

Summary
Background
The close relationship between epileptic seizure and Alzheimer's disease (AD) has been demonstrated in the past decade. Valproic acid, a traditional first‐line antiepileptic drug, exerted protective effects in transgenic models of AD. It remains uncertain whether new antiepileptic drugs could reverse neuropathology and behavioral deficits in AD transgenic mice.
Aims
APPswe/PS1dE9 transgenic mice were used in this study, which over express the Swedish mutation of amyloid precursor protein together with presenilin 1 deleted in exon 9. 7‐month‐old APPswe/PS1dE9 transgenic mice were treated daily with 20 mg/kg topiramate (TPM) and 50 mg/kg levetiracetam (LEV) for 30 days by intraperitoneal injection to explore the effects of TPM and LEV on the neuropathology and behavioral deficits.
Results
The results indicated that TPM and LEV alleviated behavioral deficits and reduced amyloid plaques in APPswe/PS1dE9 transgenic mice. TPM and LEV increased Aβ clearance and up‐regulated Aβ transport and autophagic degradation. TPM and LEV inhibited Aβ generation and suppressed γ‐secretase activity. TPM and LEV inhibited GSK‐3β activation and increased the activation of AMPK/Akt activation. Further, TPM and LEV inhibited histone deacetylase activity in vivo.
Conclusions
Therefore, TPM and LEV might have the potential to treat AD effectively in patient care.
Keywords: Alzheimer's disease, Behavior, Histone deacetylase inhibition, Levetiracetam, Neuropathology, Topiramate
Introduction
Alzheimer's disease (AD) is the most common cause of dementia in the elderly, which is characterized clinically by progressive cognitive decline and pathologically by the formation of amyloid plaques and neurofibrillary tangles in the brain 1, 2. Amyloid β‐peptide (Aβ) is the central component of amyloid plaques. Amyloid precursor protein is first cleaved by α‐secretase or β‐secretase producing C83 or C99, respectively. Subsequently, γ‐secretase cleaves C83 or C99 into peptide P3 or Aβ, respectively 3. Neurofibrillary tangles are composed of highly phosphorylated tau (p‐tau) 4. Accumulation of Aβ in the brain might be responsible for the pathogenesis of AD 5. To date, no “disease‐modifying” drug has shown therapeutic benefits and acceptable safety. Some newly developed drugs have failed in clinical trials 6.
The close relationship between epilepsy and AD has been demonstrated in the past years. Some clinical studies have noted a significantly higher incidence of seizures in patients with AD than that in age‐matched population 7, 8, 9, 10. Experimental study has shown that hAPP mice have spontaneous nonconvulsive seizure activity in cortical and hippocampal networks 11. Similarly, video‐electroencephalographic recording has showed high incidence of seizures in APPswe/PS1dE9 transgenic mice 12. Furthermore, seizure susceptibility was also found in mice overexpressing the APP intracellular domain 13. On the other hand, aberrant increases in network excitability may contribute to Aβ‐induced neurological deficits in hAPP mice 11. Further, it is suggested that fluctuations in neural activity regulate the vulnerability to Aβ deposition in transgenic mice models 14. Hence, aberrant excitatory neuronal activity has been regarded as primary upstream mechanism of AD 10. In vivo study has shown that reduction of hippocampal hyperactivity by antiepileptic drugs could improve cognition in patients with amnestic mild cognitive impairment 15. The findings support regulation of neural activity as a new therapeutic strategy for AD.
As a traditional first‐line antiepileptic drug, valproic acid (VPA) could significantly inhibit production of Aβ and p‐tau and improve behavioral performance in AD mouse models 16, 17. It remains uncertain whether new antiepileptic drugs could reverse neuropathology and cognitive deficits in AD transgenic mice. The therapeutic effects of VPA have been partly ascribed to inhibition of histone deacetylase (HDAC) 16. The activity of topiramate (TPM) and levetiracetam (LEV) as HDAC inhibitor has been demonstrated 18. Additionally, TPM and LEV could exert positive effects on neuroprotection and anti‐inflammation, both of which are involved in AD pathogenesis 19, 20, 21. Hence, it is significant and of interest to investigate the effects of antiepileptic drugs (TPM and LEV) on the behavioral performance and neuropathology in an AD mouse model.
APPswe/PS1dE9 transgenic mice over express the Swedish mutation of amyloid precursor protein together with presenilin 1 deleted in exon 9 22. The mice show increase in Aβ plaques from the age of 4 months, glial activation, and deficits in cognitive functions from the age of 6 months. Hence, APPswe/PS1dE9 transgenic mice were wildly used in research of AD 23. In the present study, we used the APPswe/PS1dE9 transgenic mouse model to provide direct evidence for the protective effects of TPM/LEV on behavior and neuropathology. VPA (a traditional antiepileptic drug and a well‐known HDAC inhibitor), which could exert therapeutic effects in AD mouse models, was used as a positive control.
Materials and Methods
Transgenic Mice and Drugs Treatment
APPswe/PS1dE9 transgenic mice (7 months old) were obtained from the Institute of Zoology, Chinese Academy of Sciences and were housed in an air‐conditioned room under a 12‐h light/12‐h dark cycle (lights on, 8:00 am through 8:00 pm). Food and water were provided ad libitum. Mice were injected with 20 mg/kg TPM, 50 mg/kg LEV, or 30 mg/kg VPA i.p., respectively, at the same time once daily, for 30 days. Control mice were injected with PBS. All animal procedures were carried out in accordance with Chinese Association for Laboratory Animal Sciences Guide for Care and Use of Laboratory Animals and were approved by the Nanjing Medical University Experimental Animal Care and Use Committee. All efforts were made to minimize the suffering of the animals.
Morris Water Maze
The Morris water maze test was performed as described elsewhere 16. The water maze test was performed in a 1.5‐m diameter pool, and a 10‐cm diameter platform was placed in the southeastern quadrant in the hidden trials. The procedure consisted of 4 days of hidden platform tests and a probe trial 24 h after the last hidden platform test. In the hidden platform tests, mice were trained for four trials, with an intertrial interval of 1 h.
Immunohistochemical Staining
Mice were anesthetized and sacrificed. Then, the brains were removed and cut into halves. One half was used for Western blotting, and the other for immunohistochemistry. The latter for immunohistochemistry was fixed in 4% paraformaldehyde for 20 h. After dehydration with alcohol, the brains were embedded in paraffin and cut into 3–4‐μm sections. Sections were deparaffinized, hydrated in distilled water, treated with 3% H2O2 for 10 min to remove residual peroxidase activity, and rinsed again with PBS. Sections were permeabilized with 1% NP‐40 and 0.1% Triton X‐100 for 10 min, rinsed in PBS, blocked with 10% normal goat serum, and incubated with primary antibody (rabbit anti‐β‐amyloid1‐42, 1:600) overnight. After being rinsed in PBST (PBS containing 0.05% Tween‐20), sections were further incubated with secondary antibody. The immunoreactivity was developed using 3,3′‐Diaminobenzidine (DAB) for 3–10 min. Plaques were counted under a microscope at 40× magnification. Plaques were quantified, and the mean plaque count per slice was recorded for each mouse.
