

Summary
Background
Glutamate homeostasis plays a critical role in mediating the addiction‐related behaviors. Therefore, preventing the disruption or reestablishing of it is a novel strategy for the treatment of addiction. Glutamate transporters are responsible for clearing extracellular glutamate and maintaining glutamate homeostasis. Our previous work demonstrated that aquaporin‐4 (AQP4) deficiency attenuated morphine dependence, but the mechanisms are unclear. According to the recent evidence that AQP4 might form a functional complex with glutamate transporter‐1 (GLT‐1), this study focused on whether AQP4 participates in the modulation of GLT‐1 and glutamate homeostasis in morphine‐dependent mice.
Results
We found that AQP4 knockout prevented the down‐regulations of GLT‐1 expression and glutamate clearance when mice were repeatedly treated with morphine. Further study revealed that inhibition of GLT‐1 by dihydrokainic acid (DHK) initiated morphine dependence in AQP4 knockout mice. In addition, AQP4 knockout abolished both decreases and increases in the extracellular glutamate levels in the prefrontal cortex during repeated morphine treatment and naloxone‐precipitated withdrawal.
Conclusion
AQP4 deficiency suppresses the down‐regulation of GLT‐1, and the disruption of glutamate homeostasis caused by repeated exposure to morphine, pointing to a strategy for maintaining glutamate homeostasis and thereby treating addiction through the modulation of AQP4 function and expression.
Keywords: Aquaporin‐4, Astrocyte, Glutamate homeostasis, Glutamate transporter‐1, Opioid dependence
Introduction
Repeated exposure and withdrawal from opioids or other drugs of abuse produces neuroadaptations that result in addiction‐related behaviors, such as withdrawal syndrome, craving, and relapse. Evidence supports a crucial role for glutamate transmission in both the development and the expression of these behaviors 1. Preventing the disruption of glutamate homeostasis or facilitating its recovery may be effective approaches to treating addiction. The homeostasis of extracellular glutamate is regulated by a number of factors, including group II metabotropic glutamate receptors, excitatory amino acid transporters (EAATs), and cystine–glutamate exchangers. Among these factors, EAATs are primarily responsible for clearing extracellular glutamate. Five subtypes of EAATs have been cloned: EAAT1 (glutamate/aspartate transporter, GLAST), EAAT2 (glutamate transporter‐1, GLT‐1), EAAT3 (excitatory amino acid‐1, EAAC1), EAAT4, and EAAT5 2. Notably, these transporter subtypes have different cellular and brain regional localizations. GLAST is a glial transporter that is widely distributed throughout the brain and mainly found in Bergmann glial cells in the cerebellum. GLT‐1 is an astrocyte‐specific transporter that is widely distributed throughout the entire forebrain, especially in the hippocampus, cerebral cortex, and striatum. EAAC1 is a neuronal transporter that is abundantly expressed in the hippocampus, striatum, and cerebellum. EAAT4 is a neuronal transporter that is present in Purkinje cell dendrites, and EAAT5 is present in photoreceptors and bipolar cells in the retina 3, 4. Although both neurons and astrocytes contain EAATs, GLT‐1 accounts for more than 90% of total glutamate transport activity throughout the central nervous system (CNS) 5. Recent studies have reported that the expression of GLT‐1 mRNA, but not GLAST mRNA, was decreased in several brain regions of morphine‐dependent rats 6, 7. Furthermore, coadministration of the glutamate transporter activator MS‐153 and chronic morphine treatment significantly attenuated the development of morphine tolerance and dependence 8, 9. These results indicate that GLT‐1 is involved in morphine tolerance and dependence.
Aquaporins (AQPs) are a family of water‐selective channels that are ubiquitously distributed throughout numerous tissues and regulate the movement of water through cell membranes 10. Aquaporin‐4 (AQP4) is the most abundant isoform of AQPs in the CNS, and it exhibits a polarized distribution in the astrocytic end‐feet that directly contact blood vessels and synapses. There is now compelling evidence for the involvement of AQP4 in a variety of physiological and cellular functions such as water balance, astrocyte migration, and neural signal transduction 11. Owing to the lack of a specific, nontoxic AQP4 inhibitor, the role of AQP4 in the brain has largely been analyzed using AQP4 knockout mice. Recently, we showed that AQP4 deficiency attenuates the opioid dependence induced by repeated exposure to morphine 12. We also found that AQP4 deficiency decreases the affinity and increases the density of opioid receptors and has no effect on chronic morphine‐induced alterations of opioid receptor characteristics 13, but further work is needed to reveal the mechanisms underlying the role of AQP4 in morphine dependence.
In astrocytes, GLT‐1 exists in a macromolecular complex with AQP4 14, and both its expression and glutamate uptake are down‐regulated by AQP4 deficiency in primary cultured astrocytes 15. These data indicate that AQP4 not only coexists with GLT‐1, but also regulates GLT‐1 function. Given the importance of GLT‐1 in maintaining glutamate homeostasis and morphine dependence and given that it is present with AQP4 in astrocytes, it is reasonable to hypothesize that GLT‐1 may play a critical role in the maintenance of glutamate homeostasis, which contributes to AQP4 deficiency attenuates morphine dependence.
