as a maintenance treatment and experience several side effects [4, 5]. Thus, there is a clear need for more effective and less toxic medications that may result from alternative strategies, such as adding a second therapeutic agent to the treatment protocols of patients with unsatisfactory responses to monotherapy [6]. Of note, the development of novel pharmacological therapeutic agents for BD and a better understanding of the neurobiology of the disorder may be hindered by the lack of valid animal models [7, 8].
Because evidence suggests a relationship between sleep and manic states exists [9, 10, 11, 12], preclinical studies have tried to establish an appropriate animal model that recapitulates core pathophysiological aspects of the disease, such as sleep disruption [7]. Indeed, sleep deprivation exacerbates manic attacks or may cause a switch from depression to mania in BD patients [13, 14, 15, 16, 17]. In this sense, sleep loss is both a cause and a consequence of mania, and thus, continued sleep loss can perpetuate the manic state. Gessa et al. [18] demonstrated that rats deprived of paradoxical sleep present a brief period of hyperactivity, which is one facet of mania‐like behavior, that is responsive to lithium treatment. Later, Benedetti et al. [19] reported that paradoxical sleep deprivation (PSD) increased locomotor activity and aggressive behavior in mice. These authors adopted a protocol based on the length of treatment and progressive sensitization to the effects of repetitive PSD sessions.
Increasing evidence also implicates abnormal protein kinase C (PKC) activity and distribution in the etiology of BD [20, 21, 22]. Biochemical data support changes in PKC and its substrates [23, 24], and an altered PKC signaling pathway after treatment with mood stabilizers [23, 25] in BD patients. Preclinical findings also support that alterations in PKC and its substrates are induced by amphetamine [26] or sleep deprivation [27]. In animals, PKC function was also attenuated by mood stabilizing treatment [28, 29]. Collectively, both clinical and preclinical data suggest that PKC activation might be involved in the manic state, whereas PKC inhibition could be related to antimanic properties.
In this context, tamoxifen, a PKC inhibitor that is widely used in the prophylactic treatment of breast cancer [30], emerges as a promising treatment for manic episodes in BD. Its properties have been investigated in preliminary clinical trials that demonstrated its antimanic effects [31, 32, 33, 34]. In rodents, tamoxifen significantly normalized amphetamine‐induced hyperactivity [35] and reduced hedonia‐like, amphetamine‐induced conditioned place preference [36].
Given that sleep disruption and abnormal intracellular pathways are involved in the etiology of BD, it seems reasonable to suppose that they may share a common mechanism in the pathophysiology of this disorder, which could potentially arise from their differential effects on PKC. To date, however, the possible involvement of PKC inhibition by tamoxifen treatment has not been fully assessed in animal models of mania induced by PSD. Moreover, its potential effects in combination with lithium need to be evaluated to provide relevant information on the use of tamoxifen as an adjunct therapy. Thus, we aimed to investigate the possible involvement of PKC inhibition by tamoxifen as monotherapy, or in combination with lithium, in PSD‐induced hyperactivity in mice.
Material and Methods
Animals
Adult male C57BL/6J mice from the Instituto Nacional de Farmacologia e Biologia Molecular (INFAR, São Paulo, Brazil) were housed in standard polypropylene cages, five animals per cage. They were given free access to food and water and were maintained on a 12‐h light–dark cycle (lights on at 7:00 am) at a temperature of 22 ± 2°C. All experimental procedures were carried out in accordance with the guidelines established by the Ethical and Practical Principles of the Use of Laboratory Animals [37] with approval of the University's Ethical Committee for animal experimentation (#1258/08).
Drugs
Immediately before use, lithium carbonate (Eurofarma, Brazil) was dissolved in 0.9% sterile saline at concentrations of 50, 100, and 150 mg/kg. The pH was adjusted to approximately 7.4 by adding 2N HCl. The PKC inhibitor, tamoxifen citrate (Sigma Co, USA), was prepared immediately before use by dissolving in propylene glycol at concentrations of 0.5, 1.0, and 2.0 mg/kg. The solvent, dose, timing, and mode of administration were chosen based on those used previously in published reports [35,36], and on a pilot study that was formerly performed in our laboratory. Half of the mice that underwent vehicle treatment (v) received saline (n = 3 animals), while the other half received propylene glycol (n = 4 animals).
