

To die or not to die—that is the question. Up to date, there is accumulating evidence that in addition to necrosis, the programmed cell death plays a significant role in acute or delayed neuronal death in the central nervous system (CNS) disorders, such as neurodegenerative diseases and ischemic stroke. After focusing on necrosis and apoptosis as the major causes of neuronal death in the CNS disorders for dozens of years, it has been recognized that elaborating the mechanisms underlying neuronal death is not as easy as it looks. In recent years, autophagy has become one of the most attractive topics in the research of neuronal death of CNS disorders. The term autophagy comes from the Greek roots “auto” (self) and “phagy” (eating) and means self‐digestion. Previously, autophagy has been viewed as type II programmed cell death with a random process because it appears to engulf cytosol indiscriminately; however, it is now recognized as a tightly regulated endogenous biological response to degrade damaged and dysfunctional cellular organelles and protein aggregates.
In neuronal cells, approximately 1% of cellular proteins are catabolized per hour by autophagy, even under nutrient‐rich condition. Although it is not fully understood how much the basal autophagy contributes to macromolecule synthesis and energy production in the steady state by supplying amino acids, glucose, and free fatty acids, the role of basal autophagy acting as the quality‐control machinery for cytoplasmic components is widely accepted 1. Many neurodegenerative disorders, such as Parkinson disease (PD) and Alzheimer's disease (AD), result from proteins that increase their propensity to form aggregates. While autophagy can eliminate aggregated proteins, defective mitochondria, and excessive reactive oxygen species (ROS) that could induce DNA damage and cell death, inadequate or defective autophagy promotes neuronal cell death in most of neurodegenerative disorders. It is becoming increasingly evident that the cell response may shift gradually from elimination of damaged proteins/organelles by autophagy, which leads to its recovery, to the induction of apoptotic or necrotic pathways determining cellular demise under both neurodegenerative disorders and brain ischemia (Figure 1).
Figure 1.
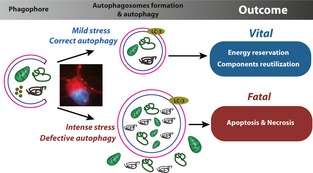
A proposed working model of autophagy in central nervous system disorders.
p62, a hot topic in cell death research in recent years, is a scaffolding protein that binds to polyubiquitin and associated with the nuclear envelope. Previous works reveal that p62 acts as a signaling hub through its ability to recruit and oligomerize important signaling molecules in cytosolic speckles to control cell apoptosis 2. Thus, p62 is thought to be a detrimental player in the cell life 2. However, this opinion is being challenged. Recently, p62 was found to be localized to autophagic compartments via preferential binding with LC3‐II form, which may assist in removal of cellular proteins damaged by dopaminergic neurotoxin 3 and is involved in the decrease of the neuronal cell number under hypoxic stress 4. p62 was also reported to be a potential diagnostic tool for Alzheimer's disease 5. Further, p62 is recruited to mitochondrial clusters and is essential for the clearance of mitochondria in the pathogenesis of Parkinson's disease 6, 7, 8, 9, although there are minor disputes regarding the detailed mechanisms underlying the effect of p62 on the dysfunctional mitochondrial clustering and mitophagy 6, 7, 8, 9. Thus, whether p62 is a detrimental or a beneficial factor for the survival of neuronal cell depends on the pathological insults and the tissue microenvironment and the switch between autophagy and apoptosis, which is regulated by p62.
