

Summary
Aim
The purpose of this investigation was to further explore the mechanism(s) underlying the amelioration in EAE caused by Fasudil, particularly focusing on anti‐inflammatory effect.
Methods
We induced a chronic‐progressive experimental autoimmune encephalomyelitis (EAE) in B6 mice immunized with myelin oligodendrocyte glycoprotein35–55 and performed Fasudil intervention in early and late stages of the disease.
Results
The administration of Fasudil (40 mg/kg, i.p) had a therapeutic effect in delaying the onset and ameliorating the severity of EAE, accompanied by the improvement in myelination and the decrease in inflammatory cells in spinal cords. Fasudil inhibited TLR‐4, p‐NF‐kB/p65, and inflammatory cytokines (IL‐1β, IL‐6, and TNF‐α) and enhanced IL‐10 production in spinal cords. The ratio of arginase/iNOS was enhanced mainly in the spinal cords of EAE mice treated with Fasudil, reflecting a shift toward the M2 (antiinflammation) macrophage/microglia phenotype. The administration of Fasudil also induced the upregulation of CB2 receptor in spinal cords, but did not significantly trigger CB1 receptor. Levels of neurotrophic factors NGF, BDNF, and GDNF in the CNS were not altered by Fasudil.
Conclusion
Fasudil ameliorates disease progression in EAE, acting possibly through antiinflammatory pathway.
Keywords: Antiinflammation, Experimental autoimmune encephalomyelitis, Fasudil
Introduction
Multiple sclerosis (MS) is an inflammatory demyelinating and neurodegenerative disease in the central nervous system (CNS). The etiology of MS is still not known, a combination of several factors may be involved, including genetics and environment 1. During development of MS, the involvement of CNS inflammation is associated with demyelination and axonal loss that leads to neurodegeneration 2. In MS and experimental autoimmune encephalomyelitis (EAE), autoreactive T cells are activated in the periphery and migrated through the blood–brain barrier. Upon entering the CNS, the T cells are reactivated by antigen‐presenting cells (APCs) such as infiltrating macrophages and resident microglia, in which chronic inflammation is propagated by continuous generation of APCs and diversification of the myelin‐specific T‐cell response 3, 4.
Rho‐kinase (ROCK), a serine/threonine kinase of molecular mass 160 kDa, is activated by binding to the active GTP‐bound form of the small GTPase Rho and is expressed both centrally and peripherally where it is implicated in fundamental cellular processes including migration, proliferation, and survival 5. A series of studies have demonstrated that ROCK inhibitor Y‐27632 reduced leukocyte infiltration in several models of inflammation such as ischemic heart injury 6, endotoxic liver 7, and lung injury 8. Fasudil, a selective ROCK inhibitor, has been used to treat cerebral vasospasm 9, stroke 10, and traumatic spinal cord injury 11. Recent studies have highlighted the potential of Fasudil in the treatment of EAE or MS. In addition, the inhibition of ROCK enhanced oligodendrocyte precursor maturation into the oligodendrocyte lineage 12, 13. The blockade of Rho/ROCK is considered to be beneficial for CNS inflammatory demyelination 14. Thus, targeting ROCK inhibition is a promising strategy to achieve neuroprotection, axonal regeneration, oligodendrocyte generation, and suppression of inflammation 14.
The goal of this study was to explore novel potential mechanism(s) of Fasudil in suppressing the development of EAE in mice. Here, Fasudil delayed EAE onset and ameliorated its clinical severity, accompanied by the improvement in myelination and the decrease in inflammatory foci and cells that infiltrated into the CNS during disease. Most importantly, Fasudil inhibited expression of TLR‐4 and p‐NF‐kB/p65 and production of inflammatory cytokines in the CNS, which may be related to the shift of macrophage/microglia phenotype from M1 to M2 and the upregulation of antiinflammatory CB2 receptor (CB2R) in the CNS, but still remains to be determined.
Materials and Methods
Animals
Female C57BL/6 mice, 8–10 weeks old and 20–22 g, were purchased from Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). All experiments were conducted in accordance with the guidelines of the International Council for Laboratory Animal Science. The study was approved by the Ethics Committee of Shanxi Datong University, Datong, China. All mice were housed under pathogen‐free conditions, received food and water ad libitum, and maintained in a reversed 12:12‐h (h) light/dark cycle in a temperature‐controlled room (25 ± 2°C) for 1 week prior to experimental manipulation.
Induction and Clinical Evaluation of EAE
Mouse myelin oligodendrocyte glycoprotein peptide35–55 (MOG35–55, MEVGWYRSPFSRVVHLYRNGK) was produced in an automatic synthesizer (CL. Bio‐Scientific Company, Xian, China). The purity of the peptide was >95% as determined by HPLC.
Chronic EAE was induced by subcutaneous immunization on the upper dorsal flanks with 300 μg of MOG35–55 in Freund's complete adjuvant (Sigma, St. Louis, MO, USA) supplemented with 3 mg/mL of M. Tuberculosis H37Ra (BD Difco, Detroit, MI, USA) (400 μg/mice). Mice were injected intraperitoneally with 400 ng of pertussis toxin (Enzo Life Sciences, Ann Arbor, MI, USA) at the same time of immunization and again 48 h later. Animals were weighed and evaluated for clinical score every other day in a blinded fashion by at least two investigators. Once clinical score of EAE was 3, we gave special care, which is necessary to soften the food with water in dish, add nutrients such as egg, and put dish in the bottom of the cage, making it easy to obtain food, water and nutrition.