Drug Administration
Animals were injected intraperitoneally (i.p.) twice a day and all injection volumes were 0.01 mL/g of body weight. For all procedures, each animal received only one treatment type. In the first experiment, for longer term administration, lithium doses were injected at 8:00 am and 4:00 pm. Mice subjected to vehicle treatment were also given vehicle at 8:00 am and 4:00 pm. Animals that underwent tamoxifen treatment were given vehicle at 8:00 am and tamoxifen at 4:00 pm. In the second experiment, mice that underwent lithium and tamoxifen co‐administration were given lithium at 8:00 am and lithium and tamoxifen at 4:00 pm. Mice subjected to vehicle treatment were given vehicle at 8:00 am and 4:00 pm. Therefore, animals from both experiments underwent 9 days of drug administration. On the test day, animals from both experiments received only one injection of the respective drug treatment 35 min prior to behavioral assessment.
Paradoxical Sleep Deprivation
PSD was induced by placing five animals inside a tiled water cage for 24 h, the cages (38 × 31 × 17 cm) contained 12 platforms (3.5 cm in diameter) that were surrounded by water up to 1 cm beneath the surface. In this modified multiple platform method, the animals were capable of moving inside the cage by jumping from one platform to another. When an animal entered the paradoxical phase of sleep, due to the muscle atonia it fell into the water and was awakened. Food and water were made available through a grid placed on top of the water cage. Control mice (CTRL) were exposed for 24 h to the same home‐cage conditions, except that the water was replaced by sawdust. The duration of PSD was determined based on pilot experiments in which 24 h of PSD was shown to be effective in inducing behavioral alterations. We also previously demonstrated that this protocol of PSD significantly suppressed paradoxical sleep in mice [38]. During the entire experimental period, the room was maintained under a 12‐h light–dark cycle (lights on at 7 am) at 22 ± 2°C, and water in the cages was replaced at least 6 h prior to locomotor activity assessment.
Locomotor Activity
The locomotor activity was evaluated in the open‐field task [39]. The open‐field apparatus was a circular wooden arena (40 cm in diameter and 50 cm high) with an open top, and a floor that was divided into 19 squares. A video camera was placed 1.8 m above the center of the apparatus and was designed to record locomotor and exploratory behavior, which were appropriately evaluated by hand‐operated counters. It should be noted that the scorer was blind to the treatment groups. Animals were gently placed in the open‐field for 5 min to score peripheral locomotion (12 squares close to the wall), central locomotion (7 squares not contiguous with the wall), and total locomotion frequencies. An entry into a square was counted once, provided that the mouse had entered it with all four paws. The rearing frequency, latency time for the first movement and immobility time during the trial were also measured. It should be noted that the 5 min length of the assessment session was used because this period of time allowed for an accurate and reliable evaluation of the effects of paradoxical sleep deprivation on mice locomotor activity under our experimental conditions [40, 41].
After each mouse was removed, the arena was cleaned using 5% alcohol to avoid odor traits. To mitigate circadian effects on the animal's behavior, the time of day of each animal's observation was kept constant, alternating with animals from different experimental groups.
Experimental Procedure
Two experiments were performed. The aim of the first experiment was to verify the effects of 50, 100, and 150 mg/kg of lithium and 0.5, 1.0, and 2.0 mg/kg of tamoxifen, in the prevention of PSD‐induced hyperactivity. Based on this experiment, the second experiment was performed to investigate the effects of subeffective doses of tamoxifen as adjunct therapy, allowing a reduction in the lithium dose requirements for treating PSD‐induced hyperactivity. Thus, lithium and tamoxifen were co‐administrated at the doses of 50 and 0.5 mg/kg, respectively. In both experiments, animals were subjected to a protocol, which was based on repetition (three times) of treatment conditions (24‐h PSD) and length of drug administration, according to an experimental design proposed by Benedetti et al. [19]. As depicted in Figure 1(A), the first 3 days of the experimental procedure promoted open‐field habituation and basal locomotor activity assessment, and allowed random distribution of the animals in different groups, 7 animals per group, based on distinct sleep conditions and drug administration (Figure 1B). From the fourth day, animals were administered one of the different drugs until the end of the experimental procedure. On the test day, mice in the water cages (PSD) or home cages (CTRL) received an injection of the respective drug. Locomotor activity was measured 35 min after the last injection, and mice were returned to their home cages and allowed undisturbed and spontaneous sleep for 48 h. This interval between injection and testing was based on previous experiments performed in our laboratory. This procedure was repeated three times and all experiments were performed during the light cycle, from 1:00 pm to 4:00 pm.
Figure 1.