Therefore, this study focused on determining the role of GLT‐1 and glutamate homeostasis in the AQP4 deficiency‐induced attenuation of morphine dependence. We first investigated the changes in glutamate transporters expression and glutamate uptake in response to repeated morphine treatment. In addition, using a selective inhibitor of GLT‐1, we further elucidated the participation of GLT‐1 in the development of morphine dependence in AQP4 knockout mice. We also performed an in vivo microdialysis study to evaluate the levels of extracellular glutamate in the course of morphine dependence and withdrawal.
Materials and Methods
Animals
AQP4‐deficient mice were generated according to previously described methods 16. The mice were housed at a constant temperature (22 ± 1°C) and relative humidity (50%) with a regular light–dark schedule. Food and water were freely available. The animals were treated in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals (1996) and in agreement with the guidelines of the local ethics committee.
Induction of Morphine Dependence and Naloxone‐Precipitated Withdrawal
Male wild‐type and AQP4 knockout mice repeatedly received morphine (30 mg/kg; Qinghai Pharmaceutic Factory, Xining, China) or saline (10 mL/kg) subcutaneous injections three times daily for 5 consecutive days. Five hours after the last morphine or saline injection, naloxone (5 mg/kg, i.p.; Sigma, St. Louis, MO, USA) was injected, and withdrawal symptoms were scored for 60 min. The number of bouts of jumping, rearing, and writhing was counted, and each behavioral sign was assigned a withdrawal score as follows: 0 = no occurrence; 1 = 1–4 occurrences; 2 = 5–9 occurrences; 3 ≥ 10 occurrences 17, 18. The loss of body weight 60 min after administration of naloxone was also measured.
Western Blotting
The mice were sacrificed about 5 h after the final injection of morphine, and the prefrontal cortex, striatum, hippocampus, and cerebellum were rapidly dissected. The preparation of membrane proteins and Western blot were performed as previously described 12, 19.
[3 H] Glutamate Uptake
Glutamate uptake was measured about 4 h after the final morphine injection. The mice were decapitated, and 400 μm slices of the prefrontal cortex, striatum, hippocampus, and cerebellum were obtained using a vibratome slicer (Campden Instruments, Sileby, UK). The slices were incubated for 30 min at 37°C in oxygenated artificial cerebrospinal fluid (ACSF, NaCl 140 mm, CaCl2 1.4 mm, MgCl2 1.2 mm, KCl 2.7 mm, glucose 5 mm, and 0.5% phosphate‐buffered saline, pH 7.4). Glutamate uptake measurements were initiated by adding l‐[3H] glutamate (100 nm, 49.9 Ci/mmol; PerkinElmer, Boston, MA, USA) in oxygenated buffer. After incubation at 37°C for 15 min, the uptake was terminated by washing the slices three times in ice‐cold phosphate‐buffered saline. The slices were then lysed with ice‐cold 0.3 m NaOH, and the level of radioactivity was determined using a liquid scintillation counter (LS6500; Beckman Coulter, Fullerton, CA, USA). The protein concentration of the lysate was measured using BCA assay (Pierce, Rockford, IL, USA).
Microdialysis Procedure
The probe implantation surgery was performed under anesthesia with chloral hydrate (400 mg/kg, i.p.), and the mice were stereotaxically implanted with a stainless steel guide cannula in the middle prefrontal cortex (A + 3.2 mm, L − 0.5 mm, V − 2.5 mm, from the bregma and skull) 20. After surgery, the mice were given at least 5 days to recover and then repeatedly treated with morphine as described earlier.
The night before microdialysis experiments, the mice were implanted with dialysis probe (MD‐2211, 1 mm membrane; Bioanalysis system Inc., West Lafayette, IN, USA) and placed into individual boxes with access to food and water ad libitum. The next morning, the probes were perfused for 4 h with ACSF at a rate of 1 μL/min. Subsequently, morphine (30 mg/kg, s.c.) was injected after two baseline samples were collected (30 min/sample), and samples were then continuously collected for 3 h. The procedure of sample collection for naloxone (5 mg/kg, i.p.) injection was similar to that described above. At the end of the experiment, the probe locations were histologically examined using hematoxylin and eosin (H&E) staining. The animals with misplaced probes were excluded.
High‐Performance Liquid Chromatography
The content of the microdialysis samples was determined using a HP 1100 series HPLC system (Agilent Technologies, Palo Alto, CA, USA) with fluorescence detection after precolumn derivatization with o‐phthaldialdehyde (OPA; Sigma). The procedure was performed as previously described 21, 22.
Intracerebroventricular Injection of DHK and Naloxone‐Precipitated Withdrawal
For microinjection, the cannula was implanted into the lateral ventricle (A − 0.2 mm, L + 1.0 mm, V − 2.5 mm, from the bregma and skull). The surgical procedure was similar to that described above, and the mice were given at least 5 days to recover.