Clinical scores of EAE were graded according to the following criteria: 0. healthy; 1. limp tail; 2. ataxia and/or paresis of hindlimbs; 3. paralysis of hindlimbs and/or paresis of forelimbs; 4. tetraparalysis; and 5. moribund or death.
Administration of Fasudil
Fasudil (Tianjin Chase Sun Pharmaceutical Co., Ltd., Tianjin, China) dissolved in normal saline (NS) was injected intraperitoneally at 40 mg/kg/day every other day on day 3 postimmunization (p.i.) (Fasudil early treatment, n = 23) or at onset of clinical symptoms of EAE (Fasudil late treatment, n = 23) till day 27 p.i. The injection of NS was set up as control (EAE control, n = 26) in a similar manner.
Histology and Immunohistochemistry
On day 21 p.i., mice were anesthetized and perfused with PBS and 4% buffered paraformaldehyde. The spinal cords of EAE mice in different groups were sliced (10 μm), and the pathological changes were detected by luxol fast blue and hematoxylin/eosin (H&E) staining. For immunohistochemistry, nonspecific binding was blocked with 3% bovine serum (Serotec, Bicester, UK) and permeabilized with 0.3% Triton X‐100 in 1% BSA‐PBS for 30 min. The sections were incubated at 4°C overnight with anti‐CD4 (1:1000; clone A60, Sigma), anti‐CD68 (1:1000, Serotec), anti‐CD11b (1:1000, eBiosence, San Diego, CA, USA), anti‐GFAP (1:1000, Millipore, Bedford, MA, USA), and anti‐p‐NF‐kB/p65 (1:1,000, Cell Signaling, Boston, MA, USA), then incubated with corresponding secondary antibodies at room temperature (RT) for 2 h. As a negative control, additional sections were treated similarly, but the primary antibodies were omitted. Total white matter in luxol fast blue was manually outlined, and color photograph in H&E stain was automatically converted to black and white. Pixel area (%) of demyelination in total white matter and number of inflammatory foci (>20 mononuclear cells/focus) in whole spinal cord were calculated by Image‐Pro Plus software. Six mice × 3 slides (6 × 3) in spinal cords were examined in a blinded fashion.
Cytokine ELISA
Mice were anesthetized and perfused with PBS only, and spinal cords were homogenized on ice with a extraction kit supplemented with protease inhibitors (Millipore) in a microcontent motor‐operated tissue homogenizer (Kimble Kontes Glass Co., Vineland, NJ, USA). Lysates were centrifuged at 10,000 × g for 20 min at 4°C, and protein concentration was determined by a Bradford protein assay. The levels of IL‐1β (Invitrogen Inc., Carlsbad, CA, USA), IL‐6, TNF‐α, and IL‐10 (Pepro tech Inc., Hawthorne, NJ, USA) were measured by a sandwich ELISA kits following the manufacturer's instructions. Determinations were performed in duplicate in three independent experiments. The results were expressed as pg/10 mg protein.
Western Blot Analysis
Mice were anesthetized and perfused with PBS only, proteins from brains and spinal cords were extracted using the above‐mentioned protocol. Extract of proteins (30 μg) was separated by SDS‐PAGE and electroblotted onto nitrocellulose membrane (Immobilon‐P, Millipore). After nonspecific antibody binding was blocked with 5% nonfat dry milk, membranes were incubated at 4°C overnight with anti‐iNOS (1:200, Cayman Chemicals Company, Ann Arbor, MI, USA), anti‐Arginase‐1 (Arg‐1) (1:300, Cayman Chemicals Company), anti‐CB1R (1:500, Cayman Chemicals Company), anti‐CB2R (1:500, Cayman Chemicals Company), anti‐TLR‐4 (1:2,000, Epitomics Inc., Burlingame, CA, USA), anti‐p‐NF‐kB/p65, and anti‐GAPDH (1:1,000, Epitomics). After washing in TBST, the immunoblots were incubated with horseradish peroxidase‐conjugated secondary antibodies (Cell Signaling Technology) at RT for 1 h. The immunoblots were developed with an enhanced chemiluminescence (ECL) system (GE Healthcare Life Sciences, Niskayuna, NY, USA) and measured with Quantity Software (Bio‐rad, Hercules, CA, USA). To compare protein loading, antibody directed against GAPDH was used.
Statistical Analysis
All the experiments were repeated two or three times, and GraphPad Prism software was used for statistical analysis. One‐way ANOVA, where applicable, was performed to determine whether an overall statistically significant change existed before Student's t‐test to analyze the difference between any two groups. Correlation was analyzed with Spearman rank correlation analysis. A statistically significant difference was assumed at P < 0.05.
