

Summary
Aim
Postoperative cognitive dysfunction (POCD) is a growing and largely underestimated problem without defined etiology. Herein, we sought to determine the relationship between cognitive decline, blood–brain barrier (BBB) permeability, and inflammation, namely high mobility group box‐1 (HMGB1), after surgery in aged rats.
Methods
Aged rats were randomly assigned as surgery group (n = 45, splenectomy under general anesthesia), anesthesia (n = 45, 2% isoflurane for 2 h), and naïve control (n = 15). Markers of inflammation were measured in plasma and brain. Blood–brain barrier ultrastructure and permeability were measured by transmission electron microscope (TEM) and IgG immunohistochemistry. Cognitive function was assessed in a reversal learning version of the Morris water maze (MWM).
Results
Surgical trauma under general anesthesia caused distinct changes in systemic and central proinflammatory cytokines. Levels of HMGB1 and the receptor for advanced glycation end products (RAGE) were significantly upregulated in the hippocampus of operated animals. Immunohistochemistry and TEM showed BBB disruption induced by surgery and anesthesia. These molecular changes were associated with cognitive impairment in latency with the MWM up to postoperative day 3.
Conclusions
HMGB1 and RAGE signaling appear pivotal mediators of surgery‐induced cognitive decline and may contribute to the changes in BBB permeability after peripheral surgical trauma.
Keywords: Aging, Blood–brain barrier (BBB), High mobility group box‐1 (HMGB1), Postoperative cognitive dysfunction (POCD), Surgery
Introduction
Postoperative cognitive dysfunction (POCD) is a common complication following surgery and hospitalization, frequently seen among elderly patients 1. With a steady increase in the surgical geriatric population, postoperative cognitive decline is rapidly becoming a major global health burden 2. 25–50% of hospitalized patients experience confusion and cognitive disturbances affecting multiple domains including learning and memory, orientation, language comprehension, abstract thinking, executive functioning, social integration, and concentration following major surgical procedures 3, 4. About 40% of hospitalized patients retain cognitive impairment after 5 years from the operation, and a link between surgery, anesthesia, and permanent dementia has been recently proposed 5, 6, 7.
Although the mechanisms underlying the pathophysiology of POCD are not well understood, some risk factors including advanced age, low level of education, comorbidities, duration of anesthesia, second operation, postoperative pain, and hospitalization have been reported from clinical studies, with age the only univocal risk factor 3, 8. Recently, a relation between cognitive decline and inflammation has been suggested. In preclinical models, surgical trauma has been associated with neuroinflammation, cytokine release, and memory dysfunction 9, 10, 11, 12, 13. In the present study, we have focused on the role of high mobility group box‐1 (HMGB1), a classical danger‐associated molecular pattern (DAMP) with a pivotal role in mediating acute damage response and subsequent inflammatory processes 14, in response to anesthesia and aseptic surgical trauma. Modulation of HMGB1 and its cognate receptor for advanced glycation end products (RAGE) have been reported in several disease states, ranging from infection to chronic inflammation and cancer 15, 16. Studies have shown that HMGB1 might play a role in neuroinflammation 17, 18, yet there is limited evidence on how HMGB1 contributes to CNS inflammation, blood–brain barrier (BBB) dysfunction, and cognitive decline.
In this study, we used a surgical model in aged rats to define the role of HMGB1 in mediating surgery‐induced cognitive dysfunction and further define the immune‐to‐brain signaling, in particular changes in the BBB, that may associate with the process of cognitive decline.
Methods
Animals
Experiments were performed in accordance with the guidelines for experimental animal use established by the ethics committee of the Central South University. A total of 105 female Sprague Dawley (SD) aged rats (22–23 months old, 450–560 g) were used in the study. Animals were purchased from the Dongchuang Laboratory animal center (Changsha, Hunan, China) and housed in standard cages under controlled laboratory conditions (temperature of 23 ± 3°C, 12‐h light/12‐h dark cycle) with free access to water and laboratory chow. All rats were allowed to adapt to their new environment for 7 days before beginning the experiments. Rats were randomly divided into seven groups: control (C, n = 15), anesthesia only (A1, A3, A7 n = 15/group), anesthesia plus surgery (S1, S3, S7 n = 15/group). Rats were sacrificed at posttreatment days 1, 3, and 7.
Experimental Procedures
Rats were exposed to isoflurane anesthesia (3% isoflurane in 100% oxygen for induction followed by 2% for maintenance) for 2 h with endotracheal intubation with a 14‐gauge catheter. Animals breathed spontaneously and were extubated as the loss of righting reflex (LORR) was regained. Gas concentrations and respiratory rate were continuously monitored with a multi‐function monitor (Datex‐Ohmeda, Helsinki, Finland), rectal temperature was controlled to 37 ± 0.5°C, and heart rate (HR) and mean arterial blood pressure (MAP) were monitored non invasively using a rat tail sphygmomanometer (Midwest Co. Ltd., Beijing, China). Pulse oximeter oxygen saturation (SpO2) was measured continuously (GE Healthcare, Finland). Animals in the anesthesia plus surgery groups underwent splenectomy under aseptic conditions as previously described 19. This standardized procedure delivered similar injury to every animal in order to assess remote effects of trauma to the CNS. Briefly, a small incision about 2–3 cm was made in the upper left quadrant through the skin and muscle wall. The spleen was visualized, isolated, and removed. The wound was infiltrated with 0.25% bupivacaine then closed with sterile sutures. Rats in the control group were exposed to 100% oxygen for 2 h in a gas chamber.