Autophagy also participates in the cerebral ischemia‐induced injury. Accumulating data supported the critical role of autophagy in cerebral ischemia as well as in neurodegenerative diseases 10, 11, 12. Nevertheless, autophagy may be a double‐edged sword for cerebral ischemia 10, 11, 12. We recently reviewed the beneficial and harmful effects of autophagy in cerebral ischemia 13. In this issue of CNS Neuroscience & Therapeutics, Qi et al. describe a new role for Akt/glycogen synthase kinase 3β (GSK‐3β)‐dependent autophagy in the neuroprotection by limb remote ischemic postconditioning in a transient cerebral ischemic rat model 14. Their study is the first report on autophagy in remote ischemic postconditioning. Ischemic preconditioning is a short period of ischemia followed by a brief period of reperfusion before a sustained ischemic insult, which was found to be a powerful method for limiting cerebral ischemia‐induced tissue damage. Previously, autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning, because blockade of autophagy abolished the neuroprotection of cerebral ischemic preconditioning 15. Moreover, the preactivation of autophagy by ischemic preconditioning boosts endogenous defense mechanisms to upregulate molecular chaperones, and hence reduces excessive endoplasmic reticulum stress during fatal ischemia 16. Qi et al. found that limb remote ischemic postconditioning significantly enhanced the autophagy in neurons of rats, with an increase of the Akt/GSK3β signaling phosphorylation. To examine the role of autophagy induced by limb remote ischemic postconditioning, they blocked the Akt/GSK3β pathway via the LY294002, and found it suppressed the enhancement of autophagy and the neuroprotection by limb remote ischemic postconditioning, suggesting a critical role for Akt/GSK3β‐dependent autophagy in reducing neuronal cell death by limb remote ischemic postconditioning. Nevertheless, many questions remain to be answered in this study. For example, how does the ischemic postconditioning in limb cause the autophagy induction in brain? Akt/GSK3β inhibitor LY294002 blocked Akt/GSK3β signaling in neuronal cells, but to what extent does limb remote ischemic postconditioning contribute to the Akt/GSK3β signaling activation in neuronal cells? Based on the current situation that the experimental results from basic studies directly targeting on autophagy may be hard to translate into clinical practice, the prompting effect of pre‐ or postconditionings on autophagy in brain may be a good therapeutic potential of cerebral ischemia because this method is easy to apply clinically. Thus, the role of autophagy in pre‐ or postconditionings is an attractive issue needed to be intensively investigated in the future.
It is undeniable that autophagy could be an ideal target for new therapies to combat CNS disorders; however, more studies are warranted to understand the exact role and elegant control of autophagy in CNS. A profound understanding of neuronal autophagy will provide novel insights into the pathogenic mechanisms of dysfunctional autophagy that underlie common CNS disorders. Moreover, how to translate the current knowledge of autophagy into clinical treatment for CNS disorders is still challenging. Academic and industry efforts are underway to develop tools that will enable high‐throughput screening of chemical libraries to identify novel candidate compounds modulating autophagy at various environments.
References
- 1. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728–741. [DOI] [PubMed] [Google Scholar]
- 2. Moscat J, Diaz‐Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009;137:1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lim J, Kim HW, Youdim MB, et al. Binding preference of p62 towards LC3‐ll during dopaminergic neurotoxin‐induced impairment of autophagic flux. Autophagy 2011;7:51–60. [DOI] [PubMed] [Google Scholar]
- 4. Tanabe F, Yone K, Kawabata N, et al. Accumulation of p62 in degenerated spinal cord under chronic mechanical compression: functional analysis of p62 and autophagy in hypoxic neuronal cells. Autophagy 2011;7:1462–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bartlett BJ, Isakson P, Lewerenz J, et al. p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy 2011;7:572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Geisler S, Holmstrom KM, Skujat D, et al. PINK1/Parkin‐mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010;12:119–131. [DOI] [PubMed] [Google Scholar]
- 7. Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin‐induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010;6:1090–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Larsen KB, Lamark T, Overvatn A, et al. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy 2010;6:784–793. [DOI] [PubMed] [Google Scholar]
- 9. Clausen TH, Lamark T, Isakson P, et al. p62/SQSTM1 is required for Parkin‐induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010;6:330–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carloni S, Girelli S, Scopa C, et al. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia‐ischemia. Autophagy 2010;6:366–377. [DOI] [PubMed] [Google Scholar]
- 11. Tian F, Deguchi K, Yamashita T, et al. In vivo imaging of autophagy in a mouse stroke model. Autophagy 2010;6:1107–1114. [DOI] [PubMed] [Google Scholar]
- 12. Wang P, Guan YF, Du H, et al. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy 2012;8:77–87. [DOI] [PubMed] [Google Scholar]
- 13. Wei K, Wang P, Miao CY. A double‐edged sword with therapeutic potential: an updated role of autophagy in ischemic cerebral injury. CNS Neurosci Ther 2012;18:879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qi ZF, Luo YM, Liu XR, Wang RL. Akt/GSK3β dependent autophagy contributes to the neuroprotection of limb remote ischemic postconditioning in the transient cerebral ischemic rat model. CNS Neurosci Ther 2012;18:doi: 10.1111/cns.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sheng R, Zhang LS, Han R, et al. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 2010;6:482–494. [DOI] [PubMed] [Google Scholar]
- 16. Sheng R, Liu XQ, Zhang LS, et al. Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy 2012;8:310–325. [DOI] [PubMed] [Google Scholar]