Results
Fasudil Delays Onset and Ameliorates Severity of EAE
To observe the therapeutic effect of Fasudil in induction and onset phases of EAE, Fasudil was injected intraperitoneally on day 3 p.i. or at onset of clinical symptoms of EAE. As shown in Supplement 1 and Figure 1(A), the incidence of EAE (8/23, 34.7%) in Fasudil early‐treated mice was decreased as compared with EAE control (25/26, 96.1%) and Fasudil late‐treated mice (18/23, 78.3%). In EAE group, mean onset date was 12.76 ± 1.86, mean maximum score was 2.41 ± 1.20. The treatment of Fasudil at onset phase of EAE slightly delayed onset (14.00 ± 2.09, P < 0.05) and declined maximum clinical score (1.35 ± 0.98, P < 0.01). Obviously, the administration of Fasudil in induction phase of EAE is able to delay its onset (16.50 ± 3.16, P < 0.01) and declined maximum clinical score (0.45 ± 0.78, P < 0.001). There is a relationship between loss of body weight and severity of clinical score during the development of EAE. The treatment of Fasudil at induction or onset phases of EAE had less body weight loss as compared with that of EAE mice (Figure 1B). In our experiments, no mortality was observed most likely due to our special care in mice with clinical score 3.
Figure 1.
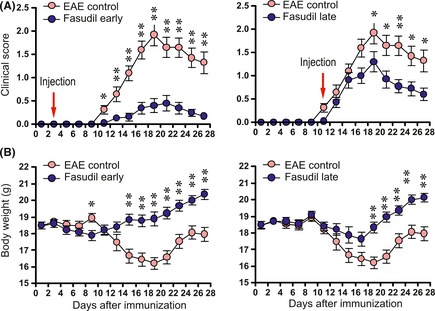
Fasudil delays onset and ameliorates severity of EAE. Fasudil was injected (intraperitoneally) at 40 mg/kg/day every other day on day 3 p.i. (Fasudil early treatment, n = 23) or at onset of clinical symptoms of EAE (Fasudil late treatment, n = 23). The injection of normal saline was set up as control (EAE control, n = 26) in a similar manner. (A) clinical score of EAE mice treated with Fasudil early or late treatment. (B) body weight of EAE mice treated with Fasudil early and late treatment. *P < 0.05; **P < 0.01.
Fasudil Inhibits Inflammation and Improves Myelination
We evaluated the pathology of CNS inflammation and demyelination. As shown in Figure 2(A) and (B), mice with EAE showed extensive myelin loss (demyelination) associated with infiltration of immune cells (inflammation) in spinal cord. When compared with EAE control, Fasudil‐treated mice had a significant improvement in the extent of demyelination (Figure 2A, P < 0.01 as compared with late and early treatment, respectively) and inflammation (Figure 2B, P < 0.05 and 0.01 as compared with late and early treatment, respectively) in spinal cords.
Figure 2.
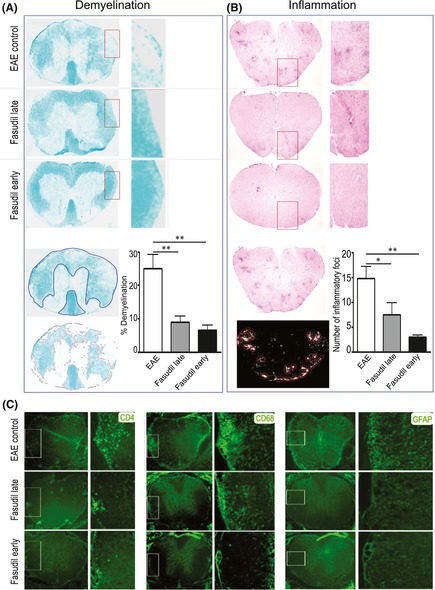
Fasudil inhibits inflammation and improves myelination in spinal cord. On day 21 p.i., spinal cords were used for luxol fast blue, H&E and immunohistochemistry staining. (A) Demyelination stained with luxol fast blue. Total white matter in luxol fast blue was manually outlined, and pixel area (%) of demyelination in total white matter was calculated by Image‐Pro Plus software. (B) Inflammation stained with H&E stain. Color photograph in H&E stain was automatically converted to black and white, and a number of inflammatory foci (>20 mononuclear cells/focus) in whole spinal cord were calculated by Image‐Pro Plus software. (C) CD4 T cells, CD68 macrophage/microglia and GFAP astrocytes. Quantitative results are analyzed for 6 mice in each group and are representative of three experiments with similar results. *P < 0.05, **P < 0.01.
As shown in Figure 2(C), mice with EAE showed extensive infiltration of inflammatory cells (CD4+ T cells and CD68+ macrophages) and relative activation of microglia and astrocytes (CD68+ microglia and GFAP+ astrocytes) in spinal cords. The ability of Fasudil to reduce the formation of inflammatory foci in spinal cords (Figure 2B) was paralleled by a similar reduction in the infiltration of CD4 T cells and macrophage as well as the activation of microglia and astrocytes (Figure 2C).