Plasma Cytokine Measurement (ELISA)
Blood samples were collected transcardially after thoracotomy, following centrifugation at 1346 g for 10 min at 4°C. Plasma samples were stored at −80°C before use. Levels of tumor necrosis factor (TNF‐α) and HMGB1 were measured by ELISA following manufacturer's instructions (R&D Systems, Minneapolis, MN, USA).
Brain Cytokine mRNA Extraction and Reverse‐Transcription PCR (RT‐PCR)
Hippocampi were quickly dissected and frozen in liquid nitrogen for further use. For PCR, total RNA was extracted from homogenized hippocampal tissues using Trizol (Invitrogen, Carlsbad, CA, USA) following manufacturer's instruction. RT‐PCR was performed using standard reagents and protocols (Fermentas, Glen Burnie, MD, USA). Gene‐specific primers, forward and reverse, were as follows: 5′‐GAAGCCGAGAGGCAAAATGTCC‐3′ and 5′‐TAGCTCTGTAGGCAGCAATATC‐3′ for HMGB1, 5′‐CCGAGTCTACCAGATTCCTG‐3′ and 5′‐TTCACGAGTGTTTCTTTGCC‐3′ for RAGE, 5′‐TGACCCCCATTACTCTGACC‐3′ and 5′‐GGCCACTACTTCAGCGTCTC‐3′ for TNF‐α, 5′‐CTCCATGAGCTTTGTACAAGG‐3′ and 5′‐TGCTGATGTACCAGTTGGGG‐3′ for IL‐1β, 5′‐GTGGGGCGCCCCAGGCACCA‐3′ and 5′‐CTTCCTTAATGTCACGCACGATT‐3′ for β‐actin. RT‐PCR was performed at 95°C/4 min and then cycling 94°C/30 s, 60°C ± 10°C/30 s, 72°C/30 s, terminated by heating to 72°C/5 min. The products were amplified for 30 cycles. Band densities were quantified using Quality One Software (Bio‐Rad, Hercules, CA, USA). The amount of mRNA was expressed as a ratio of the densitometric measurement derived from target mRNA to that from β‐actin.
Western Blot
Hippocampi were homogenized in cold RIPA buffer and centrifuged at 4°C at 13350 g for 5 min. The quantity of protein in the supernatants was determined using a BCA protein assay kit (Wellbio, China) according to the manufacturer's instructions. Equal amounts of protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred to polyvinylidene fluoride membranes. Membranes were blocked with 5% skim milk TBS buffer for 1 h and then incubated with primary antibodies: mouse anti‐HMGB1 monoclonal (1:1000; Abcam, Cambridge, UK), rabbit anti‐RAGE polyclonal (1:500; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), rabbit anti‐TNF‐α polyclonal (1:1000; Abcam, Cambridge, UK), rabbit anti‐IL‐1β polyclonal (1:1000; Santa Cruz Biotechnology Inc.) overnight at 4°C. After thorough washing, membranes were incubated with goat anti‐mouse (1:3000; Proteintech Group, Wuhan, China) and goat anti‐rabbit (1:3000; Proteintech Group) secondary antibodies at room temperature. Membranes were then treated with enhanced chemiluminescence detection kit (Pierce; Thermo Scientific, Shanghai, China), and the intensity of each band was quantified by densitometry. Relative expression was normalized to β‐actin (1:4000; Proteintech Group).
Immunohistochemistry
Blood–brain barrier damage was assessed by immunoglobulin G immunostaining as previously described 20, 21. Five rats in each group were anesthetized with chloral hydrate (10%) and perfused transcardially with 0.01 M PBS (4°C) followed by 200 mL of ice‐cold 4% paraformaldehyde. The brain was removed and postfixed in 4% paraformaldehyde overnight, rehydrated, and embedded in paraffin. Sections (5 μm) were deparaffinized, preincubated with 3% H2O2 at room temperature for 5 min to block endogenous peroxidase activity, and then washed twice with PBS. Sections were blocked in 3% normal goat serum at room temperature for 1 h followed by incubation with rabbit anti‐rat IgG antibody (1:200; Vector Laboratories, Burlingame, CA, USA) at 4°C overnight and incubated with avidin–biotin–HRP complex (ABC) reagent (1:200; Vector Laboratories) for 2 h and developed using DAB method. The sections were rinsed and mounted.
For HMGB1 immunofluorescence, sections were blocked in 3% normal horse serum and incubated with a mouse anti‐HMGB1 antibody (1:200; Abcam, Cambridge, UK) at 4°C overnight. Sections were then incubated with horse anti‐mouse secondary antibodies (Vector Laboratories) and Cy3‐streptavidin (1:100; Jackson Immuno Labs, West Grove, PA, USA) for 2 h. Images were acquired with a Nikon eclipse 80i microscope (Nikon, Tokyo, Japan). Assessments were performed in a blinded fashion.