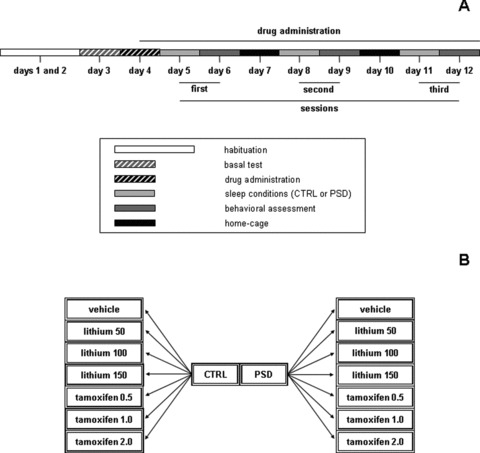
Schematic representation of the experimental design. A time‐line illustration of the sequence of events throughout the experiments shows the animals designated for behavioral tests (Panel A). Panel (B) illustrates the subdivisions of home‐cage control (CTRL) and paradoxical sleep deprived (PSD) groups treated with vehicle, lithium, or tamoxifen.
Statistical Analysis
This study included one dependent variable (total locomotion frequency) and three independent factors: sleep conditions (PSD vs. control conditions), drug administration (lithium, tamoxifen or vehicle), and repetition session (first, second, and third). Analysis was conducted using three‐way Analysis of Variance (ANOVA) with repeated measures, followed by the Newman–Keuls post hoc test. Statistical significance was set at P < 0.01. All data are presented as mean across the three repetition sessions ± standard error mean (SEM).
Results
Effects of Lithium and Tamoxifen in PSD‐Induced Hyperacitivity
Three‐way ANOVA with repeated measures detected significant PSD [F(1,84)= 254.134; P < 0.001] and drug treatment effect [F(6,84)= 5.365; P < 0.001], and a significant PSD × drug treatment interaction [F(6,84)= 7.726; P < 0.001], related to locomotor activity. PSD for 24 h significantly increased locomotor activity in mice subjected to vehicle treatment, during all behavior assessments. These PSD‐induced behavioral changes were prevented by lithium administration at the doses of 100 and 150 mg/kg, and by 1.0 and 2.0 mg/kg of tamoxifen, compared with vehicle‐treated PSD mice across first, second and third behavior sessions (Figure 2).
Figure 2.
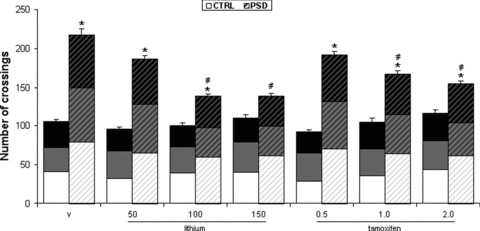
Effects of lithium and tamoxifen on PSD‐induced hyperactivity. Each bar represents the mean of first, second and third repetition sessions (white, gray, and black bars, respectively) ± SEM across the three repetition sessions for 7 animals per group. *P < 0.01 compared with control conditions and the same pharmacological treatment. #P < 0.01 compared with vehicle group subjected to the same sleep conditions (three‐way ANOVA with repeated measures followed by the Newman–Keuls post hoc test). PSD, paradoxical sleep deprivation conditions; CTRL, control conditions; v, vehicle.
Significant repetition session [F(2,168)= 25.842; P < 0.001], and repetition session × PSD interaction [F(2,168)= 8.710; P < 0.001] were also revealed by three‐way ANOVA with repeated measures, with respect to locomotor activity. Repetitions of session lead to a decreased pattern of locomotor activity in both groups (control or PSD animals). Post hoc comparisons revealed a significantly increased locomotor activity in PSD mice when compared with control mice in all three behavior assessments. In addition, repetitions of sessions significantly decreased locomotor activity in PSD animals during the second and third sessions when compared to PSD animals during the first session (Figure 3).
Figure 3.
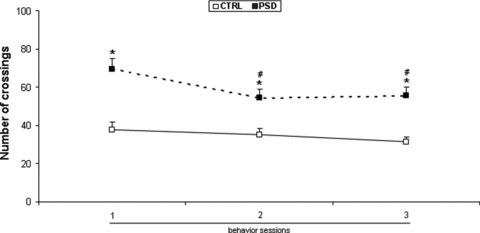
Effects of locomotor activity after repeated control or PSD conditions in the first experiment. Data are reported as the mean ± SEM for 7 animals per group. *P < 0.01 compared with control conditions at the first, second and third repetition sessions. #P < 0.01 compared with PSD group at the first repetition session (three‐way ANOVA with repeated measures followed by the Newman–Keuls post hoc test). PSD, paradoxical sleep deprivation conditions; CTRL, control conditions; v, vehicle.