Thioflavin T Staining
Thioflavin T staining was performed as follows. Sections were deparaffinized and hydrated in distilled water. They were then stained in Mayer's hematoxylin for 1 min, washed in running water for 5 min, rinsed in distilled water, and placed in 1% thioflavin T for 5 min. Sections were then differentiated in 70% alcohol for 5 min, rinsed in distilled water twice, and mounted in glycerin jelly. The thioflavin T staining was examined using a fluorescence microscope.
Cell Culture and Drug Treatments
Human SH‐SY5Y neuroblastoma cells were obtained from ATCC. The SH‐SY5Y cells were maintained in DMEM supplemented with 10% FBS, 100 g/mL streptomycin, and 100 IU/mL penicillin. Then, 0.25 mM TPM, 0.25 mM LEV, or 1.0 mM VPA was added into the culture media, with PBS used as a control.
Okadaic acid (Sigma‐Aldrich, St. Louis, MO, USA) was used to induce tau hyperphosphorylation 17. The cells were treated with okadaic acid at 20 nM for 12 h prior to the treatment of anti‐epilepsy drugs.
Western Blotting
The following primary antibodies were used: mouse anti‐LRP1 antibody [5A6] (1:3000; Merck, Darmstadt, Germany), rabbit anti‐RAGE antibody (1:1000; Cell Signaling Technology, Denver, MA, USA), rabbit anti‐ phospho‐tau‐pSer396 antibody (1:1000; Abcam, Cambridge, MA, USA), rabbit anti‐tau antibody (1:400; Abcam), rabbit anti‐APP carboxyl terminal antibody (1:4000; Sigma), rabbit anti‐BACE1 antibody (1:1000; Cell Signaling), rabbit anti‐phospho‐GSK3β‐pSer9 (1:1000; Cell Signaling), rabbit anti‐GSK3β (1:1000; Cell Signaling), rabbit anti‐phospho‐Akt‐pSer473 (1:1000; Cell Signaling), rabbit anti‐Akt (1:1000; Cell Signaling), rabbit anti‐phospho‐AMPKα‐pThr172 (1:1000; Cell Signaling), rabbit anti‐AMPKα (1:1000; Cell Signaling), rabbit anti‐Acetyl‐Histone H4 (Lys5) (1:1000; Cell Signaling), rabbit anti‐beclin 1 antibody (1:1000; Cell Signaling), rabbit anti‐LC3 antibody (1:1000; Sigma), and mouse anti‐β‐actin antibody (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Western blotting was performed as described elsewhere 24. Brain tissue or cells were lysed in Radio‐Immunoprecipitation Assay (RIPA) lysis buffer supplemented with protease inhibitors (Complete; Roche, Indianapolis, IN, USA). The lysates were resolved by SDS‐PAGE. Supernatants and the final pellets from each sample were heat‐blocked for 5 min in loading buffer (125 mM Tris–HCl, 20% glycerol, 10% 2‐mercaptoethanol, 4% SDS, 0.02% bromophenol blue, pH 6.8) and then subjected to electrophoresis on 10–20% Tris–glycine SDS‐PAGE gels. Proteins were then electrically transferred to a transfer membrane (Bio‐Rad, Hercules, CA, USA) and blocked for 1 h in Tris–HCl‐buffered saline containing 5% skim milk and 0.1% Tween. Membranes were incubated in primary antibodies at 4°C overnight in TBS buffer containing 5% bovine albumin. Membranes were then rinsed with TBS buffer containing 0.1% Tween 20, incubated with Horseradish peroxidase (HRP)‐labeled secondary antibody for 2 h, and then stained with detection reagents. Finally, membranes were developed using the enhanced chemiluminescence (ECL) system. Immunoreactivity was quantified using ImageJ software (Rasband, MD, USA) 25.
Aβ42 ELISA
Tissue extracts from transgenic mouse hippocampal and neocortical regions, and conditioned cell culture media were collected. The levels of soluble and insoluble Aβ42 in brain samples were quantified using ELISA, as described elsewhere 26. Briefly, brain tissue was homogenized in extraction buffer consisting of 50 mM Tris (pH 7.4), 2 mM EDTA, 400 mM NaCl, and Complete protease inhibitor cocktail (Roche). The homogenates were centrifuged at 20,000 × g for 5 min at 4°C. The resulting supernatants were analyzed for soluble Aβ. The pellets were homogenized in 70% formic acid and centrifuged at 44,000 × g for 5 min at 4°C. The resulting supernatants were neutralized with 1 M Tris and then diluted in ELISA buffer for the measurement of insoluble Aβ. Samples were prepared from five animals in each group. All samples were analyzed in triplicate. Standard curves were made using human Aβ42 standards provided in the ELISA kit (Invitrogen, Camarillo, CA, USA).
Statistical Analysis
Statistical analyses were performed by an individual blinded to the groups. All results were expressed as means ± SD and examined for the homogeneity of variance. Statistical analysis was performed using SPSS 13.0 software (SPSS Inc., Chicago, IL, USA). Group differences in the escape latency and path length during the Morris water maze test were analyzed using two‐way analysis of variance (ANOVA) with repeated measures followed by Bonferroni multiple comparison test with day and treatment as the sources of variation. All other data were analyzed with a one‐way ANOVA followed by LSD procedure (if variances were equal) or Games–Howell procedure (if variances were unequal). A value of P < 0.05 was considered to be statistically significant.
Results
Both TPM and LEV Decrease Neuritic Plaque Burden in APPswe/PS1dE9 Transgenic Mice
We first examined whether TPM/LEV could inhibit amyloid plaque deposition in APPswe/PS1dE9 transgenic mice. Brain tissues from vehicle‐, TPM‐, LEV‐, and VPA‐treated mice were subjected to Aβ immunohistochemistry. TPM, LEV, and VPA reduced amyloid plaque deposition in the cortex and hippocampus compared with vehicle treatment (Figure 1A). Semi‐quantitative analysis revealed that TPM, LEV, and VPA treatment reduced plaque number by 45.8, 51.9%, and 53.1%, respectively (14.1 ± 2.9, 12.5 ± 3.7 and 12.2 ± 3.3 vs. 26.0 ± 4.9, P < 0.05; Figure 1C). Post hoc analysis revealed no significant differences between TPM and VPA (P = 0.265) or between LEV and VPA (P = 0.859). To confirm the neuropathological changes in vivo, thioflavin T staining was used to detect neuritic plaques. Consistent with the observations following Aβ immunohistochemistry, we observed a marked reduction in the numbers of senile plaques in the cortex and hippocampus in the TPM‐, LEV‐, and VPA‐treated groups (Figure 1B).
Figure 1.