Transmission Electron Microscopy (TEM)
Hippocampi were quickly dissected, fixed in 2.5% glutaraldehyde in 0.1 mol/L cacodylic acid buffer (pH 7.3) overnight, and postfixed in the 2% Osmic Acid for 2 h. The fixed brain was dehydrated through an ethanol series soaked in the mixed liquor of epoxy resin and acetone for 24 h, 37°C, following embedding in Epon812, DDSA, DMP30 for 24 h, cut into semithin sections, and stained with toluidine blue to find the position. Slice were then cut into ultra‐thin sections with an ultramicrotome (LKB‐III, Sweden) and mounted on copper grids, stained with uranyl acetate and plumbi nitras. Specimens were observed under a transmission electron microscope (Hitachi H‐7500; Hitachi, Tokyo, Japan).
Morris Water Maze (MWM)
Spatial learning and memory were evaluated in the MWM using a computerized video track system (Smart Junior Software, Panlab, Spain) as previously described 11. Briefly, a transparent round platform of 10 cm in diameter was placed 1 cm below the water surface and located in the center of the SE quadrant in a circular pool. The rats were placed on the platform for 30 s preceding the start of each trial and then released into the water facing the tank wall in one of the three present entry locations. Rats were trained for 6 days with three trials per day (60 s maximum for a trial, 30 s spent on the platform). If the platform was not located within 60 s, the rats were gently guided to the platform and remained for 15 s. On day 7, animals underwent surgery and/or general anesthesia; on posttreatment days 1, 3 and 7, rats were subjected to a reversal test in which the platform was moved to the opposite quadrant of the pool (NW). The time to reach the platform (latency) and distance was recorded by a video track system (Logitech, Suzhou, China).
Statistical Analysis
Results are expressed as mean ± SD, two‐way repeated measures ANOVA was used to analyze training behavioral parameters, and a separate two‐way ANOVA was used for reversal testing. ELISA, RT‐PCR, and Western blot were analyzed with two‐way ANOVA, in which time and treatment were dependent variables. Post hoc Bonferroni was performed when ANOVA showed significance. All statistical data were analyzed using SPSS 17.0. (SPSS, Shanghai, China) A probability value of 0.05 was considered to be statistically significant.
Results
Surgery Upregulates Systemic Levels of HMGB1
To evaluate the effects of general anesthesia and surgical trauma on aged rats, we measured systemic levels of TNF‐α and HMGB1. No changes in plasma TNF‐α were observed on postoperative days 1, 3, and 7 (Figure 1A). However, levels of HMGB1 were significantly upregulated after surgery on postoperative day 1 (0.204 ± 0.023 μg/L, P = 0.003), before returning to baseline. Analysis of HMGB1 plasma levels by two‐way ANOVA revealed significant effects of time (P = 0.001), treatment (P = 0.004), and time × treatment interaction (P = 0.170; Figure 1B). Exposure to anesthesia did not affect systemic levels of these cytokines. Animals had no signs of cyanosis, hypoxia, respiratory depression, or hypotension. Heart rates, MAP, and SpO2 were 343.5 ± 35.09 beats/min, 91.60 ± 12.3 mmHg, and 98.7 ± 1.13%, respectively.
Figure 1.
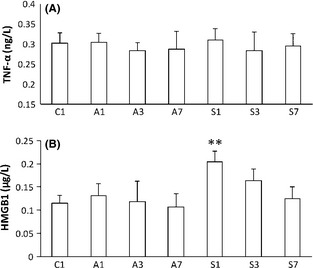
Systemic changes in proinflammatory cytokines measured by ELISA after anesthesia and surgery. (A) No changes from baseline were observed in tumor necrosis factor (TNF‐α) at all time points. (B) HMGB1 was significantly upregulated after surgery on postoperative day 1, before returning to baseline. No changes were measured after exposure to isoflurane only. Bars denote means ± SD, n = 10. *P < 0.05, **P < 0.01 versus control by two‐way ANOVA followed by Bonferroni. C: control; A1, A3, and A7: anesthesia at days 1, 3, and 7; S1, S3, and S7: surgery at days 1, 3, and 7.
Anesthesia and Surgery Impair the Blood–Brain Barrier of Aged Rats
Next, we investigated the effects of HMGB1 on the brain and the BBB. Proinflammatory cytokines modulate the CNS both via direct and indirect pathways 22. Here, we interrogated the BBB by measuring IgG expression in the brain parenchyma. IgG‐positive staining was pivotally observed in the hippocampus and was widely distributed around the CA1 area after surgical treatment, with some leakage visible also in other brain areas such as the hypothalamus (Figure 2 S1). Milder changes were seen at day 1 after exposure to isoflurane compared to naïve rats (Figure 2 A1). To assess the functionality of the neurovascular unit after surgery, we used transmission electron microscopy. Operated rats had impairments in the BBB at postoperative day 1, consisting of swollen vessels and expanded perivascular space. Furthermore, astrocytes end‐feet was enlarged, and detachment of the end‐feet plasma membrane from the basal lamina was also observed after surgery (Figure 3S). Mild changes were also noted at day 1 after exposure to anesthesia compared to naïve rats (Figure 3A and C).
Figure 2.
