

Paroxysmal dyskinesias are a rare heterogeneous group of conditions manifesting as abnormal involuntary movements that recur episodically and last only for a brief duration. The abnormal movements may be dystonic, choreic, ballistic, or a mixture of these [1, 2]. Such conditions can be divided into three types: paroxysmal kinesigenic dyskinesia (PKD), paroxysmal nonkinesigenic dyskinesia, and paroxysmal exercise‐induced dyskinesia [3]. Generally speaking, these dyskinesias are primary forms. They often occur in adolescence and respond well to antiepilepsy drugs such as phenitoin or carbamazepine. Some of these patients have a family history. Apart from these primary forms, there still are so‐called secondary (“symptomatic”) paroxysmal dyskinesias, which are often caused by environmental factors or other diseases [4].
Hashimoto encephalopathy (HE), presently known as Steroid Responsive Encephalopathy associated with Autoimmune Thyroiditis (SREAT), is a potentially fatal disease. Its manifestations are quite variable. Two distinct forms have been described: one was a vasculitic type characterized by multiple relapsing‐remitting stroke‐like episodes and mild cognitive impairment; the other was a diffuse progressive type characterized by dementia and psychiatric symptoms [5, 6]. Antineuronal antibodies were thought to be associated with the pathogenesis of HE [7]. Recently, we encountered a patient whose symptoms began with PKD and finally culminated in global confusion and dementia. He was diagnosed with HE, and recovered completely after steroid treatment.
A 75‐year‐old man was admitted to the Department of Neurology in October 2011. His wife and other family members reported that he began to develop involuntary movements on the left limbs 5 months ago. They noticed the symptoms for the first time while he was having lunch. When he wanted to take up the bowl on the table, he could not control his left arm and hand. The arm made a choreic and ballistic gesture, so he threw his bowl away. At first, this symptom occurred 2–3 times daily. Gradually, the frequency and amplitude increased day after day. They noticed that the attack was always induced by a sudden voluntary movement, for example getting up quickly to answer the telephone, or crossing the road while the traffic lights became green, but it never occurred during sleep. This condition was limited on the left side for nearly three months. He had seen doctors for several times and was prescribed carbamazepine for several days; however, this therapy could not stop the attacks. The frequency even increased to 120–150 times daily before admission. In the mean time, his family members noticed he became “stupid” and silent. He could not answer simple questions and could not even recall familiar people and places.
On admission, physical examinations showed normal body temperature, breath rate, heart rate, and blood pressure. Neurological examination was remarkable for mild lethargy and unclear speech. He was unable to cooperate when tested for comprehension, orientation, memory, and calculation. Cranial nerve functions were intact. Muscle strength was normal and Babinski sign negative. There were no signs of meningeal irritation.
Routine haematological results, including cancer markers, peripheral antineutrophil cytoplasmic antibodies, cytoplasmic antineutrophil cytoplasmic antibodies, erythrocyte sedimentation rate, antiphosphalipid antibody, antinuclear antibody, rheumatoid factor, systemic lupus erythematosus, serum Vitamin B12, and folic acid were normal or negative. His liver and kidney functions were unremarkable. Blood work for infectious diseases (Epstein–Barr virus, cytomegalovirus, simplex virus, Herpes zoster virus) was negative. The free thyroxine T3, T4, and TSH in the blood were normal. B‐type ultrasound examination showed normal gland without nodes or calcification.
His dynamic EEG for 24 h showed generalized slow waves (Figure 1), there was no focal or lateralized spike or sharp‐slow waves throughout the whole recording period. The brain MR imaging, including T1, T2, and DW were all normal (not shown).
Figure 1.
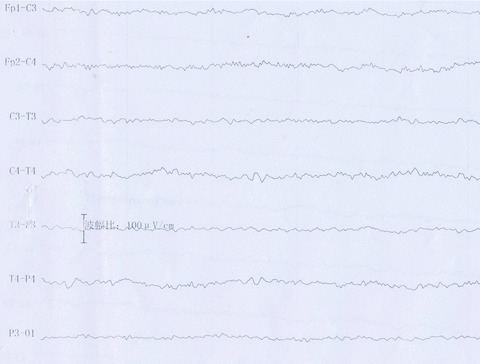
Before treatment, dynamic EEG demonstrated diffuse low and medium‐amplitude voltage, with the background of theta‐activities. There was no sharp or spike‐slow waves throughout the recording period.