There were not significant repetition session effect [F(2,48)= 0.937; NS], repetition session × PSD interaction [F(2,48)= 0.445; NS], repetition session × drug treatment interaction [F(2,48)= 0.763; NS] or repetition session × PSD × drug treatment [F(2,48)= 0.242; NS], according to three‐way ANOVA with repeated measures.
Effects of Lithium and Tamoxifen Co‐administration in PSD‐Induced Hyperacitivity
Three‐way ANOVA with repeated measures detected significant PSD [F(1,24)= 112.090, P < 0.001] and drug treatment effects [F(1,24)= 16.601; P < 0.001], and a significant PSD × drug treatment interaction [F(1,24)= 9.998; P < 0.01], related to locomotor activity. PSD for 24 h significantly increased locomotor activity in animals that underwent vehicle treatment, in all behavior assessments. The co‐administration of 50 mg/kg of lithium with 0.5 of tamoxifen prevented the PSD‐induced hyperactivity, compared with vehicle‐treated PSD mice across first, second, and third behavior sessions (Figure 4).
Figure 4.
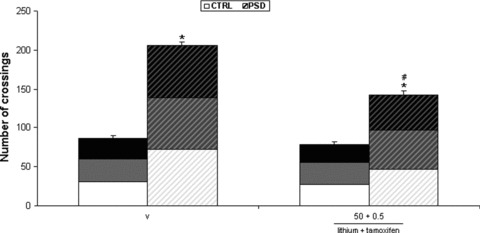
Effects of lithium and tamoxifen co‐administration on PSD‐induced hyperactivity. Each bar represents the mean of first, second and third repetition sessions (white, gray, and black bars, respectively) ± SEM across the three repetition sessions for 7 animals per group. *P < 0.01 compared with control conditions and the same pharmacological treatment. #P < 0.01 compared with vehicle group subjected to the same sleep conditions (three‐way ANOVA with repeated measures followed by the Newman–Keuls post hoc test). PSD, paradoxical sleep deprivation conditions; CTRL, control conditions; v, vehicle.
There were not significant repetition session × drug treatment interaction [F(12,168)= 1.208; NS] or repetition session × PSD × drug treatment [F(12,168)= 0.599; NS], according to three‐way ANOVA with repeated measures.
Discussion
The results of this study demonstrated that PSD increased locomotor activity. We also observed a decreased motor activity due to the effect of repetitive behavior assessments. In addition, both lithium and tamoxifen reversed PSD‐induced hyperacitivity. The reversal effect of lithium occurred under separate and combined (adjunctive treatment) administration. Taken together, these findings indicated that PSD significantly induced hyperactivity, and suggested a reversal effect of lithium and/or tamoxifen against this behavior.
The PSD paradigm is highly relevant to understanding the etiology of BD, since alterations in sleep‐wake patterns profoundly affect patients suffering from this disorder. Indeed, the manic phase is characterized by a marked decrease in the need for sleep [20]. Accordingly, the PSD model has proven valuable in the testing of potential antimanic drugs, and in the examination of neurobiological correlates between manic reactions and sleep loss, thus providing a useful tool in gaining further knowledge and a better understanding of the pathophysiology of BD. Therefore, it can be assumed that PSD is a partially valid model of mania with face, construct and predictive validities because it presents homologies with the symptoms of the disease, is interpretable in terms of neurochemical correlates, and is sensitive to the same pharmacological treatments as the clinical condition, respectively.
Hyperactivity induced by PSD was replicated in our second experiment, supporting the reproducibility of this model. Our findings are consistent with existing evidence that PSD increased locomotor activity [18, 19], which is a parameter of mania‐like behavior in rodents [18, 27, 42]. We also observed a reduction in activity level of both sleep condition groups (control or PSD) according to repetitions of session in the first experiment. This behavioral change is probably caused by a habituation to the open‐filed apparatus, leading to a decrease in exploratory behavior. Moreover, the lack of changes from progressive sensitization to the effect of repeated PSD that was observed in our study is inconsistent with previous findings [19]. Differences in experimental design involving distinct PSD methods, light‐dark cycle phase and strains of mice may account for this discrepancy. Indeed, Benedetti et al. [19] elected for the single platform method as a PSD paradigm, whereas we elected the multiple platform method. It is noteworthy that these distinct methods are associated with different alterations in corticosterone levels [43] that could be related to differences in the temporal course of PSD‐induced hyperactivity. Additionally, Benedetti et al. [19] conducted locomotor activity procedure and assessment during the dark phase of the light‐dark cycle, whereas in our study locomotor activity procedure and assessment was performed during the light phase of the light‐dark cycle. Such methodological dissimilarities may offer an interesting working hypothesis to investigate the effects of stress and light‐dark cycles on the temporal course of PSD‐induced hyperactivity. This concern notwithstanding, our data suggest that sensitization to PSD‐induced hyperactivity is not a necessary feature of the model as regards to its sensitivity to a usual therapeutic agent such as lithium.