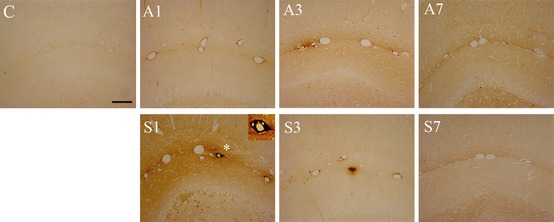
Surgery increases the permeability of the blood–brain barrier (BBB) in aged rats. Hippocampi were harvested at days 1, 3, and 7, and BBB permeability was measured with IgG immunohistochemistry. Picture show CA1 (scale bar = 400 μm) and photomicrographs were blindly scored. IgG expression was increased around the blood vessels in the hippocampus. Exposure to anesthesia caused mild changes in IgG compared to naïve rats at day 1. Significant IgG‐positive staining surrounding the lumen of blood vessels and the brain parenchyma was noted in the surgical group up to postoperative day 3. n = 5, C: control; A1, A3, and A7: anesthesia at days 1, 3, and 7; S1, S3, and S7: surgery at days 1, 3, and 7.
Figure 3.
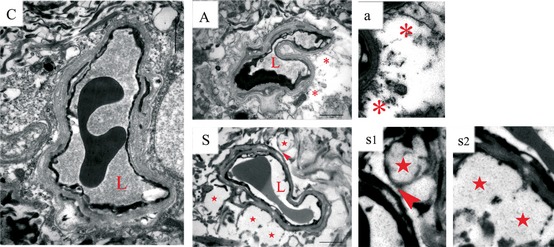
Anesthesia and surgery disrupt the structure of the blood–brain barrier (BBB) at 24 h. Ultrastructure of the neurovascular unit was observed at 24 h after surgery and/or anesthesia using transmission electron microscope. (C) Representative images from a coronal section through the hippocampus of a naïve rat show a normal BBB structure. (A) Exposure to isoflurane for 2 h caused minor changes in the BBB ultrastructure, with mild and localized swelling, expanded perivascular space (*). (S) Swelling in astrocyte end‐feet (★) and detachment (arrowhead) was observed after surgery. n = 5, C: control; A: anesthesia at day 1. S: surgery at day 1. L: lumen of blood vessel. Scale bar = 2 μm.
Blood–Brain Barrier Damage Leads to Neuroinflammation and de novo Synthesis of Cytokine in the Brain
To understand the effects of anesthesia and surgery on the brain, we measured transcription and translation of key proinflammatory cytokines including TNF‐α, IL‐1β, and HMGB1. Even though we could not detect changes in systemic TNF‐α, both mRNA levels and protein expression in hippocampal tissues were increased after exposure to anesthesia at day 1 (mRNA: 0.683 ± 0.037; protein: 0.952 ± 0.042) or surgery at day 1 (mRNA: 0.855 ± 0.035; protein: 1.045 ± 0.055) and day 3 (mRNA: 0.75 ± 0.059; protein: 0.907 ± 0.049; Figure 4A, P < 0.001). Levels of TNF‐α and IL‐1β remained elevated in the surgical group up to postoperative day 3 (IL‐1β mRNA: 0.656 ± 0.049; protein: 0.906 ± 0.042; P < 0.001 respectively), then returning to baseline (Figure 4A and B). To understand the specific effects of HMGB1 on the brain, we looked at the receptor for advanced glycation end products (RAGE), a key receptor for HMGB1 signaling. Both HMGB1 and RAGE mRNA levels and protein expression were significantly increased in the hippocampus of operated rats and anesthesia exposure after day 1 (Figure 4C and D, P < 0.001). RAGE and HMGB1 levels in operated rats remained upregulated at day 3 before returning to baseline. The variations of HMGB1 mRNA and protein levels were 0.592 ± 0.023 and 0.782 ± 0.033 at day 1 after anesthesia; 0.613 ± 0.033 and 0.943 ± 0.071 at day 1 after surgery, respectively; and 0.526 ± 0.041 and 0.799 ± 0.045 at day 3 after surgery, respectively. Levels of RAGE mRNA expression and protein were 0.491 ± 0.045 and 1.04 ± 0.096 at day 1 after anesthesia; 0.544 ± 0.033 and 1.111 ± 0.069 at day 1 after surgery; and 0.47 ± 0.035 and 0.891 ± 0.036 at day 3 after surgery, respectively. To further corroborate the increase of HMGB1 in the brain, we used immunofluorescence. HMGB1‐positive staining around the perivascular space and brain parenchyma was observed in the hippocampus of operated and anesthetized rats at day 1. Operated rats displayed higher HMGB1 in CA1 up to postoperative day 3, returning to baseline by day 7 (Figure 5).
Figure 4.
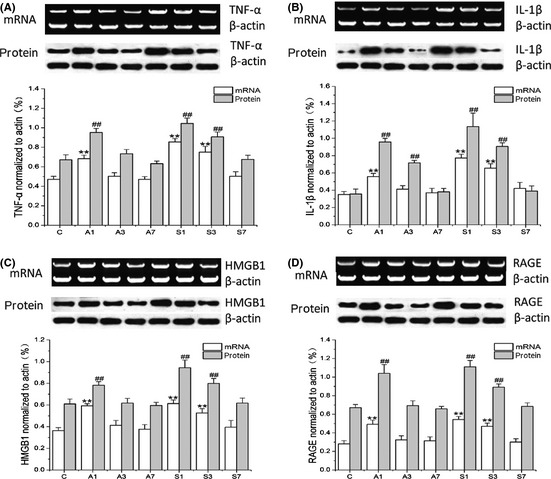
Surgery causes neuroinflammation and activates RAGE signaling. Messenger RNA and protein expressions of key proinflammatory mediators were measured in the hippocampus with reverse‐transcription polymerase chain reaction (RT‐PCR) and immunoblotting, respectively. High mobility group box‐1 (C) and its signaling receptor RAGE (D) were significantly increased after exposure to anesthesia (day 1) or surgery (days 1 and 3). Tumor necrosis factor (TNF‐α) (A) and IL‐1β (B) were also upregulated retuning to baseline at day 7. Densitometry data were compared to the respective housekeeping gene (β actin). Bars represent mean ± SD. **P < 0.01 versus control mRNA levels and ## P < 0.01 versus control protein levels by two‐way ANOVA followed by Bonferroni. n = 5, C: control; A1, A3, and A7: anesthesia at days 1, 3, and 7; S1, S3, and S7: surgery at days 1, 3, and 7.