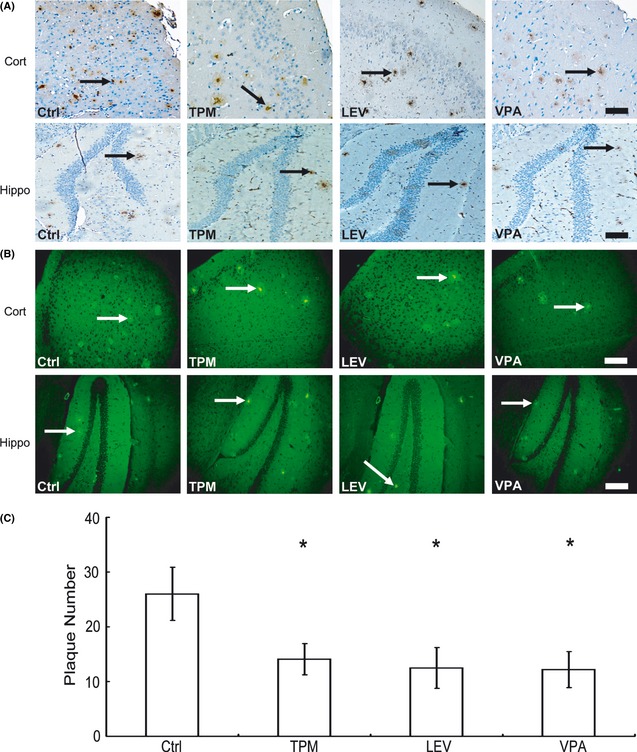
Effects of topiramate (TPM) and levetiracetam (LEV) on neuritic plaque formation in APPswe/PS1dE9 double‐transgenic mice. APPswe/PS1dE9 transgenic mice aged 7 months were treated with 20 mg/kg topiramate (TPM) or 50 mg/kg LEV for 30 days, whereas age‐matched control APPswe/PS1dE9 mice received vehicle solution (Ctrl) as a negative control or 30 mg/kg valproic acid (VPA) as a positive control. After behavioral tests, the mice were killed and the brains were dissected, fixed, and sectioned. (A) Neuritic plaques were detected by immunohistochemistry using an Aβ1‐42 antibody. The plaques were visualized by microscopy with 200× magnification. All histone deacetylase inhibitors significantly reduced the numbers of neuritic plaques in the cortex (Cort) and hippocampus (Hippo) of mice compared with controls. Black arrows point to plaques. Bars: 100 μm. (B) Neuritic plaques were further confirmed by thioflavin T staining and visualized under 200× magnification. White arrows point to plaques. Bars: 100 μm. (C) Quantification of neuritic plaques in APPswe/PS1dE9 mice in each group; the numbers represent the mean ± SD. n = 6 mice each. *P < 0.05 by one‐way ANOVA.
Both TPM and LEV Reverse Behavioral Deficits in APPswe/PS1dE9 Transgenic Mice
To investigate whether TPM and LEV could alleviate memory impairments, the Morris water maze was used to assess the effects of different drugs on spatial memory. In the hidden platform trial, mice in the TPM‐, LEV‐, and VPA‐treated groups showed significant improvements compared with the vehicle group. TPM‐, LEV‐, and VPA‐treated mice showed a shorter escape latency on the fourth day after treatment than did vehicle‐treated mice (27.97 ± 10.24, 23.22 ± 6.90, and 21.21 ± 5.95 vs. 49.04 ± 19.41, P < 0.05; Figure 2A). Similarly, TPM‐, LEV‐, and VPA‐treated mice showed a shorter swimming distance to find the platform on the fourth day after treatment than did vehicle‐treated mice (3.09 ± 1.21, 2.46 ± 0.65, and 2.35 ± 0.64 vs. 5.25 ± 2.14, P < 0.05; Figure 2B). In the probe trial, TPM‐, LEV‐, and VPA‐treated mice travelled into the southeastern quadrant, where the hidden platform was placed, significantly more times than did control mice (6.25 ± 0.71, 7.25 ± 0.50, and 5.60 ± 1.34 vs. 2.80 ± 0.45, P < 0.05; Figure 2C). In terms of escape latency/path length on the fourth day after treatment and crossing platform times in the probe trial, post hoc analysis detected no significant differences between TPM and VPA or between LEV and VPA.
Figure 2.
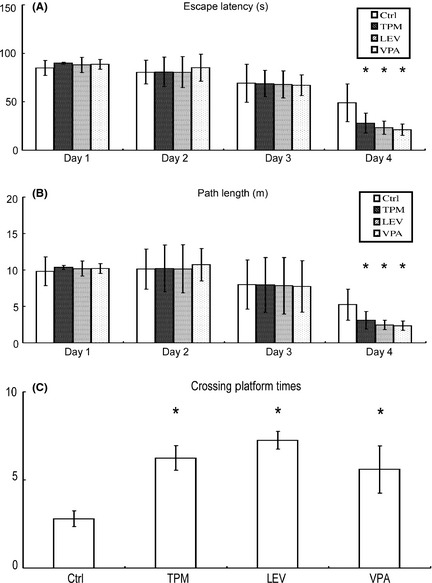
Effects of topiramate (TPM) and levetiracetam (LEV) on behavioral deficits in APPswe/PS1dE9 double‐transgenic mice. APPswe/PS1dE9 transgenic mice aged 7 months were given i.p. PBS, TPM, LEV, or valproic acid (VPA) for 30 day, and spatial memory was assessed using the Morris water maze (n = 8 per group). The Morris water maze test consists of 4 days of hidden platform tests, plus a probe trial 24 h after the last hidden platform test. (A) In the hidden platform tests, mice were trained for four trials, with an intertrial interval of 1 h. TPM‐, LEV‐, and VPA‐treated mice exhibited a significant decrease in the time needed to find the hidden platform on the fourth day. *P < 0.05 by two‐way ANOVA with repeated measures. (B) TPM‐, LEV‐, and VPA‐treated mice had a shortened swimming length before finding the hidden platform on the fourth day. *P < 0.05 by two‐way ANOVA with repeated measures. (C) Similarly, in the probe trial, TPM‐, LEV‐, and VPA‐treated mice travelled into the southeastern quadrant, where the hidden platform was placed, significantly more times than did control mice. *P < 0.05 by one‐way ANOVA.
Both TPM and LEV Up‐Regulate Aβ Transport Across the Blood–Brain Barrier in APPswe/PS1dE9 Double‐Transgenic Mice
To clarify the mechanisms by which TPM and LEV inhibited plaque deposition, the Aβ concentrations in brain and peripheral blood were examined by ELISA. As expected, TPM, LEV, and VPA reduced the brain soluble Aβ42 concentration to 28.8%, 31.1%, and 32.2%, respectively (670.03 ± 143.83 pg/mg protein, 647.91 ± 122.65 pg/mg protein, and 638.11 ± 63.45 pg/mg protein vs. 940.53 ± 105.66 pg/mg protein, P < 0.05; Figure 3A). Meanwhile, TPM, LEV, and VPA reduced brain insoluble Aβ42 concentration to 37.2%, 42.9%, and 43.3%, respectively (59.81 ± 10.17 pg/mg protein, 54.38 ± 9.80 pg/mg protein, and 54.00 ± 9.22 pg/mg protein vs. 95.22 ± 3.28 pg/mg protein, P < 0.05; Figure 3B). Post hoc analysis detected no significant differences between TPM and VPA or between LEV and VPA in brain soluble and insoluble Aβ concentrations.
Figure 3.