Fasudil Inhibits the Inflammatory Molecules
The therapeutic effect of Fasudil in EAE prompted us to investigate its possible mechanisms and to identify the target molecules through which Fasudil ameliorates the severity of EAE. It is postulated that inflammation may be primed via TLR‐4 that stimulates NF‐kB activation, which may contribute to inflammatory microenvironment 15. We first measured the expression of TLR‐4 in brains and spinal cords by Western blot. The expression of TLR‐4 in spinal cords was obviously higher than that of brains (Figure 3A). Fasudil treatment inhibited the expression of TLR‐4 in brains (P < 0.05, vs. Fasudil early treatment) and spinal cords (P < 0.05, EAE vs. Fasudil early and late treatment), especially in spinal cords (Figure 3A). These data provide strong evidence that Fasudil directly inhibits the expression of TLR‐4 as compared with EAE control mice.
Figure 3.
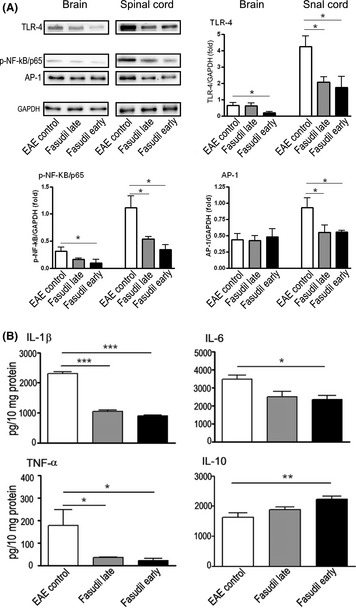
Fasudil inhibits inflammatory molecules. On day 28 p.i., brains and spinal cords were collected, and proteins were extracted by protein extraction kit. (A) Representative bands of Western blot for TLR‐4, p‐NF‐kB/p65, and AP‐1 in brains and spinal cords by Western blot. Quantitative analysis of TLR‐4, p‐NF‐KB/p65, and AP‐1 was performed from 5 to 6 mice in each group. (B) The levels of IL‐1β, IL‐6, TNF‐α, and IL‐10 in spinal cords by ELISA. Quantitative analysis was performed from 5 to 6 mice in each group. *P < 0.05; **P < 0.01, ***P < 0.001.
Accumulating evidence demonstrates that TLR‐4–NF‐kB signaling contributes to inflammatory responses 16, 17. We next examined the expression of p‐NF‐κB/p65 in brain and spinal cord of EAE, demonstrating that expression of NF‐κB/p65 in spinal cords was obviously higher than that of brains (Figure 3A). Fasudil treatment significantly inhibited the expression of p‐NF‐κB/p65 in brains and spinal cords of mice treated with Fasudil in both late‐ and early‐treated groups (Figure 3A, P < 0.05, respectively).
Besides NF‐κB, other transcription factor AP‐1 is known to affect inflammatory cascades, manifested by the ample production of inflammatory mediators. Figure 3(A) shows that the expression of AP‐1 in spinal cords was inhibited in mice treated with Fasudil in both late‐ or early‐treated groups as compared with EAE control (P < 0.05, respectively).
Because Fasudil has been shown to suppress p‐NF‐κB/p65 in the CNS (mainly in spinal cord), we next measured the levels of proinflammatory molecules such as IL‐1β, IL‐6, and TNF‐α among three groups. As expected, the results showed that IL‐1β, IL‐6, and TNF‐α were suppressed in spinal cords from mice treated with Fasudil in late‐ or early‐treated groups (Figure 3(B), P < 0.05 and 0.001, respectively). In parallel, the levels of IL‐10 were enhanced in spinal cord of EAE treated with Fasudil early treatment (Figure 3B, P < 0.05).
We further examined the cellular localization of NF expression in spinal cord,finding that p‐NF‐kB/p65 was observed in both white matter and gray matter (Figure 4). However, p‐NF‐kB/p65 was expressed mainly in white matter after Fasudil treatment (Figure 4). Double immunohistochemistry showed that astrocytes, but not microglia, overlap with p‐NF‐kB/p65 (Figure 4), suggesting that astrocytes are main cell source of p‐NF‐kB/p65 in EAE. Fasudil treatment (late and early) inhibited p‐NF‐kB/p65 in astrocytes as compared with EAE control (Figure 4).
Figure 4.
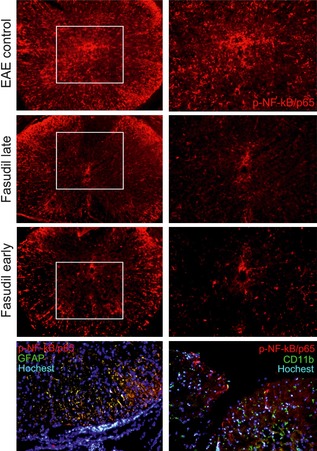
Expression and location of p‐NF‐kb/65 in spinal cord. On day 28 p.i., slices from spinal cords were stained with anti‐p‐NF‐kB/65, anti‐GFAP, and anti‐CD11b. In EAE mice, p‐NF‐kB/p65 was observed in both white matter and gray matter. Double immunohistochemistry showed that astrocytes, but not microglia, overlap with p‐NF‐kB/p65.