During the experiment, the AQP4 knockout mice received dihydrokainic acid (DHK, Sigma, 10 μg/μL) and morphine injections concurrently. Mice were treated with morphine (30 mg/kg) or saline (10 mL/kg) subcutaneous injections three times daily for 5 consecutive days. DHK or artificial cerebrospinal fluid (ACSF) was administered 30 min prior to every morphine or saline injection. DHK or ACSF was infused in a volume of 1 μL over 4 min, and injectors were left in for an additional minute to allow for diffusion. After the last injection, withdrawal was precipitated by naloxone (5 mg/kg, i.p.), and behavioral signs of withdrawal were observed continuously for 60 min. At the end of each experiment, the locations of cannulas were verified. The animals with misplaced cannulas were excluded from further analysis.
Statistical Analysis
All data are presented as mean ± SEM. The statistical significance of the microdialysis results was calculated using two‐way anova with repeated measurements followed by Bonferroni post hoc test. Other data were tested with two‐way anova followed by Bonferroni post hoc test to analyze the effect of morphine or basal level in each group. In addition, statistical analysis between the two groups was performed with Student's t‐tests. P < 0.05 was considered to indicate statistical significance.
Results
AQP4 Knockout Suppressed the Down‐Regulation of GLT‐1 Expression Induced by Repeated Morphine Treatment
Two‐way anova revealed significant effects of genotype, treatment, and interaction on the GLT‐1 expression (genotype: F prefrontal cortex (1, 28) = 11.10, P < 0.01; F striatum (1, 28) = 7.91, P < 0.01; F hippocampus (1, 24) = 9.26, P < 0.01; and F cerebellum (1, 36) = 23.84, P < 0.01, respectively; treatment: F prefrontal cortex (1, 28) = 9.54, P < 0.01; F striatum (1, 28) = 4.81, P < 0.04; and F cerebellum (1, 36) = 5.20, P < 0.03, respectively; interaction: F prefrontal cortex (1, 28) = 4.68, P < 0.04; F striatum (1, 28) = 7.34, P < 0.01; and F cerebellum (1, 36) = 14.56, P < 0.01, respectively). A reduction in basal GLT‐1 expression was observed in the AQP4 knockout mice. Compared with the wild‐type mice, the basal GLT‐1 expression in the prefrontal cortex, striatum, hippocampus, and cerebellum of AQP4 knockout mice was reduced by 14.71%, 20.43%, 22.12%, and 28.49%, respectively (P < 0.05 and P < 0.01, Figure 1A). When the wild‐type mice were repeatedly treated with morphine (30 mg/kg, s.c., t.i.d., 5 days), GLT‐1 expression levels in the prefrontal cortex, striatum, and cerebellum were reduced by 14.72%, 12.14%, and 17.53%, respectively (P < 0.05, Figure 1A). No significant reduction was observed in the hippocampus. However, repeated morphine treatment did not affect GLT‐1 expression levels in the four examined brain regions in AQP4 knockout mice (Figure 1A). Unlike GLT‐1, neither AQP4 knockout nor morphine treatment had significant effects on GLAST and EAAC1 expressions (Figure 1B, C).
Figure 1.
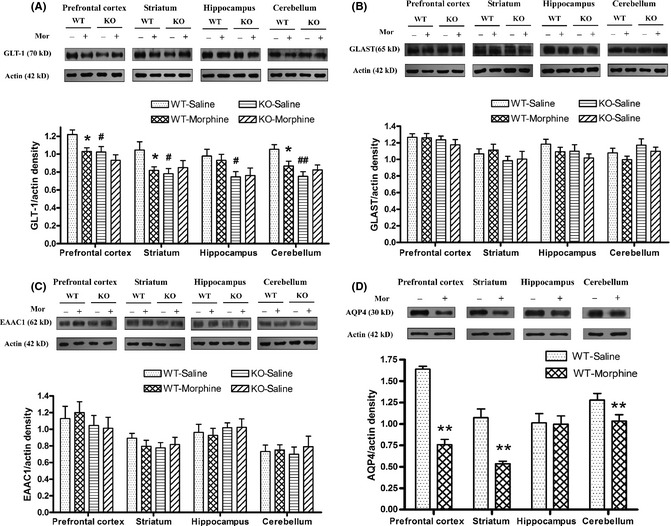
Effects of repeated morphine treatment (30 mg/kg, s.c., t.i.d., 5 days) on the expression of GLT‐1 (A), GLAST (B), EAAC1 (C), and AQP4 (D) in the prefrontal cortex, striatum, hippocampus, and cerebellum of wild‐type (WT) and AQP4 knockout (KO) mice. Data are expressed as mean ± SEM. (n = 6–10 per group). *P < 0.05, **P < 0.01 versus WT‐saline group, # P < 0.05 versus WT‐saline group, two‐way anova followed by Bonferroni post hoc test or t‐test.