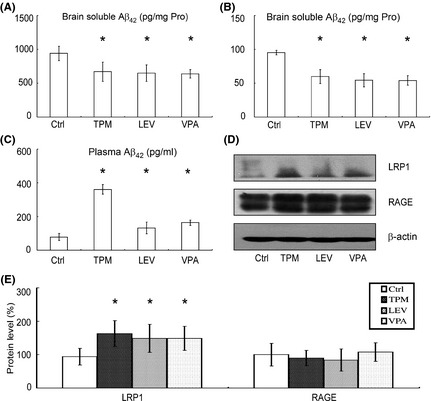
Effects of topiramate (TPM) and levetiracetam (LEV) on the transport of Aβ across the blood–brain barrier in APPswe/PS1dE9 double‐transgenic mice. An ELISA assay was conducted to measure brain soluble Aβ42 (Unit: pg/mg Protein) (A), insoluble Aβ42 (Unit: pg/mg Protein) (B), and peripheral blood Aβ42 (Unit: pg/mL) (C) levels in the brains of mice in each group. TPM, LEV, and valproic acid (VPA) significantly reduced soluble and insoluble Aβ42 levels in brain and increased the peripheral blood Aβ42 level. n = 3. *P < 0.05 by one‐way ANOVA. (D) Half brains from mice in each group were lysed in RIPA buffer. LRP1 and RAGE were detected by Western blotting using β‐actin as a loading control. (E) Quantification of LRP1 and RAGE in brains. TPM, LEV, and VPA significantly increased LRP1 expression, but exerted no significant effect on RAGE expression. n = 3. *P < 0.05 by one‐way ANOVA.
An imbalance between Aβ generation and Aβ clearance results in neuritic plaque formation. Aβ transport across the blood–brain barrier, which is mediated by LRP1 and RAGE, contributes to Aβ removal from the brain. Significantly, treatment with TPM, LEV, and VPA enhanced peripheral blood Aβ42 concentration to 362%, 168%, and 208%, respectively (360.89 ± 49.11 pg/mL, 131.44 ± 34.53 pg/mL, and 162.56 ± 16.06 pg/mL vs. 78.11 ± 21.11 pg/mL, P < 0.05; Figure 3C). Post hoc analysis revealed that the blood Aβ42 concentration in TPM‐treated mice was significantly higher than that in the LEV and VPA groups (P < 0.05). To further explore the mechanism, the levels of LRP1 and RAGE in the brains of each group were examined by Western blotting (Figure 3D). TPM, LEV, and VPA treatment significantly increased the LRP1 level in brains to 173 ± 39.2%, 158 ± 42.0%, and 159 ± 36.1%, respectively (Figure 3E). There were no significant differences in RAGE level between the drug‐treated groups and the vehicle group (Figure 3E).
Both TPM and LEV Up‐Regulate Autophagic Proteins in APPswe/PS1dE9 Double‐Transgenic Mice and SH‐SY5Y Cells
We then measured autophagy‐associated proteins in the brains of APPswe/PS1dE9 transgenic mice. Beclin 1 and LC3 levels were determined by Western blotting. As shown in Figure 4A,B, TPM, LEV, and VPA induced significant increases in the level of beclin 1 to 232 ± 36.1%, 286 ± 66.1%, and 248 ± 48.1%, respectively, and in the levels of LC3‐II/LC3‐I to 207 ± 50.1%, 170 ± 30.1%, and 175 ± 31.2%, respectively. We further examined the levels of beclin 1 and LC3 in SH‐SY5Y cells. As expected, TPM, LEV, and VPA treatment increased the level of beclin 1 to 168 ± 30.4%, 215 ± 48.8%, and 225 ± 62.0%, respectively, and that of LC3‐II/LC3‐I to 224 ± 17.6%, 254 ± 22.0%, and 333 ± 55.3%, respectively (Figure 4C,D). Collectively, these results suggest that the clearance of amyloid plaques by TPM and LEV may be mediated by up‐regulation of autophagy.
Figure 4.
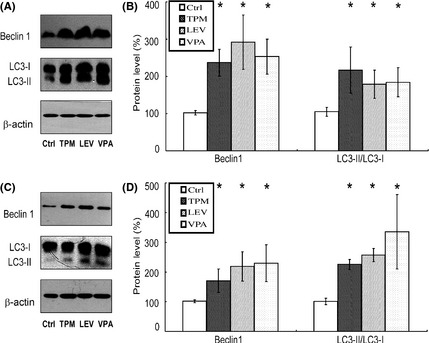
Effects of topiramate (TPM) and levetiracetam (LEV) on the levels of autophagic proteins (beclin1 and LC3) in APPswe/PS1dE9 double‐transgenic mice and SH‐SY5Y cells. (A) The levels of beclin 1 and LC3 in brain tissues from APPswe/PS1dE9 transgenic mice were determined by Western blotting, using β‐actin as a loading control. (B) Quantification of beclin 1 and LC3 in brain tissues. TPM, LEV, and valproic acid (VPA) significantly increased the levels of beclin1 and LC3. n = 3. *P < 0.05 by one‐way ANOVA. (C) The levels of beclin1 and LC3 in SH‐SY5Y cells were determined by Western blotting, using β‐actin as a loading control. (D) Quantification of beclin 1 and LC3 in SH‐SY5Y cells. TPM, LEV, and VPA significantly increased the levels of beclin1 and LC3. n = 3. *P < 0.05 by one‐way ANOVA.
Both TPM and LEV Reduce Tau Phosphorylation in APPswe/PS1dE9 Double‐Transgenic Mice and Okadaic Acid‐Treated SH‐SY5Y Cells
Abnormal hyperphosphorylation of tau is another pathological characteristic of AD. Hence, we asked whether TPM and LEV administration could impact tau phosphorylation. P‐Tau (pSer396) was detected by Western blotting in an animal model (APPswe/PS1dE9 transgenic mice) and a cell model (okadaic acid‐treated SH‐SY5Y cells) (Figure 5A). As shown in Figure 5A, the levels of total tau showed no significant differences between control and drug‐treated groups. However, TPM, LEV, and VPA reduced the expression levels of p‐tau to 48.0 ± 2.20%, 51.1 ± 1.09%, and 24.8 ± 1.24%, respectively, in the animal model, and to 39.9 ± 11.0%, 55.0 ± 16.8%, and 34.0 ± 14.7%, respectively, in the cell model (Figure 5B).
Figure 5.
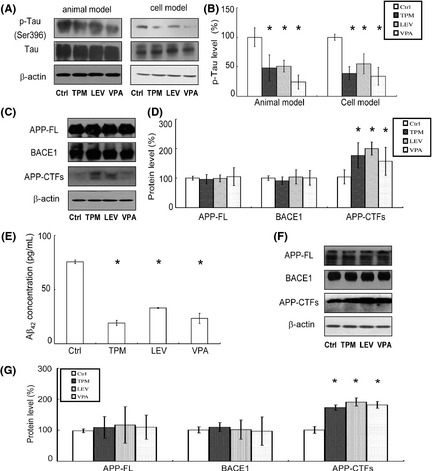
Effects of topiramate (TPM) and levetiracetam (LEV) on phosphorylated tau (p‐tau) and APP processing pathways in APPswe/PS1dE9 double‐transgenic mice and SH‐SY5Y cells. (A) Western blotting was used to detect the levels of phosphorylation of tau at Ser 396 and total tau in brain tissues from APPswe/PS1dE9 transgenic mice and OA‐treated SH‐SY5Y cells in each group. (B) Quantification of p‐tau in brain tissues and cells. TPM, LEV, and valproic acid (VPA) significantly reduced p‐tau expression. n = 3. *P < 0.05 by one‐way ANOVA. (C) Brain tissues from APPswe/PS1dE9 mice were subjected to Western blotting to determine the levels of APP‐full length (FL), BACE1, and APP‐CTFs, with β‐actin as a loading control. (D) Quantification of APP‐FL, BACE1, and APP‐CTFs in brain tissues. TPM, LEV, and VPA significantly increased APP‐CTF production. n = 3. *P < 0.05 by one‐way ANOVA. (E) ELISA assay was conducted to measure the Aβ42 level in the conditioned media of SH‐SY5Y cells. The cells were cultured for 1 week before drug treatment for 24 h. Unit: pg/mL. n = 3. *P < 0.05 by one‐way ANOVA. (F) SH‐SY5Y cells were subjected to Western blotting to determine the levels of APP‐FL, BACE1, and APP‐CTFs, with β‐actin as a loading control. The cells were cultured for 1 week before drug treatment for 24 h. (G) Quantification of APP‐FL, BACE1, and APP‐CTFs in SH‐SY5Y cells. TPM, LEV, and VPA significantly increased APP‐CTF production. n = 3. *P < 0.05 by one‐way ANOVA.