Fasudil shifts M1 to M2 phenotype
The classical M1 polarization of macrophage/microglia has been linked with promoting inflammation, whereas M2 phenotype is antiinflammatory and promotes tissue repair 18. Arg‐1 is responsible for metabolism of arginine to ornithine and polyamines in M2 macrophages, thereby diminishing expression of iNOS by M1 cells 19. Fasudil inhibited the expression of iNOS in spinal cords (Figure 5A and B) and elevated the expression of Arg‐1 in mice treated with Fasudil in both late‐ or early‐treated groups (Figure 5A and C). The ratio of Arg‐1/iNOS was significantly enhanced as compared with EAE control (Figure 5D), indicating an obvious shift toward an M2 macrophage/macrophage phenotype, which is consistent with the decrease in inflammatory cytokines as described earlier.
Figure 5.
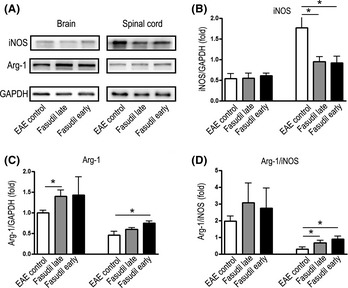
Fasudil shifts M1 to M2 phenotype. On day 28 p.i., brains and spinal cords were collected, and proteins were extracted for Western blot. (A) Representative bands of Western blot for iNOS and arginase in brains and spinal cords, (B) quantitative analysis of iNOS, (C) quantitative analysis of arginase, and (D) quantitative analysis of arginase/iNOS ratio. *P < 0.05.
Fasudil Induces the Expression of CB2 Receptor, not CB1 Receptor
Cannabinoids are involved in neuroprotection via NF‐kB inhibition 20. Thus, we detected the changes of CB1R and CB2R among three groups. After Fasudil early treatment, the expression of CB2R was upregulated in spinal cords (Figure 6A and C), but the expression of CB2R did not exhibit significant difference in Fasudil late treatment compared with EAE control. In brains, we did not find significant difference on CB2R among three groups (Figure 6A and C). As for CB1R, we did not find any significant differences in brains and spinal cords among three groups (Figure 6A and B).
Figure 6.
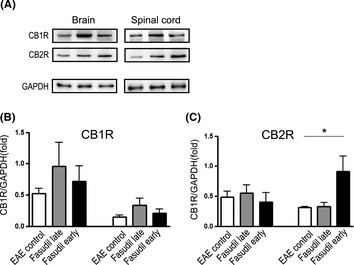
Fasudil enhances the expression of CB 2R in brains and spinal cords. On day 28 p.i., brains and spinal cords were collected, and proteins were extracted for Western blot. (A) Representative bands of Western blot for CB 1R and CB 2R in brains and spinal cords, (B) quantitative analysis of CB 1R, and (C) quantitative analysis of CB 2R. Quantitative analysis was performed from 5 to 6 mice in each group. *P < 0.05.
Fasudil Did Not Trigger The Production of Neurotrophic Factors
It has been demonstrated that NF‐kB is associated with the pathophysiology of ischemic/traumatic injuries and neurodegenerative diseases, where it modulates the expression of target genes involved in cell growth, survival, adhesion, and death 21. We finally observed the changes in neurotrophic factors in mice treated with Fasudil, in which NF‐kB was inhibited. The results indicate that the levels of NGF, BDNF, and GDNF from brains and spinal cords of mice treated with Fasudil in both late‐ or early‐treated groups were not altered by Fasudil (Supplement 2). It is possible that the regulation of these neurotrophic factors is mediated possibly via indirect mechanisms.
Discussion
In the present study, Fasudil delayed the onset of EAE and ameliorated the severity of symptoms when administered in induction or onset phases of EAE, but better therapeutic effect could be observed at the initial phase of EAE (on day 3 p.i.), which represents the immunological incipience of the disease. Consistent with previous studies, Fasudil inhibited the migration of inflammatory cells into the CNS. It has been reported that ROCK is required for efficient T‐cell polarization and migration on endothelial cells and transendothelial migration by affecting both actomyosin contractility and microtubules stability 22, 23.
Toll like receptors (TLRs) are transemembrane proteins that play an important role in mediating inflammatory response. In this sense, TLR‐4 is inducible and associated with downstream NF‐kB activation and proinflammatory cytokine release, which may contribute to neuronal death 13, 24. Constitutive activity of NF‐kB in myeloid cells drives the pathogenicity of monocytes and macrophages during EAE 25. Our results show the inhibition and the relationship of TLR‐4 and p‐NF‐kB/p65 in EAE treated with Fasudil. NF‐κB is an essential element of the TLR‐mediated response in cells of immune system. In macrophages, one of the physiological consequences of TLR stimulation is the release of inflammatory cytokines such as TNF, IL‐1β, and IL‐6 26. In spinal cords, Fasudil treatment inhibited the production of these inflammatory cytokines possibly through TLR‐4 and/or NF‐kB suppression. Although the relationship of TLR‐4 and NF‐kB in other groups has been confirmed, the TLR‐4 and NF‐kB pathway in this study is still need to be defined.