Lithium exerted reversal effects against PSD‐induced hyperactivity in mice under our experimental conditions. This result corroborates previous data on lithium's ability to reverse PSD‐induced hyperactivity in rats [18]. The PKC inhibitor tamoxifen also reversed PSD‐induced hyperactivity. These findings are also in accordance with previous clinical [34, 44] and preclinical studies [35, 36] suggesting that tamoxifen is an antimanic agent. As tamoxifen is also an estrogen receptor modulator, the possibility that estrogen receptor blockade, or other intracellular effects of this drug, may play a role in the observed behavioral changes cannot be excluded [31, 35, 36].
Both lithium and tamoxifen prevented PSD‐induced hyperactivity. Since the mode of action of lithium may be, at least in part, through attenuation of PKC function [21, 24, 25, 45, 46, 47, 48], suggesting that periods of sleep deprivation could induce PKC overactivity. Indeed, previous data showed that phosphorylation of the myristoylated alanine‐rich C kinase substrate (MARKS), a marker of PKC activity, was increased in the prefrontal cortex of sleep‐deprived rats, but was decreased in lithium‐treated rats [27]. Overactive PKC signaling might lead to alterations in neuroplasticity, which, in addition to sleep disruption‐induced cellular impairments, may contribute to the pathophysiolgy of the manic episode [27, 45, 49]. Although, the possibility that other mechanisms may underlie this relationship cannot be ruled out given that both sleep and BD promote global changes in brain function. It should be clarified that the mechanistic hypothesis to establish the correlation between sleep and PKC signaling mentioned above is based on assumptions and on previous findings, since the present study did not directly measured PKC activity.
The possible involvement of PKC signaling in the pathophysiology of BD may have relevant clinical implications, since many patients are unresponsive to standard pharmacological monotherapies [4, 20]. In fact, there is increasing evidence indicating that optimal response by most BD patients requires multiple‐drug therapy [20], supporting the strategy of adding a second agent rather than switching to a different monotherapy [6].
In support of this concept, our second experiment demonstrated that co‐administration of lithium and tamoxifen at their lowest doses also prevented PSD‐induced hyperactivity. The fact that the effective dose of lithium can be significantly decreased by co‐administration of a low dose of tamoxifen suggests a drug interaction. In this sense, given that lithium has been implicated in inositol depletion, which attenuates PKC [21, 24, 25, 45, 46, 47, 48], and that tamoxifen is a PKC inhibitor, it is tempting to speculate that the inhibitory effect of these two pharmacological agents in their subeffective doses on PSD‐induced hyperactivity may be due to their impact on this common PKC signaling pathway.
Furthermore, the co‐administration of lithium and tamoxifen significantly reversing PSD‐induced hyperactivity when compared to lithium or tamoxifen individually provides objective evidence in favor of a combination of treatments over monotherapy under our experimental conditions. In this sense, although adding treatments does increase the risk of drug–drug interactions and adverse‐effects [6], our results demonstrated that the co‐administration of lithium and tamoxifen did not affect behavioral measures in control animals. However, the establishment of safety, tolerability and efficacy of tamoxifen as an adjunct to lithium therapy should be addressed in additional studies with different design approaches using other animal models of mania.
In conclusion, we have demonstrated that tamoxifen and lithium can reverse PSD‐induced hyperactivity, when administrated separately or in combination. A plausible hypothesis to explain our findings could be that sleep disruption might induce PKC overactivity via a common pathway that mediates the relationship between sleep loss and manic states. These data could contribute to our increased understanding of the mechanisms underlying the pathophysiology of BD. In addition, although extrapolation to clinical situations from animal data must always be made with caution, tamoxifen as an adjunct to lithium may be promising, since adjunctive treatment appears to be a well‐supported management strategy for acute mania, especially in cases refractory to current treatments.