In addition, we also demonstrated that chronic morphine treatment down‐regulated AQP4 expression in wild‐type mice. Compared with control, AQP4 expression in the prefrontal cortex, striatum, and cerebellum was significantly decreased by 54, 50, and 19% respectively (P < 0.01, Figure 1D).
These results were similar to those obtained in the cerebrum previously 23. It was reasonable to note that the expressions of GLT‐1 and AQP4 were consistent during chronic morphine treatment, and they played a critical role in morphine dependence.
AQP4 Knockout Suppressed the Reduction in [3 H] Glutamate Uptake Induced by Repeated Morphine Treatment
To investigate the effect of repeated morphine exposure on glutamate transport function in the wild‐type and AQP4 knockout mice, we measured the [3H] glutamate uptake in the four brain regions. Two‐way anova showed significant effects of genotype, treatment, and interaction on the glutamate uptake (genotype: F prefrontal cortex (1, 16) = 9.24, P < 0.01; F striatum (1, 16) = 19.04, P < 0.01; F hippocampus (1, 16) = 8.32, P < 0.01; and F cerebellum (1, 16) = 130.11, P < 0.01, respectively; treatment: F prefrontal cortex (1, 16) = 4.70, P < 0.05; F striatum (1, 16) = 4.95, P < 0.05; and F cerebellum (1, 16) = 14.34, P < 0.01, respectively; interaction: F prefrontal cortex (1, 16) = 6.87, P < 0.05; and F striatum (1, 16) = 8.98, P < 0.01, respectively). Compared with the wild‐type mice, the basal glutamate uptake levels in the prefrontal cortex, striatum, hippocampus, and cerebellum of the AQP4 knockout mice were decreased by 11.02 ± 1.71%, 15.77 ± 3.29%, 5.46 ± 1.76%, and 20.34 ± 2.00%, respectively (P < 0.05 and P < 0.01, Figure 2). In the wild‐type mice, repeated morphine treatment (30 mg/kg, s.c., t.i.d., 5 days) down‐regulated the ability of [3H] glutamate uptake. Compared with the control, [3H] glutamate uptake was reduced by 9.32 ± 1.04%, 4.78 ± 1.04%, and 8.73 ± 1.65% in the prefrontal cortex, striatum, and cerebellum, respectively (P < 0.05 and P < 0.01, Figure 2A, B and D). However, no difference in the [3H] glutamate uptake was observed in the hippocampus (Figure 2C). In addition, after repeated morphine treatment, no significant differences in [3H] glutamate uptake were observed in the four observed brain regions of the AQP4 knockout mice (Figure 2A–D).
Figure 2.
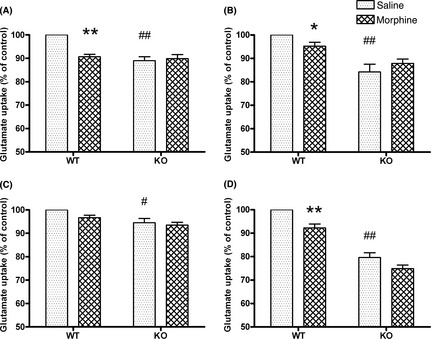
[3 H] glutamate uptake after repeated morphine treatment (30 mg/kg, s.c., t.i.d., 5 days) in the prefrontal cortex (A), striatum (B), hippocampus (C), and cerebellum (D) of wild‐type (WT) and AQP4 (KO) knockout mice. Data are expressed as mean ± SEM. (n = 5 per group). *P < 0.05, **P < 0.01 versus WT‐saline group, # P < 0.05, ## P < 0.01 versus WT‐saline group, two‐way anova followed by Bonferroni post hoc test.
These results suggest that AQP4 deficiency suppressed the down‐regulation of both GLT‐1 expression and function induced by repeated morphine exposure.
Inhibiting the GLT‐1 Function by a Selective GLT‐1 Inhibitor DHK Induced the Morphine Dependence in the AQP4 Knockout Mice
According to the above results, an additional question was raised: does reducing the expression or function of GLT‐1 induce morphine dependence in the AQP4 knockout mice? In this experiment, we used DHK, a selective GLT‐1 inhibitor, to interfere with GLT‐1 function and observed the development of morphine dependence in the AQP4 knockout mice. The mice were challenged with naloxone after repeated morphine exposure accompanying the intracerebroventricular injections of DHK. Four distinct withdrawal signs were quantified with morphine and DHK treatment. A global withdrawal score was calculated by compiling the scores of all observed signs. In Figure 3A, the global withdrawal score in morphine–DHK group was higher than another groups, suggesting that the inhibition of GLT‐1 function by DHK induced the development of morphine dependence in the AQP4 knockout mice (Two‐way anova: morphine F (1, 24) = 252.84, P < 0.01; DHK F (1, 24) = 167.87, P < 0.01; and interaction F (1, 24) = 100.23, P < 0.01). In a sign‐by‐sign analysis (Figure 3B–E), we found that a subset of the withdrawal signs was influenced by the DHK treatment. DHK significantly facilitated rearing, writhing, and weight loss (rearing: morphine F (1, 24) = 50.33, P < 0.01; DHK F (1,24) = 37.40, P < 0.01; and interaction F (1,24) = 34.73, P < 0.01; writhing: morphine F (1, 24) = 39.91, P < 0.01; DHK F (1,24) = 38.01, P < 0.01; and interaction F (1, 24) = 23.05, P < 0.01; weight loss: morphine F (1, 24) = 13.29, P < 0.01; DHK F (1, 24) = 9.54, P < 0.01; and interaction F (1,24) = 5.35, P < 0.01), but it had no significant effect on jumping behavior.