Encephalomyelitis is initiated through the peripheral activation and proliferation of myelin‐specific CD4 T cells, which then migrate into the CNS. Once inside, microglia are activated, and a large number of macrophages are recruited from the circulating blood, resulting in CNS inflammation 27, 28. In recent years, a population of myeloid cells suppressing immune responses has been described 29, 30, 31. These cells mainly exert their functions through iNOS and/or arginase activity 32, 33. In TLR‐4 deficient mice, arginase activity in response to L. major is enhanced, implicating that TLR‐4 negatively controls arginase induction 34. Arginase+‐cells were detected exclusively when the switch from proinflammatory to antiinflammatory conditions occurs, limiting the immune response 33, 35. Arginase+‐myeloid‐derived cells from the spinal cords of EAE limit the inflammation by promoting T‐cell apoptosis 36. Our data show that ratio of arginase/iNOS is enhanced in mice treated with Fasudil, which may be differentially regulated by TLR‐4/NF‐kB pathway. In turn, the upregulation of arginase significantly decreased macrophage infiltration and inflammation, whereas downregulation of arginase aggravated these adverse effects 37. In addition, cytokines can also influence the balance between arginase and iNOS in inflammation, whereas Th 1 cytokine, IFN‐γ, induces iNOS, the Th2 cytokines IL‐4 and IL‐10 induce arginase 38. Based on previous studies and our results, we postulate that the functional ramification of arginase and NOS induction and their reciprocal regulation likely depend on the disease process, TLR4 and/or NF‐kB signaling, and cytokine milieu.
After many years of continuous research, it is becoming more evident that cannabinoid compounds and the endocannabinoid signaling system offer a promising novel form of therapy for MS. This appears reasonable that, in addition to the potential of CB2R in antiinflammatory effect, CB1R may also represent an important protective target able to improve neuronal damage in the progression of MS 20, 39. CB2R knockout mice exacerbated EAE symptoms, which was associated with CD4+ T‐cell infiltration and myeloid cell recruitment into the inflamed CNS, whereas the opposite action was observed upon CB2R‐selective agonist administration 39. A selective pharmacological CB2R agonist HU‐308 improved EAE symptoms and reduced spinal cord lesions and microglial activation 40. CB2R stimulation suppressed the proliferation and attenuated proinflammatory cytokine production 41, 42, 43, indicating that CB2R reduces inflammation and could be beneficial for the treatment of MS 44. Studies in CB1R knockout mice showed more rapid development of neurodegeneration during EAE 39, 45, raising the possibility that neurotrophic factors and CBR interact to regulate multiple functions in the brain. Neuroprotective effects driven by CB1R may involve control of excitotoxic glutamate activity and toxic ion influxes, antioxidant properties and neurotrophic factors 39, 46, 47. The level of BDNF was decreased in the brain of mice lacking CB1R 48, whereas activation of these receptors increased BDNF production in rodents 49 and in humans 50. Furthermore, BDNF release triggered by CB1R stimulation mediated the neuroprotective effects 51. It is by now established that MS is not simply an autoimmune disease, and that in addition to inflammation and demyelination, axonal injury and neuronal loss underlie the accumulation of disability and the disease progression. Thus, we try to explore whether Fasudil‐mediated amelioration of clinical score in EAE is related with neurotrophic factors. In this study, Fasudil early treatment upregulated the expression of CB2R in spinal cords. However, the role of CB2R in Fasudil‐treated mice still needs further exploration.
In conclusion, Fasudil delays the onset and ameliorates the severity in different phases of EAE, accompanied by the improvement in myelination and the inhibition of inflammatory responses in the CNS. Fasudil suppresses CNS inflammatory responses possibly through inhibiting TLR‐4 and/or NF‐kB activation, which may be related to the shift of macrophage/microglia phenotype from M1 to M2 and the upregulation of neuroprotective CB2R in the CNS, but still remains to be determined.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Table S1. The clinical evaluation of EAE mice.
Fig S1. Fasudil did not influences the levels of neurotrophic factors.
Acknowledgment
This work was supported by grant from National Natural Science Foundation of China (No. 81070957, No. 30972715 and No. 81070956).