Both TPM and LEV Regulate APP Processing in APPswe/PS1dE9 Double‐Transgenic Mice and SH‐SY5Y cells
We have shown that TPM and LEV inhibit amyloid plaque deposition in APPswe/PS1dE9 transgenic mice. To investigate the underlying mechanisms, the levels of some key proteins in the APP processing pathway were determined by Western blotting, including APP‐full length (FL), BACE1, and APP C‐terminal fragments (CTFs) (Figure 5C). TPM, LEV, and VPA had no influence on APP‐FL or BACE1 levels in animals (Figure 5C,D). Significantly, they increased APP‐CTF levels in mouse brains to 170 ± 33.1%, 192 ± 24.2%, and 151 ± 36.9%, respectively (Figure 5C,D). To further confirm the effects of TPM and LEV on Aβ production, we measured Aβ42 concentration in the conditioned media of cultured SH‐SY5Y cells. TPM, LEV, and VPA significantly reduced Aβ42 concentration compared with vehicle (19.23 ± 2.22 pg/mL, 32.98 ± 0.45 pg/mL, and 23.41 ± 4.69 pg/mL vs. 75.48 ± 1.63 pg/mL, P < 0.05) (Figure 5E). Furthermore, TPM, LEV, and VPA significantly increased APP‐CTF level in SH‐SY5Y cells to 171 ± 9.02%, 189 ± 13.1%, and 180 ± 11.2%, respectively (Figure 5F,G), with no impact on the levels of APP‐FL and BACE1 (Figure 5F,G). Together, these findings show that TPM and LEV decrease Aβ42 production and increased the levels of APP‐CTFs in both APPswe/PS1dE9 transgenic mice and SH‐SY5Y cells. These results suggested that TPM and LEV inhibited the APP amyloidogenic pathway and down‐regulated γ‐secretase cleavage of APP.
Both TPM and LEV Inhibit GSK3β Activation and Increased the Activation of Akt and AMPK in APPswe/PS1dE9 Double‐Transgenic Mice and SH‐SY5Y Cells
We have shown that TPM and LEV decrease Aβ production by inhibiting γ‐secretase cleavage of APP. In addition, TPM and LEV reduced the levels of p‐tau in vivo and in vitro. GSK 3β is a key protein, with a pivotal role in both Aβ production and phosphorylation of tau. To determine whether the protective effects of TPM and LEV were mediated by GSK 3β, the level of p‐GSK 3β (pSer9) (the inhibitory phosphorylation site) in the brains of APPswe/PS1dE9 transgenic mice was measured by Western blotting (Figure 6A). As shown in Figure 5, there were no significant differences in the level of total GSK 3β between vehicle‐ and drug‐treated mouse brains. However, TPM, LEV, and VPA induced significant increases in the level of p‐GSK 3β (pSer9) to 185 ± 43.8%, 190 ± 33.5%, and 237 ± 41.3%, respectively (Figure 6B). Furthermore, the levels of p‐Akt (pSer473) in TPM, LEV, and VPA‐treated mice were increased to 132 ± 4.91%, 140 ± 9.28%, and 128 ± 14.8%, respectively, while those of p‐AMPKα (pThr172) were increased to 296 ± 45.6%, 379 ± 80.6%, and 279 ± 41.1%, respectively (Figure 6B). Consistent with the transgenic mice data, TPM, LEV, and VPA treatment in vitro also increased the level of p‐GSK 3β (pSer9) to 154 ± 14.1%, 188 ± 44.5%, and 208 ± 35.9%, respectively (Figure 6C,D). As expected, the levels of p‐Akt (pSer473) in TPM, LEV, and VPA‐treated SH‐SY5Y cells were increased to 263 ± 54.5%, 244 ± 48.7%, and 298 ± 55.3%, respectively, while those of p‐AMPKα (pThr172) were increased to 161 ± 43.4%, 174 ± 47.2%, and 195 ± 57.9%, respectively (Figure 6C,D). Collectively, our data indicate that both TPM and LEV inhibit GSK3β activation and increased the activation of Akt and AMPK.
Figure 6.
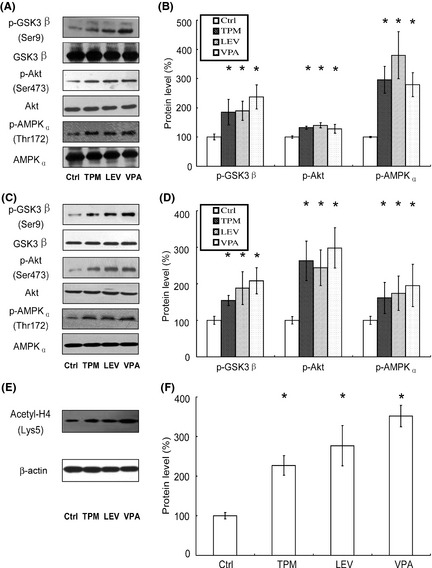
Effects of topiramate (TPM) and levetiracetam (LEV) on p‐GSK 3β, p‐Akt, and p‐AMPK acetylated histone H4 levels in APPswe/PS1dE9 double‐transgenic mice and SH‐SY5Y cells. (A) The levels of p‐GSK 3β, p‐Akt, and p‐AMPK, relative to the levels of total GSK 3β, Akt and AMPK, respectively, in the brain tissues from APPswe/PS1dE9 transgenic mice, were determined by Western blotting. (B) Quantification of p‐GSK 3β, p‐Akt, and p‐AMPK in brain tissues. TPM, LEV, and valproic acid (VPA) significantly increased the ratios of p‐GSK 3β/GSK 3β, p‐Akt/Akt, and p‐AMPK/AMPK. n = 3. *P < 0.05 by one‐way ANOVA. (C) The levels of p‐GSK 3β, p‐Akt, and p‐AMPK relative to the levels of total GSK 3β/Akt/AMPK, respectively, in SH‐SY5Y cells, were determined by Western blotting. (D) Quantification of p‐GSK 3β, p‐Akt, and p‐AMPK in SH‐SY5Y cells. TPM, LEV, and VPA significantly increased the ratios of p‐GSK 3β/GSK 3β, p‐Akt/Akt, and p‐AMPK/AMPK. n = 3. *P < 0.05 by one‐way ANOVA. (E) The levels of acetylated histone H4, relative to the level of β‐actin in the brain tissues from APPswe/PS1dE9 transgenic mice, were determined by Western blotting. (F) Quantification of acetylated histone H4 in brain tissues. TPM, LEV, and VPA significantly increased the ratio of acetylated histone H4/β‐actin. n = 3. *P < 0.05 by one‐way ANOVA.