Figure 5.
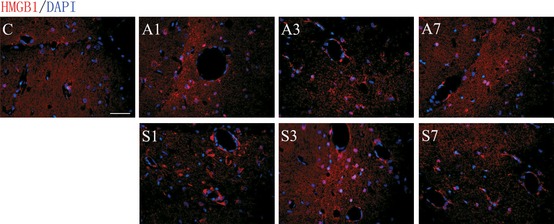
Expression of HMGB1 in the CA1 region of hippocampus in operated rats. HMGB1 was measured using immunofluorescence at days 1, 3, and 7 after surgery and anesthesia. (C) Representative photomicrographs show a low expression of HMGB1 in naïve rats. (A1) Isoflurane induced HMGB1 in the CA1 region of the hippocampus at day 1, returning to baseline thereafter. (S1, S3) Operated rats displayed higher HMGB1 staining in the brain parenchyma and around the vasculature up to postoperative day 3, returning to baseline at day 7. n = 5, C: control; A1, A3, and A7: anesthesia at days 1, 3, and 7; S1, S3, and S7: surgery at days 1, 3, and 7. Scale bar = 100 μm
Anesthesia and Surgery Cause Distinct Neurocognitive Impairments
To elucidate the effect of surgery and anesthesia on spatial learning and reference memory, we used the Morris water maze (Figure 6A). No significant differences in latency and swimming speed were reported throughout the groups, and all animals progressively improved over time during the acquisition phase (Figure 6B and C). Repeated measures ANOVA revealed that the latency significantly improved over time (P < 0.001). A separate two‐way ANOVA examined the effects of time and treatment on spatial memory performance. Analysis of the latency to the platform revealed significant effects of time (P < 0.001), treatment (P < 0.001), and time × treatment interaction (P = 0.037). No changes were reported in swimming speed (P = 0.605; Figure 6D and E). Notably, latency was impaired at day 1 after anesthesia (35.99 ± 7.70 s, P = 0.001) and up to postoperative day 3 in the surgical group (44.54 ± 6.63 s at day 1 and 38.30 ± 6.25 s at day 3, P < 0.001 respectively; Figure 6D and E).
Figure 6.
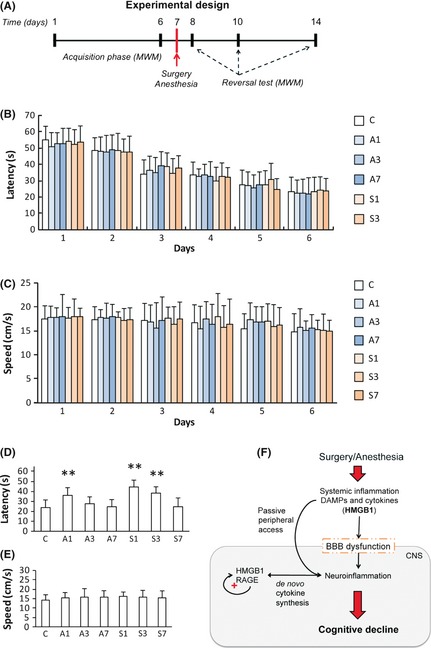
Spatial memory and learning are impaired after anesthesia and surgery. (A) Diagram of the experimental design for the neurocognitive assessment with the Morris water maze. (B) The time required for each animal to reach the hidden platform (latency) and (C) speed during the 6 days of training in the acquisition phase. (D) Anesthesia induced spatial learning and memory impairment in the reversal test at day 1. Similarly, operated rats required significantly longer time to locate the submerged platform at postoperative days 1 and 3. (E) Average swimming speed during reversal testing did not change across groups. (F) Working model: surgical trauma with general anesthesia evokes a systemic inflammatory response in aged rats. Damage‐associated molecular pattern (DAMPs), including HMGB1, are released in the systemic circulation contributing to the permeability of the blood–brain barrier (BBB). Opening of the BBB allows peripheral cytokines and alarmins to enter the brain and the CNS, causing neuroinflammation. In the hippocampus, HMGB1 activates RAGE signaling thus perpetuating the inflammatory response and further contributing to neuroinflammation, including de novo synthesis of cytokines like TNF‐α and IL‐1β. Activation of this positive feedback loop accounts for memory dysfunction and possibly localized synaptic/neuronal deficits. Bars denote mean ± SD. **P < 0.01 versus control by two‐way ANOVA followed by Bonferroni. n = 15, C: control; A1, A3, and A7: anesthesia at days 1, 3, and 7; S1, S3, and S7: surgery at days 1, 3, and 7.