Figure 3.
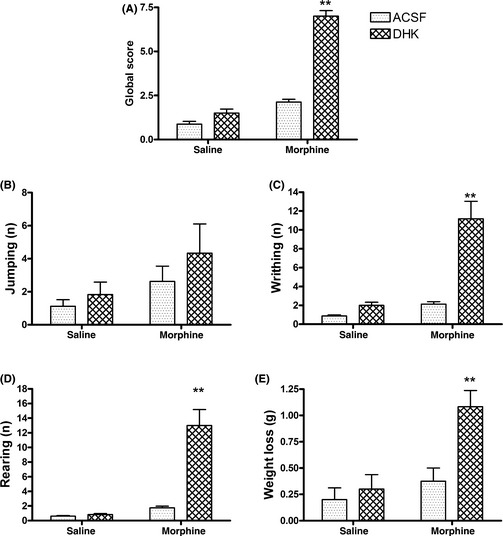
Effects of chronic DHK treatment (10 μg, i.c.v., t.i.d, 5 days) with morphine (30 mg/kg, s.c., t.i.d., 5 days) on behavioral withdrawal signs in AQP4 knockout mice. Global scores (A), jumping,(B), writhing (C), rearing (D), and weight loss (E) were determined for 60 min after naloxone precipitation. Data are expressed as mean ± SEM. (ACSF group, n = 8; DHK group, n = 6). **P < 0.01 versus ACSF–morphine group, two‐way anova followed by Bonferroni post hoc test.
AQP4 Knockout Prevented the Disruption of the Prefrontal Cortical Glutamate Homeostasis Induced by Repeated Morphine Treatment
GLT‐1 is primarily responsible for clearing extracellular glutamate, which is an important role in the maintenance of glutamate homeostasis. With evidence that AQP4 knockout suppressed the down‐regulation of GLT‐1 expression and function induced by repeated morphine exposure, the question remains as to whether it can also prevent the disruption of glutamate homeostasis. The prefrontal cortex not only receives glutamatergic afferents from the mediodorsal thalamus, but it also sends glutamatergic efferents to the nucleus accumbens and the ventral tegmental area. We used the in vivo microdialysis method to evaluate the extracellular glutamate levels in the prefrontal cortex. Compared with the saline‐treated control group, repeated morphine treatment (30 mg/kg, s.c., t.i.d., 5 days) decreased the glutamate levels in the prefrontal cortical of wild‐type mice (interaction (treatment × genotype) F (1, 5) = 28.96, P < 0.01), and this reduction continued during the postmorphine‐injection period, reaching a maximal response of 60.89 ± 3.39% of the baseline value (Figure 4A). After repeated morphine treatment, naloxone precipitation produced a significant increase in the glutamate levels in the prefrontal cortex of the wild‐type mice (interaction (treatment × genotype) F (1, 5) = 11.14, P < 0.05), and it reached a maximum value of 152.05 ± 7.38% of the baseline level 30–60 min after naloxone injection (Figure 4B). However, neither repeated morphine treatment nor naloxone precipitation modulated the extracellular glutamate levels in the prefrontal cortex of the AQP4 knockout mice (Figure 4).
Figure 4.
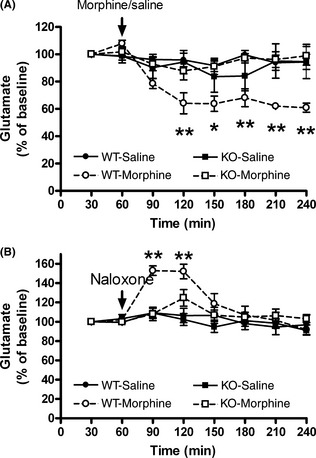
Effects of repeated morphine treatment (30 mg/kg, s.c., t.i.d., 5 days) (A) and naloxone precipitation (5 mg/kg, i.p.) (B) on the extracellular levels of glutamate in the prefrontal cortex of wild‐type (WT) and AQP4 (KO) knockout mice. Data are expressed as mean ± SEM. (n = 6 per group). *P < 0.05, **P < 0.01 versus WT‐saline group; two‐way repeated measures anova followed by Bonferroni post hoc test. The first basal values of extracellular glutamate are as following: (A) WT‐saline: 1.07 ± 0.28 μm; WT‐morphine: 1.03 ± 0.17 μm; KO‐saline: 1.09 ± 0.24 μm; KO‐morphine: 1.11 ± 0.17 μm. (B) WT‐saline: 1.05 ± 0.22 μm; WT‐morphine: 1.10 ± 0.19 μm; KO‐saline: 1.17 ± 0.21 μm; KO‐morphine: 1.11 ± 0.13 μm.