Both TPM and LEV Increase Histone H4 Acetylation in APPswe/PS1dE9 Double‐Transgenic Mice
To further explore potential mechanism, we investigated the level of acetylated histone H4 in the brains of APPswe/PS1dE9 double‐transgenic mice. As expected, TPM, LEV, and VPA induced significant increases in the level of acetylated histone H4 (Lys5) to 227 ± 1.81%, 277 ± 1.58%, and 351 ± 7.50%, respectively (Figure 6E,F). Our data indicate that protective effects of TPM and LEV in APPswe/PS1dE9 transgenic mice might be associated with inhibition of HDAC.
Discussion
In this study, the new antiepileptic drugs and potential HDAC inhibitors TPM and LEV showed protective effects against behavioral impairment and neuropathology in APPswe/PS1dE9 transgenic mice. First, TPM and LEV alleviated behavioral deficits in APPswe/PS1dE9 transgenic mice. Second, TPM and LEV reduced amyloid plaque burden and tau phosphorylation. Third, TPM and LEV enhanced Aβ transport across the blood–brain barrier in APPswe/PS1dE9 double‐transgenic mice. Fourth, TPM and LEV increased autophagy in vivo and in vitro. Fifth, TPM and LEV inhibited amyloidogenic APP processing at the γ‐secretase site and regulated the activation of AMPK/Akt/GSK3β in vivo and in vitro. At last, TPM and LEV inhibited HDAC activity in vivo.
Histone deacetylase inhibitors have recently been regarded as a new promising therapy for neurodegenerative disorders, because of their neuroprotective, neurotrophic, and anti‐inflammatory properties 27, 28. The classical HDAC inhibitor VPA has shown therapeutic effects in models of cancer, stroke, AD, Parkinson's disease, and experimental autoimmune encephalomyelitis 16, 17, 28, 29, 30, 31, 32, 33, 34. The mechanisms involved in its protective effects against AD might include suppression of spontaneous epileptiform discharges, regulation of the GSK‐3β/β‐catenin/Wnt pathway, and inhibition of inflammation 35, 36. Considering the significant toxic effects in patients with AD treated with VPA in a clinical trial, more effective and safer HDAC inhibitors were needed 37, 38. The present study showed that TPM and LEV inhibited HDAC activity in APPswe/PS1dE9 transgenic mice.
As new antiepileptic drugs, the safety of TPM and LEV is accepted because of fewer drug interactions and simpler pharmacokinetics 39. More importantly, it was shown that TPM increased survival rate and saved hippocampal neurons by protecting hippocampal mitochondria against external calcium challenge 19. In neonatal rat model of right carotid artery ligation, TPM extended the therapeutic window for hypothermia‐mediated neuroprotection 40. TPM promoted neurological recovery in rats after traumatic brain injury 41. In rat model of cervical spinal cord injury, TPM increased tissue sparing and preserved oligodendrocytes 42. In the rat middle cerebral artery occlusion model, LEV significantly reduced the infarct volume 43. In experimental status epilepticus, LEV protected against mitochondrial dysfunction 44. Another study suggested that LEV reduced the decrease in CA3 neuron and attenuated Brain‐derived neurotrophic factor (BDNF) expression in the dentate gyrus, striatum radiatum and CA3 45. Together, Both TPM and LEV have been proven to be neuroprotective in the animal models of other central nervous system diseases. In the present study, our data showed that TPM and LEV ameliorated neuropathology and water maze performance deficits in APPswe/PS1dE9 transgenic mice.
Both Aβ generation and Aβ clearance are pivotal processes in the deposition of amyloid plaques. GSK‐3β regulates Aβ generation by inhibition of γ‐secretase. Meanwhile, autophagy is a key clearance pathway involved in the removal of aberrant aggregates of Aβ. AMPK, which inhibits GSK‐3β activity and decreases mTOR signaling activity to facilitate autophagic degradation of Aβ, has been a focus of recent research 46. It is suggested that AMPK activation might decrease Aβ generation through regulating APP processing 47, 48, 49. Furthermore, AMPK was shown to be involved in the effects of GSK‐3β inhibitors in decreasing Aβ production 50. Consistent with studies from other groups, our results suggested that TPM and LEV reduced Aβ generation, inhibited γ‐secretase activity, and modulated the activity of the AMPK/Akt/GSK‐3β pathway in vivo and in vitro. On the other hand, AMPK activation could control mTOR signaling, autophagy, and Aβ clearance 51. Autophagy has been shown to be dysfunctional in patients with AD, and modulation of autophagy has become a new therapeutic target for the treatment of AD 52, 53, 54. In the present in vivo and in vitro study, TPM and LEV increased activation of AMPK, which might enhance autophagy. Further work is needed to explore whether the effects of TPM and LEV were mediated by regulation of AMPK using AMPK activators/inhibitors or small interfering RNAs.
In the classical Aβ cascade hypothesis, AD is caused by an imbalance between Aβ production and clearance, which results in amyloid plaques in brain. A recent study suggested that an impairment of Aβ clearance might be a major cause of sporadic AD 55. Indeed, an animal model of AD showed defective brain‐to‐blood transport of Aβ, which is an important mode of Aβ clearance 56. The most pivotal receptors involved in Aβ transport across the blood–brain barrier are LRP1 (Aβ transport from brain to blood) and RAGE (Aβ transport from blood to brain), which have been regarded as promising new therapeutic targets for treatment of AD 57. Our results indicated that TPM and LEV increased Aβ transport from brain to blood through the up‐regulation of LRP1 expression, with no significant impact on RAGE expression. Further work is needed to investigate the detailed mechanisms involved in the regulation of LRP1 by TPM and LEV.
A recent study showed that chronic treatment with LEV reversed aberrant neural network activity, behavior deficits, and synaptic deficits in hAPPJ20 mice 58. In line with the findings, our study suggested similar protective effects on behavior deficits in APPswe/PS1dE9 transgenic mice. However, prolonged LEV treatment (s.c. for 20 days) did not change Aβ level in the hippocampus of hAPPJ20 mice at 3 month of age (before formation of amyloid plaques) 58. Alteration of Aβ level in the cortex of hAPPJ20 mice was not included. In our study, we found that chronic LEV treatment (i.p. for 30 days) reduced soluble and insoluble Aβ42 level in the brains (including cortex and hippocampus) of APPswe/PS1dE9 mice at 7 month of age (formation of amyloid plaques). The paradoxical effects of LEV on Aβ in different studies might be ascribed to different animal strains and various routes of drug administration. In addition, the different age‐month of the mice in the two studies might also cause the contradictory results.