Discussion
In the present study, we have explored the role of HMGB1 in the development of postoperative cognitive decline. General anesthesia, but more evidently surgical trauma, caused distinct changes in levels of HMGB1 leading to BBB disruption, subsequent neuroinflammation, and memory impairment in aged rats. Overall, these results suggest a novel role for DAMPs signaling in mediating surgery‐induced cognitive decline after aseptic trauma and anesthesia (Figure 6F).
Postoperative cognitive dysfunction is currently a poorly understood disorder with undefined etiology and no treatment. Anesthesia has been historically associated with cognitive decline and neurotoxicity, for example during development 23. Isoflurane, a commonly used anesthetic agent, was shown to increase cytokine expression and cell injury in brain areas such as the hippocampus, thus causing cognitive dysfunction in adult rats 24, 25. In the present study, we found that a single exposure to a clinically relevant concentration of isoflurane with airway intubation impaired learning and memory, suggesting that anesthesia per se may play a role in the development of cognitive decline, especially in aged animals. In a limited study of 438 patients, no significant changes in the incidence of POCD were reported when comparing general versus regional anesthesia; however, regional anesthesia decreased mortality and acute cognitive failure in elderly patients 26. Aside anesthesia, cytokine release, and neuroinflammation have been associated with sickness behavior and cognitive disturbances, for example after infection and more recently following aseptic surgery 27, 28. Cytokines have been described as a “double‐edged sword” with a pivotal role in protecting against harmful organism/pathogens and supporting homeostasis, but with potentially lethal effects when they become uncontrolled and dysregulated, for example in septic shock 29. Proinflammatory molecules are promptly released in the systemic circulation after tissue damage and trauma; TNF‐α systemic levels peak within minutes after orthopedic surgery, 10 but following splenectomy, we did not observe significant changes that persisted over days (Figure 1A). Yet, we found increased transcription and translation in the hippocampus of operated rats up to postoperative day 3, confirming a pivotal role for cytokines, including TNF‐α and IL‐1β in mediating cognitive dysfunction (Figure 4A and B). Furthermore, levels of HMGB1 and its cell‐surface receptor RAGE were also increased (Figure 4C and D), suggesting a possible role for DAMPs signaling in mediating neuroinflammation, BBB disruption, and cognitive decline after surgery.
High mobility group box‐1 is a highly conserved nuclear protein with a pivotal role in mediating acute damage response 30. In stroke models, HMGB1 was induced in activated astrocytes and microvascular structure, inducing alterations in endothelia cell function and increasing BBB permeability 31. Astrocytes and endothelial cells integrity play a key role in maintaining a patented BBB by controlling its permeability and overall integrity. Administration of HMGB1 neutralizing antibody in brain ischemia–reperfusion injury also limits the damage on the BBB and may represent a viable therapeutic target also after peripheral trauma 32. Recently, Chavan et al. described a role for HMGB1 in mediating cognitive decline among sepsis survivor 33; changes in systemic HMGB1 and cognitive impairments were also observed after lipopolysaccharide administration in mice 34. HMGB1 can interact with various receptors including RAGE but also toll‐like receptor (TLR)‐2 and TLR‐4 to mediate chemotaxis and release of proinflammatory cytokines 35. Interestingly, HMGB1 has been reported to cause lethality in TLR4‐defective mice 36, but on the other hand, TLR4‐dependent signaling does not appear to be required for the establishment of cognitive decline after aseptic surgery 10. Here, we described a key role for RAGE signaling and its upregulation in the brain and hippocampal tissue suggesting a pivotal role in causing neuroinflammation and cognitive dysfunction. Remarkably, RAGE has been described as an amplifier of the immune and inflammatory response 37; hence, it may also play a key role in the pathophysiology of neuroinflammation and cognitive decline.
Animal studies consistently show that systemic proinflammatory cytokines communicate with the brain, possibly by impairing BBB function or via neuronal signaling 38, 39, 40. The BBB is a complex cellular gateway that regulates the transport of molecules into the brain and CNS; loss of BBB function is an hallmark of many neurological diseases such as Alzheimer's disease (AD) and multiple sclerosis (MS) 41. A recent review indicates that aging, hypertension, diabetes, cholesterol, and inflammation can impair and disrupt the BBB, ultimately resulting in neurodegeneration and cognitive decline 42, 43. Recent studies have also implicated that aging deteriorates the structure and function of the BBB and together with an proinflammation systemic milieu may account for the neuronal loss and cognitive decline during aging 44, 45.
To date, there is little direct evidence whether HMGB1 and BBB dysfunction could contribute to the cognitive impairment, especially when the injury is on a peripheral organ and not directly to the brain. Our results indicate that surgical trauma, and to a smaller extent anesthesia, induced spatial and learning impairment in aged rats (Figure 6D). It is possible that other factors, including general anesthesia, adjuvate this process by opening the BBB, thus facilitating neuroinflammatory processes and cognitive decline as we observed after day 1 (Figure 3) and up to day 3 (Figure 5). Other groups have reported a progressive breakdown of the BBB after isoflurane exposure 46, 47. The effects of anesthetics, surgical trauma, and preexisting comorbidities may actively contribute to the perioperative cognitive failure as often seen in our intensive care units (ICU) 48.