Role of Funding Source
This work was supported by grants from the Associação Fundo de Incentivo à Psicofarmacologia (AFIP), CNPq, and FAPESP (CEPID #98/14303–3 to ST). All the authors listed in the present study reported no biomedical financial interests or potential conflicts of interest.
Author Contributions
Fernanda Armani, Monica Levy Andersen, Roberto Andreatini, Sergio Tufik and José Carlos Fernandes Galduróz designed the study and wrote the protocol. Fernanda Armani, Roberto Andreatini and José Carlos Fernandes Galduróz managed the literature searches. Fernanda Armani and Roberto Frussa‐Filho performed acquisition of data and managed analyses and interpretation of data. Fernanda Armani and Monica Levy Andersen undertook the statistical analysis. All authors contributed to and have approved the final manuscript.
Conflict of Interest
For this review, the authors do not have conflict of interest to declare.
Acknowledgments
We would like to thank the staff of our animal facility for taking care of the mice used in this study. The authors would also like to express their cordial thanks to Thaís Fernanda Trombini, Waldermaks Leite, Marilde Costa, Theotila R.R. Amaral, and Cleomar S. Ferreira for their assistance during the project.
References
References
- 1. Angst J. Epidemiology of the bipolar spectrum. Encephale 1995;21:37–42. [PubMed] [Google Scholar]
- 2. Kupfer DJ. The increasing medical burden in bipolar disorder. JAMA 2005;293:2528–2530. [DOI] [PubMed] [Google Scholar]
- 3. Krishnan KR. Psychiatric and medical comorbidities of bipolar disorder. Psychosom Med 2005;67:1–8. [DOI] [PubMed] [Google Scholar]
- 4. Judd LL, Akiskal HS. The prevalence and disability of bipolar spectrum disorders in the US population: Re‐analysis of the ECA database taking into account subthreshold cases. J Affect Disord 2003;73:123–131. [DOI] [PubMed] [Google Scholar]
Introduction
- 5. Tohen M, Zarate CA, Jr. , Hennen J, et al The McLean‐Harvard First‐Episode Mania Study: Prediction of recovery and first recurrence. Am J Psychiatry 2003;160:2099–2107. [DOI] [PubMed] [Google Scholar]
- 6. Sachs GS, Gardner‐Schuster EE. Adjunctive treatment of acute mania: A clinical overview. Acta Psychiatr Scand Suppl 2007;116:27–34. [DOI] [PubMed] [Google Scholar]
- 7. Machado‐Vieira R, Kapczinski F, Soares JC. Perspectives for the development of animal models of bipolar disorder Prog Neuropsychopharmacol Biol Psychiatry 2004;28:209–224. [DOI] [PubMed] [Google Scholar]
- 8. Gould TD, Einat H. Animal models of bipolar disorder and mood stabilizer efficacy: A critical need for improvement. Neurosci Biobehav Rev 2007;31:825–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harvey AG. Sleep and circadian rhythms in bipolar disorder: Seeking synchrony, harmony, and regulation. Am J Psychiatry 2008;165:820–829. [DOI] [PubMed] [Google Scholar]
- 10. Plante DT, Winkelman JW. Sleep disturbance in bipolar disorder: Therapeutic implications. Am J Psychiatry 2008;165:830–843. [DOI] [PubMed] [Google Scholar]
- 11. Giglio LM, Andreazza AC, Andersen M, Cereser KM, Walz JC, Sterz L, Kapczinski F. Sleep in bipolar patients. Sleep Breath 2009;13:169–173. [DOI] [PubMed] [Google Scholar]
- 12. Hudson JI, Lipinski JF, Keck PE, Jr. , et al Polysomnographic characteristics of young manic patients. Comparison with unipolar depressed patients and normal control subjects. Arch Gen Psychiatry 1992;49:378–383. [DOI] [PubMed] [Google Scholar]
- 13. Wehr TA, Goodwin FK, Wirz‐Justice A, Breitmaier J, Craig C. 48‐hour sleep‐wake cycles in manic‐depressive illness: Naturalistic observations and sleep deprivation experiments. Arch Gen Psychiatry 1982;39:559–565. [DOI] [PubMed] [Google Scholar]