Discussion
Previous studies have demonstrated that the central glutamate system is not only involved in the development of morphine dependence and the expression of morphine withdrawal but also implicated in the craving and relapse to morphine. The extracellular glutamate released from nerve terminals is counterbalanced by the actions of glutamate transporters 23, and the alteration of glutamate transporters expression should impact the clearance of extracellular glutamate. In the present study, we observed that repeated morphine treatment induced the reductions in GLT‐1 expression and glutamate uptake in the wild‐type mice, rather than in the AQP4 knockout mice, suggesting that an AQP4 mechanism may underlie chronic morphine‐induced reduction in GLT‐1 expression and opioid dependence. Now, that the clearance of extracellular glutamate is one of the important mechanisms in the maintenance of glutamate homeostasis, it is reasonable to propose that AQP4 deficiency might be helpful in preventing the disruption of glutamate homeostasis caused by repeated exposure to morphine. In fact, we found that AQP4 knockout abolished both the decrease and the increase in extracellular glutamate levels in the prefrontal cortex under repeated morphine treatment and naloxone‐precipitated withdrawal, respectively, indicating the maintenance of glutamate homeostasis. These findings might provide a potential approach to preventing the disruption of glutamate homeostasis through the modulation of AQP4 function or expression.
AQP4, a member of the water channels in the CNS, coexists with GLT‐1 in astrocytes, and the interaction between these two molecules is believed to involve an intimate spatial relationship between these two components of the same supramolecular complex 24. Evidence has shown that AQP4 deficiency down‐regulates the basal GLT‐1 expression and reduces glutamate uptake in primary cultured astrocytes 15, which mirrors our observations that AQP4 knockout decreased the basal levels of GLT‐1 expression and inhibited glutamate uptake in the brain. In the present study, we also found that both AQP4 and GLT‐1 expressions were concomitantly down‐regulated in the prefrontal cortex, striatum, and cerebellum, but not the hippocampus, of morphine‐treated wild‐type mice. Previous study reported that AQP4 and GLT‐1, but not GLAST, interact as a complex and simultaneously internalize into endosome to down‐regulate their expressions in the plasma membrane 24. Additionally, GLT‐1 protein is up‐regulated in differentiating astrocyte progenitors and in nonneural cells expressing AQP4 transgenically 24. These indicated a potential correlation between the reductions in GLT‐1 expression and AQP4 expression induced by chronic exposure to morphine. It should be hypothesized that AQP4 may be effect morphine dependence through regulating GLT‐1 expression and function. However, further study is needed to clarify this correlation and demonstrate why the decrease in AQP4 expression induces GLT‐1 reduction.
The present study showed that chronic exposure to morphine decreased GLT‐1 expression and function in the wild‐type mice, which should cause an increase in extracellular glutamate levels in principle. However, we found that the last morphine injection produced a reduction in extracellular glutamate level in the wild‐type mice, but not in the AQP4 knockout mice. Although these findings may appear to conflict; in fact, they do not. It is well known that the extracellular glutamate levels depend on both the glutamate release and its uptake. Besides reducing glutamate uptake 25, morphine suppresses glutamate release as well. For instance, morphine decreases capsaicin or K+ ‐evoked release of glutamate in rat spinal dorsal or cerebral cortical slices 26, 27. Additionally, morphine suppresses glutamate release evoked by stimulation of the cerebral cortex through neural pathways in vivo 28. Thus, we observed that extracellular glutamate was decreased after the last morphine injection, which was due to the integrated actions of morphine to glutamate release and uptake. This result is consistent with previous report 29.
DHK, a selective GLT‐1 inhibitor, can reduce GLT‐1 function both in vitro and in vivo 30, 31, 32. A large body of evidence implicates the locus ceruleus (LC) in the expression of morphine withdrawal syndrome. Electrophysiological studies of the LC have demonstrated that LC neurons exhibit hyperactivity during naloxone‐precipitated withdrawal. One mechanism underlying this phenomenon is that increased glutamatergic afferent input induces LC hyperactivity during opioid withdrawal 18. Local over‐expression of GLT‐1 in the LC, the systemic injection of glutamate transporter activators, or the intracerebroventricular injection of glutamate could inhibit or enhance the development of morphine dependence and the expression of withdrawal signs 8, 21, 33. When we coadministered intracerebroventricular injections of DHK and morphine in the AQP4 knockout mice, mimicking the inhibition of GLT‐1 function in the wild‐type mice, we observed somatic signs of naloxone‐precipitated withdrawal, including weight loss, rearing, and writhing, indicating that morphine dependence developed in the AQP4 knockout mice. These data further supported that preventing the reduction in the GLT‐1 function was important to the attenuation of morphine dependence that results from AQP4 deficiency.