Some limitations of our study must be pointed out. The relationship between HMGB1 and cognitive dysfunction cannot be completely ascertained as we did not selectively block this molecule, for example using monoclonal neutralizing antibodies. Also, the mechanisms whereby the BBB is impaired after surgery require further understanding, and in this context, neuronal pathways may also contribute to the neuroinflammatory response. Recently, infiltration of peripheral macrophages through a damaged BBB was observed after orthopedic surgery, and stimulation of the cholinergic antiinflammatory pathway improved cognitive outcome 40. Indeed, animal models cannot completely reproduce the clinical situation and its complexity; it remains essential to confirm whether similar changes occur in patients after surgery.
Conclusion
We have demonstrated that proinflammatory cytokines including HMGB1, TNF‐α, IL‐β, and RAGE signaling were upregulated by anesthesia and surgical trauma. This appears to contribute to the neuronal damage and cognitive dysfunction in aged rats. Increased BBB permeability seems a key hallmark in the development of neuroinflammation and cognitive decline. HMGB1 may represent a novel viable target to interrupt and modulate the inflammatory response underlying the pathogenesis of POCD.
Conflict of Interest
The authors have no conflict of interest.
Acknowledgments
This work was funded by National Natural Science Foundation of China (No. 30871306), the 125 Personnel Training Foundation of Third Xiangya Hospital, and the Science‐Technology Foundation of Hunan Province, China (2010sk3111).
References
- 1. Terrando N, Brzezinski M, Degos V, et al. Perioperative cognitive decline in the aging population. Mayo Clin Proc 2011;86:885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Newman S, Stygall J, Hirani S, Shaefi S, Maze M. Postoperative cognitive dysfunction after noncardiac surgery: a systematic review. Anesthesiology 2007;106:572–590. [DOI] [PubMed] [Google Scholar]
- 3. Moller JT, Cluitmans P, Rasmussen LS, et al. Long‐term postoperative cognitive dysfunction in the elderly ISPOCD1 study. ISPOCD investigators. International Study of Post‐Operative Cognitive Dysfunction. Lancet 1998;351:857–861. [DOI] [PubMed] [Google Scholar]
- 4. Monk TG, Weldon BC, Garvan CW, et al. Predictors of cognitive dysfunction after major noncardiac surgery. Anesthesiology 2008;108:18–30. [DOI] [PubMed] [Google Scholar]
- 5. Lyketsos CG, Toone L, Tschanz J, et al. A population‐based study of the association between coronary artery bypass graft surgery (CABG) and cognitive decline: the Cache County study. Int J Geriatr Psychiatry 2006;21:509–518. [DOI] [PubMed] [Google Scholar]
- 6. Tang JX, Baranov D, Hammond M, Shaw LM, Eckenhoff MF, Eckenhoff RG. Human Alzheimer and inflammation biomarkers after anesthesia and surgery. Anesthesiology 2011;115:727–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xie Z, Tanzi RE. Alzheimer's disease and post‐operative cognitive dysfunction. Exp Gerontol 2006;41:346–359. [DOI] [PubMed] [Google Scholar]
- 8. Krenk L, Rasmussen LS, Kehlet H. New insights into the pathophysiology of postoperative cognitive dysfunction. Acta Anaesthesiol Scand 2010;54:951–956. [DOI] [PubMed] [Google Scholar]
- 9. Cibelli M, Fidalgo AR, Terrando N, et al. Role of interleukin‐1beta in postoperative cognitive dysfunction. Ann Neurol 2010;68:360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Terrando N, Monaco C, Ma D, Foxwell BM, Feldmann M, Maze M. Tumor necrosis factor‐alpha triggers a cytokine cascade yielding postoperative cognitive decline. Proc Natl Acad Sci USA 2010;107:20518–20522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rosczyk HA, Sparkman NL, Johnson RW. Neuroinflammation and cognitive function in aged mice following minor surgery. Exp Gerontol 2008;43:840–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vizcaychipi MP, Lloyd DG, Wan Y, Palazzo MG, Maze M, Ma D. Xenon pretreatment may prevent early memory decline after isoflurane anesthesia and surgery in mice. PLoS One 2011;6:e26394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kamer AR, Galoyan SM, Haile M, et al. Meloxicam improves object recognition memory and modulates glial activation after splenectomy in mice. Eur J Anaesthesiol 2012;29:332–337. [DOI] [PubMed] [Google Scholar]
- 14. Yang H, Wang H, Czura CJ, Tracey KJ. The cytokine activity of HMGB1. J Leukoc Biol 2005;78:1–8. [DOI] [PubMed] [Google Scholar]
- 15. Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 2011;29:139–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol 2010;28:367–388. [DOI] [PubMed] [Google Scholar]
- 17. Maroso M, Balosso S, Ravizza T, et al. Toll‐like receptor 4 and high‐mobility group box‐1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med 2010;16:413–419. [DOI] [PubMed] [Google Scholar]
- 18. Pedrazzi M, Patrone M, Passalacqua M, et al. Selective proinflammatory activation of astrocytes by high‐mobility group box 1 protein signaling. J Immunol 2007;179:8525–8532. [DOI] [PubMed] [Google Scholar]
- 19. Wan Y, Xu J, Ma D, Zeng Y, Cibelli M, Maze M. Postoperative impairment of cognitive function in rats: a possible role for cytokine‐mediated inflammation in the hippocampus. Anesthesiology 2007;106:436–443. [DOI] [PubMed] [Google Scholar]