Topiramate is approved for partial onset and generalized epilepsy in children and adults and for migraine prophylaxis in adults. A randomized double‐blind study showed that TPM‐induced cognitive impairment is dose‐dependent with significant effects at 192 and 384 mg/day, but not at 64 or 96 mg/day 59. Another randomized, double‐blind, placebo‐controlled, multicenter study showed that 100 mg/day TPM exerted no effects on learning, memory, and executive function in migraine patients aged 12 through 17 years 60. It suggested that the effects of TPM on cognition might associate with different dose and various diseases. An experimental study suggested that 20 mg/kg but not 100 mg/kg significantly ameliorated water maze performance deficits in the rat model of pilocarpine‐induced temporal lobe epilepsy 61. To date, there is little information about the effects of TPM on cognition in AD transgenic mice. To avoid side effects associated with large dose, we chose 20 mg/kg TPM in the present study. The present study provided direct evidence for the protective effects of TPM against behavioral impairment in APPswe/PS1dE9 transgenic mice.
To confirm the effect of antiepileptics in vivo, neuron‐like SH‐SY5Y cells were used to obtain the in vitro data. Okadaic acid, a potent inhibitor of PP1 and PP2A phosphatases, was used to induce tau hyperphosphorylation 17. Consistent with previous study, potent HDAC inhibitor TPM and LEV reduced hyperphosphorylated tau in okadaic acid‐treated SH‐SY5Y cells. The effects and mechanisms of antiepileptics on Aβ clearance and production pathway were investigated in vivo, which were further confirmed in vitro SH‐SY5Y cells.
In conclusion, the current study reveals neuroprotective effects of TPM and LEV against the neuropathology and behavioral impairment in APPswe/PS1dE9 transgenic mice. TPM and LEV increased Aβ clearance and up‐regulated Aβ transport and autophagic degradation. TPM and LEV inhibited Aβ generation and suppressed γ‐secretase activity. TPM and LEV inhibited GSK‐3β activation and increased the activation of AMPK/Akt activation. Further, TPM and LEV inhibited HDAC activity in vivo. Therefore, TPM and LEV might have the potential to treat AD effectively in patient care. However, their efficacy and safety should be further investigated in clinical trials.
Conflict of Interest
The authors declare no conflict of interest
Acknowledgments
This work was supported by National Natural Science Foundation of China grant (81271418). We thank Laboratory Animal Center of Nanjing First Hospital for breeding the animals and providing animal experimental facility. This manuscript has been edited and proofread by Edanz Group China.
References
- 1. Cummings JL, Cole G. Alzheimer disease. JAMA 2002;287:2335–2338. [DOI] [PubMed] [Google Scholar]
- 2. Hardy J. A hundred years of Alzheimer's disease research. Neuron 2006;52:3–13. [DOI] [PubMed] [Google Scholar]
- 3. O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci 2011;34:185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med 2012;2:a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hardy JA, Higgins GA. Alzheimer's disease: The amyloid cascade hypothesis. Science 1992;256:184–185. [DOI] [PubMed] [Google Scholar]
- 6. Delrieu J, Ousset PJ, Caillaud C, et al. ‘Clinical trials in Alzheimer's disease’: Immunotherapy approaches. J Neurochem 2012;120(Suppl 1):186–193. [DOI] [PubMed] [Google Scholar]
- 7. Hauser WA, Morris ML, Heston LL, et al. Seizures and myoclonus in patients with Alzheimer's disease. Neurology 1986;36:1226–1230. [DOI] [PubMed] [Google Scholar]
- 8. Hesdorffer DC, Hauser WA, Annegers JF, et al. Dementia and adult‐onset unprovoked seizures. Neurology 1996;46:727–730. [DOI] [PubMed] [Google Scholar]
- 9. Amatniek JC, Hauser WA, DelCastillo‐Castaneda C, et al. Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia 2006;47:867–872. [DOI] [PubMed] [Google Scholar]
- 10. Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol 2009;66:435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Palop JJ, Chin J, Roberson ED, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron 2007;55:697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Minkeviciene R, Rheims S, Dobszay MB, et al. Amyloid beta‐induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci 2009;29:3453–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vogt DL, Thomas D, Galvan V, et al. Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiol Aging 2011;32:1725–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bero AW, Yan P, Roh JH, et al. Neuronal activity regulates the regional vulnerability to amyloid‐β deposition. Nat Neurosci 2011;14:750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bakker A, Krauss GL, Albert MS, et al. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 2012;74:467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qing H, He G, Ly PT, et al. Valproic acid inhibits Abeta production, neuritic plaque formation, and behavioral deficits in Alzheimer's disease mouse models. J Exp Med 2008;205:2781–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu JP, Xie JW, Wang CY, et al. Valproate reduces tau phosphorylation via cyclin‐dependent kinase 5 and glycogen synthase kinase 3 signaling pathways. Brain Res Bull 2011;85:194–200. [DOI] [PubMed] [Google Scholar]
- 18. Eyal S, Yagen B, Sobol E, et al. The activity of antiepileptic drugs as histone deacetylase inhibitors. Epilepsia 2004;45:737–744. [DOI] [PubMed] [Google Scholar]
- 19. Kudin AP, Debska‐Vielhaber G, Vielhaber S, et al. The mechanism of neuroprotection by topiramate in an animal model of epilepsy. Epilepsia 2004;45:1478–1487. [DOI] [PubMed] [Google Scholar]
- 20. Belcastro V, Pierguidi L, Tambasco N. Levetiracetam in brain ischemia: Clinical implications in neuroprotection and prevention of post‐stroke epilepsy. Brain Dev 2011;33:289–293. [DOI] [PubMed] [Google Scholar]
- 21. Thöne J, Ellrichmann G, Faustmann PM, et al. Anti‐inflammatory effects of levetiracetam in experimental autoimmune encephalomyelitis. Int Immunopharmacol 2012;14:9–12. [DOI] [PubMed] [Google Scholar]
- 22. Jankowsky JL, Fadale DJ, Anderson J, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta‐amyloid peptide in vivo: Evidence for augmentation of a 42‐specific gamma secretase. Hum Mol Genet 2004;13:159–170. [DOI] [PubMed] [Google Scholar]
- 23. Malm T, Koistinaho J, Kanninen K. Utilization of APPswe/PS1dE9 Transgenic Mice in Research of Alzheimer's Disease: Focus on Gene Therapy and Cell‐Based Therapy Applications. Int J Alzheimers Dis 2011;2011:517160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shi JQ, Shen W, Chen J, et al. Anti‐TNF‐α reduces amyloid plaques and tau phosphorylation and induces CD11c‐positive dendritic‐like cell in the APP/PS1 transgenic mouse brains. Brain Res 2011;1368:239–247. [DOI] [PubMed] [Google Scholar]
- 25. Rasband WS. ImageJ v1. Bethesda, Maryland: U. S. National Institutes of Health, 1997–2011. http://rsb.info.nih.gov/ij/.