- 14. Wehr TA, Sack DA, Rosenthal NE. Sleep reduction as a final common pathway in the genesis of mania. Am J Psychiatry 1987;144:201–204. [DOI] [PubMed] [Google Scholar]
- 15. Wehr TA. Sleep‐loss as a possible mediator of diverse causes of mania. Br J Psychiatry 1991;159:576–578. [DOI] [PubMed] [Google Scholar]
- 16. Barbini B, Bertelli S, Colombo C, Smeraldi E. Sleep loss, a possible factor in augmenting manic episode. Psychiatry Res 1996;65:121–125. [DOI] [PubMed] [Google Scholar]
- 17. Colombo C, Benedetti F, Barbini B, Campori E, Smeraldi E. Rate of switch from depression into mania after therapeutic sleep deprivation in bipolar depression. Psychiatry Res 1999;86:267–270. [DOI] [PubMed] [Google Scholar]
- 18. Gessa GL, Pani L, Fadda P, Fratta W. Sleep deprivation in the rat: An animal model of mania. Eur Neuropsychopharmacol 1995;5(Suppl):89–93. [DOI] [PubMed] [Google Scholar]
- 19. Benedetti F, Fresi F, Maccioni P, Smeraldi E. Behavioural sensitization to repeated sleep deprivation in a mice model of mania Behav Brain Res 2008;187:221–227. [DOI] [PubMed] [Google Scholar]
- 20. Berns GS, Nemeroff CB. The neurobiology of bipolar disorder. Am J Med Genet C Semin Med Genet 2003;123C:76–84. [DOI] [PubMed] [Google Scholar]
- 21. Ackenheil M. Neurotransmitters and signal transduction processes in bipolar affective disorders: A synopsis. J Affect Disord 2001;62:101–111. [DOI] [PubMed] [Google Scholar]
- 22. Kato T. Molecular neurobiology of bipolar disorder: A disease of ‘mood‐stabilizing neurons’? Trends Neurosci 2008;31:495–503. [DOI] [PubMed] [Google Scholar]
- 23. Wang HY, Friedman E. Enhanced protein kinase C activity and translocation in bipolar affective disorder brains. Biol Psychiatry 1996;40:568–575. [DOI] [PubMed] [Google Scholar]
- 24. Wang H, Friedman E. Increased association of brain protein kinase C with the receptor for activated C kinase‐1 (RACK1) in bipolar affective disorder. Biol Psychiatry 2001;50:364–370. [DOI] [PubMed] [Google Scholar]
- 25. Hahn CG, Umapathy , Wang HY, Koneru R, Levinson DF, Friedman E. Lithium and valproic acid treatments reduce PKC activation and receptor‐G protein coupling in platelets of bipolar manic patients. J Psychiatr Res 2005;39:355–363. [DOI] [PubMed] [Google Scholar]
- 26. Iwata S, Hewlett GH, Gnegy ME. Amphetamine increases the phosphorylation of neuromodulin and synapsin I in rat striatal synaptosomes. Synapse 1997;26:281–291. [DOI] [PubMed] [Google Scholar]
- 27. Szabo ST, Machado‐Vieira R, Yuan P, et al Glutamate receptors as targets of protein kinase C in the pathophysiology and treatment of animal models of mania. Neuropharmacology 2009;56:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang HY, Friedman E. Lithium inhibition of protein kinase C activation‐induced serotonin release. Psychopharmacology (Berl) 1989;99:213–218. [DOI] [PubMed] [Google Scholar]
- 29. Manji HK, Etcheberrigaray R, Chen G, Olds JL. Lithium decreases membrane‐associated protein kinase C in hippocampus: Selectivity for the alpha isozyme. J Neurochem 1993;61:2303–2310. [DOI] [PubMed] [Google Scholar]
- 30. Jordan VC. A current vew of tamoxifen for the treatment of breast cancer. Br J Pharmacol 1994;110:507–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bebchuk JM, Arfken CL, Dolan‐Manji S, Murphy J, Hasanat K, Manji HK. A preliminary investigation of a protein kinase C inhibitor in the treatment of acute mania. Arch Gen Psychiatry 2000;57:95–97. [DOI] [PubMed] [Google Scholar]
- 32. Kulkarni J, Garland KA, Scaffidi A, et al A pilot study of hormone modulation as a new treatment for mania in women with bipolar affective disorder. Psychoneuroendocrinology 2006;31:543–547. [DOI] [PubMed] [Google Scholar]