- 20. Bake S, Friedman JA, Sohrabji F. Reproductive age‐related changes in the blood brain barrier: expression of IgG and tight junction proteins. Microvasc Res 2009;78:413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Greenhalgh AD, Galea J, Denes A, Tyrrell PJ, Rothwell NJ. Rapid brain penetration of interleukin‐1 receptor antagonist in rat cerebral ischaemia: pharmacokinetics, distribution, protection. Br J Pharmacol 2010;160:153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther 2011;130:226–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stratmann G. Review article: Neurotoxicity of anesthetic drugs in the developing brain. Anesth Analg 2011;113:1170–1179. [DOI] [PubMed] [Google Scholar]
- 24. Lin D, Zuo Z. Isoflurane induces hippocampal cell injury and cognitive impairments in adult rats. Neuropharmacology 2011;61:1354–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Culley DJ, Yukhananov RY, Xie Z, Gali RR, Tanzi RE, Crosby G. Altered hippocampal gene expression 2 days after general anesthesia in rats. Eur J Pharmacol 2006;549:71–78. [DOI] [PubMed] [Google Scholar]
- 26. Rasmussen LS, Johnson T, Kuipers HM, et al. Does anaesthesia cause postoperative cognitive dysfunction? A randomised study of regional versus general anaesthesia in 438 elderly patients Acta Anaesthesiol Scand 2003;47:260–266. [DOI] [PubMed] [Google Scholar]
- 27. Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 2008;9:46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eckenhoff RG, Laudansky KF. Anesthesia, surgery, illness and Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry, 2012. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oppenheim JJ, Feldmann M, Durum SK. Cytokine reference: a compendium of cytokines and other mediators of host defense. San Diego: Academic Press, 2001. [Google Scholar]
- 30. Bianchi M.E. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 2007;81:1–5. [DOI] [PubMed] [Google Scholar]
- 31. Kim JB, Lim CM, Yu YM, Lee JK. Induction and subcellular localization of high‐mobility group box‐1 (HMGB1) in the postischemic rat brain. J Neurosci Res 2008;86:1125–1131. [DOI] [PubMed] [Google Scholar]
- 32. Zhang J, Takahashi HK, Liu K, et al. Anti‐high mobility group box‐1 monoclonal antibody protects the blood‐brain barrier from ischemia‐induced disruption in rats. Stroke 2011;42:1420–1428. [DOI] [PubMed] [Google Scholar]
- 33. Chavan SS, Huerta PT, Robbiati S, et al. HMGB1 Mediates Cognitive Impairment in Sepsis Survivors. Mol Med 2012;18:930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Terrando N, Rei Fidalgo A, Vizcaychipi M, et al. The impact of IL‐1 modulation on the development of lipopolysaccharide‐induced cognitive dysfunction. Crit Care 2010;14:R88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang H, Tracey KJ. Targeting HMGB1 in inflammation. Biochim Biophys Acta 2010;1799:149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang H, Bloom O, Zhang M, et al. HMG‐1 as a late mediator of endotoxin lethality in mice. Science 1999;285:248–251. [DOI] [PubMed] [Google Scholar]
- 37. Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest 2001;108:949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci 2008;28:9451–9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Serres S, Anthony DC, Jiang Y, et al. Systemic inflammatory response reactivates immune‐mediated lesions in rat brain. J Neurosci 2009;29:4820–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Terrando N, Eriksson LI, Ryu JK, et al. Resolving postoperative neuroinflammation and cognitive decline. Ann Neurol 2011;70:986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Daneman R. The blood‐brain barrier in health and disease. Ann Neurol, 2012. (in press). [DOI] [PubMed] [Google Scholar]
- 42. Farrall AJ, Wardlaw JM. Blood‐brain barrier: ageing and microvascular disease–systematic review and meta‐analysis. Neurobiol Aging 2009;30:337–352. [DOI] [PubMed] [Google Scholar]
- 43. Popescu BO, Toescu EC, Popescu LM, et al. Blood‐brain barrier alterations in ageing and dementia. J Neurol Sci 2009;283:99–106. [DOI] [PubMed] [Google Scholar]
- 44. Villeda SA, Luo J, Mosher KI, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011;477:90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zeevi N, Pachter J, McCullough LD, Wolfson L, Kuchel GA. The blood‐brain barrier: geriatric relevance of a critical brain‐body interface. J Am Geriatr Soc 2010;58:1749–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Masamoto K, Fukuda M, Vazquez A, Kim SG. Dose‐dependent effect of isoflurane on neurovascular coupling in rat cerebral cortex. Eur J Neurosci 2009;30:242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tetrault S, Chever O, Sik A, Amzica F. Opening of the blood‐brain barrier during isoflurane anaesthesia. Eur J Neurosci 2008;28:1330–1341. [DOI] [PubMed] [Google Scholar]
- 48. Uchikado H, Akiyama H, Kondo H, et al. Activation of vascular endothelial cells and perivascular cells by systemic inflammation‐an immunohistochemical study of postmortem human brain tissues. Acta Neuropathol 2004;107:341–351. [DOI] [PubMed] [Google Scholar]