- 26. Kawarabayashi T, Younkin LH, Saido TC, et al. Age‐dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci 2001;21:372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peleg S, Sananbenesi F, Zovoilis A, et al. Altered histone acetylation is associated with age‐dependent memory impairment in mice. Science 2010;328:753–756. [DOI] [PubMed] [Google Scholar]
- 28. Gräff J, Rei D, Guan JS, et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 2012;483:222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Foley AG, Gannon S, Rombach‐Mullan N, et al. Class I histone deacetylase inhibition ameliorates social cognition and cell adhesion molecule plasticity deficits in a rodent model of autism spectrum disorder. Neuropharmacology 2012;63:750–760. [DOI] [PubMed] [Google Scholar]
- 30. Liu XS, Chopp M, Kassis H, et al. Valproic acid increases white matter repair and neurogenesis after stroke. Neuroscience 2012;220:313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xuan A, Long D, Li J, et al. Neuroprotective effects of valproic acid following transient global ischemia in rats. Life Sci 2012;90:463–468. [DOI] [PubMed] [Google Scholar]
- 32. Zhang Z, Zhang ZY, Wu Y, et al. Valproic acid ameliorates inflammation in experimental autoimmune encephalomyelitis rats. Neuroscience 2012;221:140–150. [DOI] [PubMed] [Google Scholar]
- 33. Lv J, Du C, Wei W, et al. The antiepileptic drug valproic acid restores T cell homeostasis and ameliorates pathogenesis of experimental autoimmune encephalomyelitis. J Biol Chem 2012;287:28656–28665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xiong N, Jia M, Chen C, et al. Potential autophagy enhancers attenuate rotenone‐induced toxicity in SH‐SY5Y. Neuroscience 2011;199:292–302. [DOI] [PubMed] [Google Scholar]
- 35. Ziyatdinova S, Gurevicius K, Kutchiashvili N, et al. Spontaneous epileptiform discharges in a mouse model of Alzheimer's disease are suppressed by antiepileptic drugs that block sodium channels. Epilepsy Res 2011;94:75–85. [DOI] [PubMed] [Google Scholar]
- 36. Zhang XZ, Li XJ, Zhang HY. Valproic acid as a promising agent to combat Alzheimer's disease. Brain Res Bull 2010;81:3–6. [DOI] [PubMed] [Google Scholar]
- 37. Fleisher AS, Truran D, Mai JT, et al. Chronic divalproex sodium use and brain atrophy in Alzheimer disease. Neurology 2011;77:1263–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tariot PN, Schneider LS, Cummings J, et al. Chronic divalproex sodium to attenuate agitation and clinical progression of Alzheimer disease. Arch Gen Psychiatry 2011;68:853–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Perucca P, Gilliam FG. Adverse effects of antiepileptic drugs. Lancet Neurol 2012;11:792–802. [DOI] [PubMed] [Google Scholar]
- 40. Liu Y, Barks JD, Xu G, et al. Topiramate extends the therapeutic window for hypothermia‐mediated neuroprotection after stroke in neonatal rats. Stroke 2004;35:1460–1465. [DOI] [PubMed] [Google Scholar]
- 41. Kouzounias K, Kimiskidis VK, Siozos T, et al. Topiramate promotes neurological recovery in a new model of traumatic brain injury in rats. Neuroscience 2011;183:171–177. [DOI] [PubMed] [Google Scholar]
- 42. Gensel JC, Tovar CA, Bresnahan JC, et al. Topiramate treatment is neuroprotective and reduces oligodendrocyte loss after cervical spinal cord injury. PLoS ONE 2012;7:e33519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hanon E, Klitgaard H. Neuroprotective properties of the novel antiepileptic drug levetiracetam in the rat middle cerebral artery occlusion model of focal cerebral ischemia. Seizure 2001;10:287–293. [DOI] [PubMed] [Google Scholar]
- 44. Gibbs JE, Walker MC, Cock HR. Levetiracetam: Antiepileptic properties and protective effects on mitochondrial dysfunction in experimental status epilepticus. Epilepsia 2006;47:469–478. [DOI] [PubMed] [Google Scholar]
- 45. Sugata S, Hanaya R, Kumafuji K, et al. Neuroprotective effect of levetiracetam on hippocampal sclerosis‐like change in spontaneously epileptic rats. Brain Res Bull 2011;86:36–41. [DOI] [PubMed] [Google Scholar]
- 46. Cai Z, Yan LJ, Li K, et al. Roles of AMP‐activated protein kinase in Alzheimer's disease. Neuromolecular Med 2012;14:1–14. [DOI] [PubMed] [Google Scholar]
- 47. Tschäpe JA, Hammerschmied C, Mühlig‐Versen M, et al. The neurodegeneration mutant löchrig interferes with cholesterol homeostasis and Appl processing. EMBO J 2002;21:6367–6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Won JS, Im YB, Kim J, et al. Involvement of AMP‐activated‐protein‐kinase (AMPK) in neuronal amyloidogenesis. Biochem Biophys Res Commun 2010;399:487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vingtdeux V, Giliberto L, Zhao H, et al. AMP‐activated protein kinase signaling activation by resveratrol modulates amyloid‐beta peptide metabolism. J Biol Chem 2010;285:9100–9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cai Z, Li B, Li K, et al. Down‐regulation of amyloid‐β through AMPK activation by inhibitors of GSK‐3β in SH‐SY5Y and SH‐SY5Y‐AβPP695 cells. J Alzheimers Dis 2012;29:89–98. [DOI] [PubMed] [Google Scholar]
- 51. Vingtdeux V, Chandakkar P, Zhao H, et al. Novel synthetic small‐molecule activators of AMPK as enhancers of autophagy and amyloid‐β peptide degradation. FASEB J 2011;25:219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li L, Zhang X, Le W. Autophagy dysfunction in Alzheimer's disease. Neurodegener Dis 2010;7:265–271. [DOI] [PubMed] [Google Scholar]
- 53. Nixon RA, Yang DS. Autophagy failure in Alzheimer's disease–locating the primary defect. Neurobiol Dis 2011;43:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lai AY, McLaurin J. Inhibition of amyloid‐beta peptide aggregation rescues the autophagic deficits in the TgCRND8 mouse model of Alzheimer disease. Biochim Biophys Acta 2012;1822:1629–1637. [DOI] [PubMed] [Google Scholar]
- 55. Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS beta‐amyloid in Alzheimer's disease. Science 2010;330:1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Banks WA, Kumar VB, Farr SA, et al. Impairments in brain‐to‐blood transport of amyloid‐β and reabsorption of cerebrospinal fluid in an animal model of Alzheimer's disease are reversed by antisense directed against amyloid‐β protein precursor. J Alzheimers Dis 2011;23:599–605. [DOI] [PubMed] [Google Scholar]
- 57. Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta‐peptide clearance through transport across the blood‐brain barrier. Stroke 2004;35(11 Suppl 1):2628–2631. [DOI] [PubMed] [Google Scholar]
- 58. Sanchez PE, Zhu L, Verret L, et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc Natl Acad Sci USA 2012;109:E2895–E2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Loring DW, Williamson DJ, Meador KJ, et al. Topiramate dose effects on cognition: Randomized double‐blind study. Neurology 2011;76:131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pandina GJ, Ness S, Polverejan E, et al. Cognitive effects of topiramate in migraine patients aged 12 through 17 years. Pediatr Neurol 2010;42:187–195. [DOI] [PubMed] [Google Scholar]
- 61. Frisch C, Kudin AP, Elger CE, et al. Amelioration of water maze performance deficits by topiramate applied during pilocarpine‐induced status epilepticus is negatively dose‐dependent. Epilepsy Res 2007;73:173–180. [DOI] [PubMed] [Google Scholar]