- 33. Yildiz A, Guleryuz S, Ankerst DP, Ongur D, Renshaw PF. Protein kinase C inhibition in the treatment of mania: A double‐blind, placebo‐controlled trial of tamoxifen Arch Gen Psychiatry 2008;65:255–263. [DOI] [PubMed] [Google Scholar]
- 34. Zarate CA, Jr. , Singh JB, Carlson PJ, Quiroz J, Jolkovsky L, Luckenbaugh DA, Manji HK. Efficacy of a protein kinase C inhibitor (tamoxifen) in the treatment of acute mania: A pilot study. Bipolar Disord 2007;9:561–570. [DOI] [PubMed] [Google Scholar]
- 35. Sabioni P, Baretta IP, Ninomiya EM, Gustafson L, Rodrigues AL, Andreatini R. The antimanic‐like effect of tamoxifen: Behavioural comparison with other PKC‐inhibiting and antiestrogenic drugs. Prog Neuropsychopharmacol Biol Psychiatry 2008;32:1927–1931. [DOI] [PubMed] [Google Scholar]
- 36. Einat H, Yuan P, Szabo ST, Dogra S, Manji HK. Protein kinase C inhibition by tamoxifen antagonizes manic‐like behavior in rats: Implications for the development of novel therapeutics for bipolar disorder. Neuropsychobiology 2007;55:123–131. [DOI] [PubMed] [Google Scholar]
- 37. Andersen ML, D’Almeida V, Ko GM, Kawakami R, Martins PJF, Magalhães LE, Tufik S. Procedimentos Experimentais In: UNIFESP , editor. Princípios Éticos e Práticos do uso de Animais de Experimentação. São Paulo: UNIFESP, 2004;45–69. [Google Scholar]
- 38. Zager A, Andersen ML, Lima MM, Reksidler AB, Machado RB, Tufik S. Modulation of sickness behavior by sleep: The role of neurochemical and neuroinflammatory pathways in mice. Eur Neuropsychopharmacol 2009;19:589–602. [DOI] [PubMed] [Google Scholar]
- 39. Broadhust P. Experiments in psychogenetics In: Eisenk HJ, editors. Experiments in personality. London : Routledge & K Paul, 1960. [Google Scholar]
- 40. Calzavara MB, Andersen ML, Fukushiro DF, Lopez GB, Abilio VC, Tufik S, Frussa‐Filho R. Sleep rebound attenuates context‐dependent behavioural sensitization induced by amphetamine. Prog Neuropsychopharmacol Biol Psychiatry 2008;32:1277–1282. [DOI] [PubMed] [Google Scholar]
- 41. Frussa‐Filho R, Goncalves MT, Andersen ML, de Araujo NP, Chinen CC, Tufik S. Paradoxical sleep deprivation potentiates amphetamine‐induced behavioural sensitization by increasing its conditioned component. Brain Res 2004;1003:188–193. [DOI] [PubMed] [Google Scholar]
- 42. Einat H, Chen G, Manji H. Possible involvement of protein kinase C in the pathophysiology and treatment of bipolar disorder. Harefuah 2004;143:420–562. [PubMed] [Google Scholar]
- 43. Suchecki D, Lobo LL, Hipolide DC, Tufik S. Increased ACTH and corticosterone secretion induced by different methods of paradoxical sleep deprivation. J Sleep Res 1998;7:276–281. [DOI] [PubMed] [Google Scholar]
- 44. Yildis‐Yesiloglu A. Targeted treatment strategies in mania: Anti‐manic effects of a PKC inhibitor tamoxifen. Biol Psychiatry 2007;61:1S–266S. [Google Scholar]
- 45. Schloesser RJ, Huang J, Klein PS, Manji HK. Cellular plasticity cascades in the pathophysiology and treatment of bipolar disorder. Neuropsychopharmacology 2007;33:110–133. [DOI] [PubMed] [Google Scholar]
- 46. Zarate C, Singha J, Manji H. Cellular plasticity cascades: Targets for the development of novel therapeutics for bipolar disorder. Biol Psychiatr 2006;59:1006–1020 [DOI] [PubMed] [Google Scholar]
- 47. Einat H, Manji HK. Cellular plasticity cascades: Genes‐to‐behavior pathways in animal models of bipolar disorder. Biol Psychiatry 2006;59:1160–1171. [DOI] [PubMed] [Google Scholar]
- 48. Manji HK, Lenox RH. Signaling: Cellular insights into the pathophysiology of bipolar disorder. Biol Psychiatry 2000;48:518–530. [DOI] [PubMed] [Google Scholar]
- 49. Manji H. The role of synaptic and cellular plasticity cascades in the pathophysiology and treatment of bipolar disorder. Actas Esp Psiquiatr 2008;36(Suppl 1):1–2. [DOI] [PubMed] [Google Scholar]