References
- 1. Tselis A. Evidence for viral etiology of multiple sclerosis. Semin Neurol 2011;31:307–316. [DOI] [PubMed] [Google Scholar]
- 2. Stadelmann C, Wegner C, Brück W. Inflammation, demyelination, and degeneration – recent insights from MS pathology. Biochim Biophys Acta 2011;1812:275–282. [DOI] [PubMed] [Google Scholar]
- 3. Tuohy VK, Yu M, Yin L, et al. The epitope spreading cascade during progression of experimental autoimmune encephalomyelitis and multiple sclerosis. Immunol Rev 1998;164:93–100. [DOI] [PubMed] [Google Scholar]
- 4. Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol 2005;23:683–747. [DOI] [PubMed] [Google Scholar]
- 5. Guilluy C, Garcia‐Mata R, Burridge K. Rho protein crosstalk: another social network? Trends Cell Biol 2011;21:718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bao W, Hu E, Tao L, et al. Inhibition of Rho‐kinase protects the heart against ischemia/reperfusion injury. Cardiovasc Res 2004;61:548–558. [DOI] [PubMed] [Google Scholar]
- 7. Slotta JE, Laschke MW, Menger MD, Thorlacius H. Rho‐kinase signalling mediates endotoxin hypersensitivity after partial hepatectomy. Br J Surg 2008;95:976–984. [DOI] [PubMed] [Google Scholar]
- 8. Ding RY, Zhao DM, Zhang ZD, Guo RX, Ma XC. Pretreatment of Rho kinase inhibitor inhibits systemic inflammation and prevents endotoxin‐induced acute lung injury in mice. J Surg Res 2011;171:e209–e214. [DOI] [PubMed] [Google Scholar]
- 9. Pierot L, Aggour M, Moret J. Vasospasm after aneurysmal subarachnoid hemorrhage: recent advances in endovascular management. Curr Opin Crit Care 2010;16:110–116. [DOI] [PubMed] [Google Scholar]
- 10. Rikitake Y, Kim HH, Huang Z, et al. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke 2005;36:2251–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hara M, Takayasu M, Watanabe K, et al. Protein kinase inhibition by fasudil hydrochloride promotes neurological recovery after spinal cord injury in rats. J Neurosurg 2000;93:94–101. [DOI] [PubMed] [Google Scholar]
- 12. Wang L, Kamath A, Frye J, Iwamoto GA, Chun JL, Berry SE. Aorta‐derived mesoangioblasts differentiate into the oligodendrocytes by inhibition of the Rho kinase signaling pathway. Stem Cells Dev 2012;21:1069–1089. [DOI] [PubMed] [Google Scholar]
- 13. Wang H, Rusielewicz T, Tewari A, Leitman EM, Einhenber S, Melendez‐Vasquez CV. Myosin II is a negative regulator of oligodendrocyte morphological differentiation. J Neurosci Res 2012;90:1547–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mueller BK, Mack H, Teusch N. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov 2005;4:387–398. [DOI] [PubMed] [Google Scholar]
- 15. Wang Z, Liu D, Wang F, et al. Saturated fatty acids activate microglia via Toll‐like receptor 4/NF‐κB signalling. Br J Nutr 2012;107:229–241. [DOI] [PubMed] [Google Scholar]
- 16. Bhaskar S, Shalini V, Helen A. Quercetin regulates oxidized LDL induced inflammatory changes in human PBMCs by modulating the TLR‐NF‐κB signaling pathway. Immunobiology 2011;216:367–373. [DOI] [PubMed] [Google Scholar]
- 17. Woods DC, White YA, Dau C, Johnson AL. TLR4 activates NF‐κB in human ovarian granulosa tumor cells. Biochem Biophys Res Commun 2011;409:675–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 2009;29:13435–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mahbub S, Deburghgraeve CR, Kovacs EJ. Advanced age impairs macrophage polarization. J Interferon Cytokine Res 2012;32:18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Panikashvili D, Mechoulam R, Beni SM, Alexandrovich A, Shohami E. CB1 cannabinoid receptors are involved in neuroprotection via NF‐kappa B inhibition. J Cereb Blood Flow Metab 2005;25:477–484. [DOI] [PubMed] [Google Scholar]
- 21. Teng FY, Tang BL. NF‐kappaB signaling in neurite growth and neuronal survival. Rev Neurosci 2010;21:299–313. [DOI] [PubMed] [Google Scholar]
- 22. Heasman SJ, Ridley AJ. Multiple roles for RhoA during T cell transendothelial migration. Small Gtpases 2010;1:174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takesono A, Heasman SJ, Wojciak‐Stothard B, Garg R, Ridley AJ. Microtubules regulate migratory polarity through Rho/ROCK signaling in T cells. PLoS ONE 2010;5:e8774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mogi M, Kondo A. Down‐regulation of NF‐kappaB led to up‐regulation of NGF production in mouse osteoblasts. J Immunoassay Immunochem 2010;31:92–101. [DOI] [PubMed] [Google Scholar]
- 25. Ellrichmann G, Thöne J, Lee DH, Rupec RA, Gold R, Linker RA. Constitutive activity of NF‐kappa B in myeloid cells drives pathogenicity of monocytes and macrophages during autoimmune neuroinflammation. J Neuroinflammation 2012;9:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mills KH. TLR‐dependent T cell activation in autoimmunity. Nat Rev Immunol 2011;11:807–822. [DOI] [PubMed] [Google Scholar]
- 27. Prendergast CT, Anderton SM. Immune cell entry to central nervous system–current understanding and prospective therapeutic targets. Endocr Metab Immune Disord Drug Targets 2009;9:315–327. [DOI] [PubMed] [Google Scholar]
- 28. Herz J, Zipp F, Siffrin V. Neurodegeneration in autoimmune CNS inflammation. Exp Neurol 2010;225:9–17. [DOI] [PubMed] [Google Scholar]