Besides glutamate system adaptations, opioid receptor and dopamine system adaptations also play a fundamental role in opioid dependence. It has been reported that AQP4 deficiency affects extracellular dopamine levels induced by depolarizing stimuli 34 or acute and chronic cocaine exposure 21. In addition, our previous study showed that AQP4 deficiency decreases the affinity and increases the density of opioid receptors 13. However, AQP4 deficiency does not affect chronic morphine‐induced decrease in the affinity of opioid receptors. Thus, AQP4 might also modulate addiction‐related behaviors, such as withdrawal syndrome, sensitization, craving, and relapse, through its regulating dopamine neurotransmission and opioid receptors' binding. Moreover, AQP4 deficiency also enhanced morphine analgesia, attenuated morphine tolerance 12, suggesting that AQP4, as a water channel, could modulate multiple actions of opioids.
Taken together, the present study found that AQP4 deficiency suppressed the reductions in GLT‐1 expression and extracellular glutamate clearance caused by repeated exposure to morphine. Furthermore, AQP4 deficiency prevented the disruption of glutamate homeostasis. These findings may provide a potential approach to preventing the adaptation of glutamate neurotransmission and thereby treating addiction through the modulation of AQP4 function or expression.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgment
This research was supported by grants from the National Basic Research Program of China (2009CB522008) and the Beijing Nature Science Foundations (No. 7113162 and No. 7102124).
The first two authors contributed equally to this work.
References
- 1. Kalivas PW, McFarland K, Bowers S, Szumlinski K, Xi ZX, Baker D. Glutamate transmission and addiction to cocaine. Ann N Y Acad Sci 2003;1003:169–175. [DOI] [PubMed] [Google Scholar]
- 2. Danbolt NC. Glutamate uptake. Prog Neurobiol 2001;65:1–105. [DOI] [PubMed] [Google Scholar]
- 3. Tao YX, Gu J, Stephens RL Jr. Role of spinal cord glutamate transporter during normal sensory transmission and pathological pain states. Mol Pain 2005;1:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Camacho A, Massieu L. Role of glutamate transporters in the clearance and release of glutamate during ischemia and its relation to neuronal death. Arch Med Res 2006;37:11–18. [DOI] [PubMed] [Google Scholar]
- 5. Romera C, Hurtado O, Mallolas J, et al. Ischemic preconditioning reveals that GLT1/EAAT2 glutamate transporter is a novel PPARgamma target gene involved in neuroprotection. J Cereb Blood Flow Metab 2007;27:1327–1338. [DOI] [PubMed] [Google Scholar]
- 6. Ozawa T, Nakagawa T, Shige K, Minami M, Satoh M. Changes in the expression of glial glutamate transporters in the rat brain accompanied with morphine dependence and naloxone‐precipitated withdrawal. Brain Res 2001;905:254–258. [DOI] [PubMed] [Google Scholar]
- 7. Nakagawa T, Satoh M. Involvement of glial glutamate transporters in morphine dependence. Ann N Y Acad Sci 2004;1025:383–388. [DOI] [PubMed] [Google Scholar]
- 8. Nakagawa T, Ozawa T, Shige K, Yamamoto R, Minami M, Satoh M. Inhibition of morphine tolerance and dependence by MS‐153, a glutamate transporter activator. Eur J Pharmacol 2001;419:39–45. [DOI] [PubMed] [Google Scholar]
- 9. Mao J, Sung B, Ji RR, Lim G. Chronic morphine induces downregulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 2002;22:8312–8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Agre P, King LS, Yasui M, et al. Aquaporin water channels from atomic structure to clinical medicine. J Physiol 2002;542:3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Verkman AS, Binder DK, Bloch O, Auguste K, Papadopoulos MC. Three distinct roles of aquaporin‐4 in brain function revealed by knockout mice. Biochim Biophys Acta 2006;1758:1085–1093. [DOI] [PubMed] [Google Scholar]
- 12. Wu N, Lu XQ, Yan HT, et al. Aquaporin 4 deficiency modulates morphine pharmacological actions. Neurosci Lett 2008;448:221–225. [DOI] [PubMed] [Google Scholar]
- 13. Wang JF, Wang ZY, Wu N, Yan HT, Li J. Effects of aquaporin4 deficiency on opioid receptors characteristics in naive and chronic morphine‐treated mice. Neurosci Lett 2009;457:111–114. [DOI] [PubMed] [Google Scholar]
- 14. Vitellaro‐Zuccarello L, Mazzetti S, Bosisio P, Monti C, De Biasi S. Distribution of Aquaporin 4 in rodent spinal cord: relationship with astrocyte markers and chondroitin sulfate proteoglycans. Glia 2005;51:148–159. [DOI] [PubMed] [Google Scholar]