- 29. Bowen JL, Olson JK. Innate immune CD11b+Gr‐1+ cells, suppressor cells, affect the immune response during Theiler's virus‐induced demyelinating disease. J Immunol 2009;183:6971–6980. [DOI] [PubMed] [Google Scholar]
- 30. Slaney CY, Toker A, La FA, Backstrom BT, Harper JL. Naïve blood monocytes suppress T‐cell function. A possible mechanism for protection from autoimmunity. Immunol Cell Biol 2011;89:7–13. [DOI] [PubMed] [Google Scholar]
- 31. Zhu B, Bando Y, Xiao S. CD11b+Ly‐6C(hi) suppressive monocytes in experimental autoimmune encephalomyelitis. J Immunol 2007;179:5228–5237. [DOI] [PubMed] [Google Scholar]
- 32. Bronte V, Zanovello P. Regulation of immune responses by L‐arginine metabolism. Nat Rev Immunol 2005;5:641–654. [DOI] [PubMed] [Google Scholar]
- 33. Highfill SL, Rodriguez PC, Zhou Q, et al. Bone marrow myeloid‐derived suppressor cells (MDSCs) inhibit graft‐versus‐host disease (GVHD) via an arginase‐1‐dependent mechanism that is up‐regulated by interleukin‐13. Blood 2010;116:5738–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kropf P, Freudenberg MA, Modolell M, et al. Toll‐like receptor 4 contributes to efficient control of infection with the protozoan parasite Leishmania major. Infect Immun 2004;72:1920–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Greten TF, Manns MP, Korangy F. Myeloid derived suppressor cells in human diseases. Int Immunopharmacol 2011;11:802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moliné‐Velázquez V, Cuervo H, Ortega MC, Clemente D, de Castro F. Myeloid‐derived suppressor cells limit the inflammation by promoting T lymphocyte apoptosis in the spinal cord of a murine model of multiple sclerosis. Brain Pathol 2011;21:678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang XP, Chen YG, Qin WD, et al. Arginase I attenuates inflammatory cytokine secretion induced by lipopolysaccharide in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2011;31:1853–1860. [DOI] [PubMed] [Google Scholar]
- 38. Ckless K, van der Vliet A, Janssen‐Heininger Y. Arginase modulates NF‐κB activity via a nitric oxide–dependent mechanism. Am J Respir Cell Mol Biol 2007;36:645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maresz K, Pryce G, Ponomarev ED, et al. Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nat Med 2007;13:492–497. [DOI] [PubMed] [Google Scholar]
- 40. Palazuelos J, Davoust N, Julien B, et al. The CB(2) cannabinoid receptor controls myeloid progenitor trafficking: involvement in the pathogenesis of an animal model of multiple sclerosis. J Biol Chem 2008;283:13320–13329. [DOI] [PubMed] [Google Scholar]
- 41. Cabral GA, Harmon KN, Carlisle SJ. Cannabinoid‐mediated inhibition of inducible nitric oxide production by rat microglial cells: evidence for CB1 receptor participation. Adv Exp Med Biol 2001;493:207–214. [DOI] [PubMed] [Google Scholar]
- 42. Molina‐Holgado F, Molina‐Holgado E, Guaza C, Rothwell NJ. Role of CB1 and CB2 receptors in the inhibitory effects of cannabinoids on lipopolysaccharide‐induced nitric oxide release in astrocyte cultures. J Neurosci Res 2002;67:829–836. [DOI] [PubMed] [Google Scholar]
- 43. Chuchawankul S, Shima M, Buckley NE, Hartmann CB, McCoy KL. Role of cannabinoid receptors in inhibiting macrophage costimulatory activity. Int Immunopharmacol 2004;4:265–278. [DOI] [PubMed] [Google Scholar]
- 44. Pertwee RG. The therapeutic potential of drugs that target cannabinoid receptors or modulate the tissue levels or actions of endocannabinoids. AAPS J 2005;7:E625–E654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pryce G, Ahmed Z, Hankey DJ, et al. Cannabinoids inhibit neurodegeneration in models of multiple sclerosis. Brain 2003;126:2191–2202. [DOI] [PubMed] [Google Scholar]
- 46. Docagne F, Muneton V, Clemente D, et al. Excitotoxicity in a chronic model of multiple sclerosis: neuroprotective effects of cannabinoids through CB1 and CB2 receptor activation. Mol Cell Neurosci 2007;34:551–561. [DOI] [PubMed] [Google Scholar]
- 47. Croxford JL, Pryce G, Jackson SJ, et al. Cannabinoid‐mediated neuroprotection, not immunosuppression, may be more relevant to multiple sclerosis. J Neuroimmunol 2008;193:120–129. [DOI] [PubMed] [Google Scholar]
- 48. Aso E, Ozaita A, Valdizán EM, et al. BDNF impairment in the hippocampus is related to enhanced despair behavior in CB1 knockout mice. J Neurochem 2008;105:565–572. [DOI] [PubMed] [Google Scholar]
- 49. Butovsky E, Juknat A, Goncharov I, et al. In vivo up‐regulation of brain‐derived neurotrophic factor in specific brain areas by chronic exposure to Delta‐tetrahydrocannabinol. J Neurochem 2005;93:802–811. [DOI] [PubMed] [Google Scholar]
- 50. D'Souza DC, Pittman B, Perry E, Simen A. Preliminary evidence of cannabinoid effects on brain‐derived neurotrophic factor (BDNF) levels in humans. Psychopharmacology 2009;202:569–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Khaspekov LG, Brenz Verca MS, Frumkina LE, Hermann H, Marsicano G, Lutz B. Involvement of brain‐derived neurotrophic factor in cannabinoid receptor‐dependent protection against excitotoxicity. Eur J Neurosci 2004;19:1691–1698. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The clinical evaluation of EAE mice.
Fig S1. Fasudil did not influences the levels of neurotrophic factors.