- 15. Zeng XN, Sun XL, Gao L, Fan Y, Ding JH, Hu G. Aquaporin‐4 deficiency down‐regulates glutamate uptake and GLT‐1 expression in astrocytes. Mol Cell Neurosci 2007;34:34–39. [DOI] [PubMed] [Google Scholar]
- 16. Fan Y, Zhang J, Sun XL, et al. Sex‐and region‐specific alterations of basal amino acid and monoamine metabolism in the brain of aquaporin‐4 knockout mice. J Neurosci Res 2005;82:458–464. [DOI] [PubMed] [Google Scholar]
- 17. Dehpour AR, Sadr SS, Nouroddini M, et al. Comparison of simultaneous administration of lithium with L‐NAME or L‐arginine on morphine withdrawal syndrome in mice. Hum Psychopharmacol 2000;15:87–93. [DOI] [PubMed] [Google Scholar]
- 18. Sullivan ME, Hall SR, Milne B, Jhamandas K. Suppression of acute and chronic opioid withdrawal by a selective soluble guanylyl cyclase inhibitor. Brain Res 2000;859:45–56. [DOI] [PubMed] [Google Scholar]
- 19. Zeng XN, Xie LL, Liang R, Sun XL, Fan Y, Hu G. AQP4 knockout aggravates ischemia/reperfusion injury in mice. CNS Neurosci Ther 2012;18:388–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paxions G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates, 2nd edn San Diego: Academic Press, 2001. [Google Scholar]
- 21. Li Z, Gao L, Liu Q, et al. Aquaporin‐4 knockout regulated cocaine‐induced behavior and neurochemical changes in mice. Neurosci Lett 2006;403:294–298. [DOI] [PubMed] [Google Scholar]
- 22. Huang X, Kong H, Tang M, Lu M, Ding JH, Hu G. D‐Serine Regulates Proliferation and Neuronal Differentiation of Neural Stem Cells from Postnatal Mouse Forebrain. CNS Neurosci Ther 2012;18:4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ozawa T, Nakagawa T, Sekiya Y, Minami M, Satoh M. Effect of gene transfer of GLT‐1, a glutamate transporter, into the locus coeruleus by recombinant adenoviruses on morphine physical dependence in rats. Eur J Neurosci 2004;19:221–226. [DOI] [PubMed] [Google Scholar]
- 24. Hinson SR, Roemer SF, Lucchinetti CF, et al. Aquaporin‐4‐binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down‐regulating EAAT2. J Exp Med 2008;205:2473–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu NJ, Bao L, Fan HP, et al. Morphine withdrawal increases glutamate uptake and surface expression of glutamate transporter GLT1 at hippocampal synapses. J Neurosci 2003;23:4775–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ueda M, Sugimoto K, Oyama T, Kuraishi Y, Satoh M. Opioidergic inhibition of capsaicin‐evoked release of glutamate from rat spinal dorsal horn slices. Neuropharmacology 1995;34:303–308. [DOI] [PubMed] [Google Scholar]
- 27. Nicol B, Rowbotham DJ, Lambert DG. mu‐ and kappa‐opioids inhibit K+ evoked glutamate release from rat cerebrocortical slices. Neurosci Lett 1996;218:79–82. [DOI] [PubMed] [Google Scholar]
- 28. Coutinho‐Netto J, Abdul‐Ghani AS, Bradford HF. Morphine suppression of neurotransmitter release evoked by sensory stimulation in vivo. Biochem Pharmacol 1982;31:1019–1023. [DOI] [PubMed] [Google Scholar]
- 29. Hao Y, Yang JY, Guo M, Wu CF, Wu MF. Morphine decreases extracellular levels of glutamate in the anterior cingulate cortex: an in vivo microdialysis study in freely moving rats. Brain Res 2005;1040:191–196. [DOI] [PubMed] [Google Scholar]
- 30. Zschocke J, Bayatti N, Clement AM, et al. Differential promotion of glutamate transporter expression and function by glucocorticoids in astrocytes from various brain regions. J Biol Chem 2005;280:34924–39232. [DOI] [PubMed] [Google Scholar]
- 31. Rasmussen BA, Baron DA, Kim JK, Unterwald EM, Rawls SM. β‐Lactam antibiotic produces a sustained reduction in extracellular glutamate in the nucleus accumbens of rats. hippocampal synapses. J Neurosci 2003;23:4775–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thomson LM, Zeng J, Terman GW. Differential effect of glutamate transporter inhibition on EPSCs in the morphine naïve and morphine tolerant neonatal spinal cord slice. Neurosci Lett 2006;407:64–69. [DOI] [PubMed] [Google Scholar]
- 33. Tokuyama S, Wakabayashi H, Ho IK. Direct evidence for a role of glutamate in the expression of the opioid withdrawal syndrome. Eur J Pharmacol 1996;295:123–129. [DOI] [PubMed] [Google Scholar]
- 34. Ding JH, Sha LL, Chang J, Zhou XQ, Fan Y, Hu G. Alterations of striatal neurotransmitter release in aquaporin‐4 deficient mice: an in vivo microdialysis study. Neurosci Lett 2007;422:175–180. [DOI] [PubMed] [Google Scholar]