A lumbar puncture showed mild increase in protein 900 mg/L (normally, 150–450 mg/L), with normal cell count and glucose. The IgG, IgA, and IgM in the cerebrospinal fluid was 116 mg/L (normally, 10–50 mg/L), 18.9 mg/L (normally, 0–11.1 mg/L), and 1.4 mg/L (normally, 0–11.1 mg/L), respectively.
The antithyroidglobulin antibody and the anti‐TSH antibody were normal in the blood; however, the antithyroid peroxidase antibody was significantly elevated (more than 340 IU/mL), which decreased to 179.82 IU/mL after 1‐month treatment.
After excluding infectious diseases and Creutzfeldt–Jacob disease, HE was considered. He was administered methyprednisolone 500 mg, IV, once a day, for 15 days, 250 mg for another fortnight, and then tapered gradually till 20 mg prednisone, thrice a day.
Ten days after steroid treatment, his consciousness began to improve, and the frequency of dyskinesia began to decrease; 20 days after treatment, the PKD symptom completely disappeared. Thereafter, progressive clinical improvement was observed. On the previous day of his discharge, the EEG showed normal (Figure 2).
Figure 2.
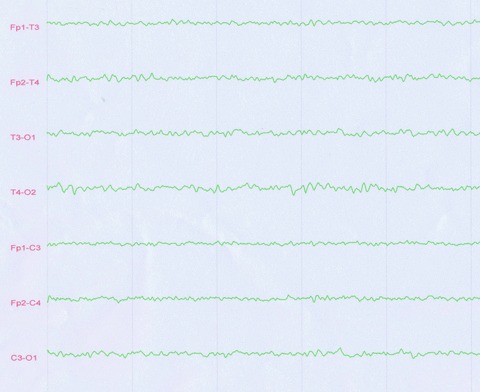
After treatment for 1 month, routine EEG demonstrated medium‐amplitude voltage, with the background of alpha‐activities.
This is the first report of HE, which begins with PKD to our knowledge. PKD is often primary (genetic or idiopathic), which occurs at the age between 5 and 20 years and responds well to antiepilepsy drugs. However, Blakeley et al. found that a specific cause had been identified in some cases. These causes include peripheral trauma, central trauma, vascular lesions, multiple sclerosis, menigovascular syphilis, and kernicterus [7]. Such secondary dyskinesias were easily controlled with anticonvulsant therapy, requiring only one agent. What is different in our patient is that his PKD could not be stopped, because it is unresponsive to carbamazepine or other antiepilepsy drugs, such as valproate, or clonazepam. Further, it is just the initial symptom of the whole course. As the disease progressed, other mental disorders such as global confusion and lethargy predominated over the clinical aspects.
The clinical features, laboratory findings, and his responsiveness to steroid treatment are in complete accordance with the revised criteria for SREAT made by Castillo et al. [8]. The pathogenesis and etiology of HE has not yet been identified. Currently, the most broadly accepted hypothesis is that an autoimmune etiology results in either cerebral vasculitis or direct injury from autoantibodies [8]. Although we could not see any abnormalities in the MR imaging, there must be some subcellular changes in the brain, at least some functional disturbance, as seen in the EEG before treatment. Zhao et al. performed brain biopsy in a patient and found reduced density of cortical tissue with neuron loss, swelling of surviving neurons, reactive gliosis, and angiogenesis. Transmission electron microscopy showed vacuolar degeneration of some neurons with swollen mitochondria and microglia activation. Formation of small cysts, demyelination, and sponge‐like changes within white matter were also found [9]. However, Castillo et al. performed brain biopsy in two patients, and found the histological findings were entirely normal in one patient, with only meningeal thicking and dural enhancement [8].
In conclusion, this report is interesting and has important clinical significance. Facing a patient with PKD, which is not responsive to antiepilepsy drugs, and especially if the patient undergoes progressive global confusion, clinicians should consider the possibility of HE. The elevated antithyroid antibodies in the serum, especially anti‐TPO antibody, are important markers in evaluating this condition.
Conflict of Interest
The authors have no conflict of interest.
References
- 1. Bhatia KP. Familial (idiopathic) paroxysmal dyskinesias: An update. Semin Neurol 2001;21:69–74. [DOI] [PubMed] [Google Scholar]
- 2. Demirkiran M, Jankovic J. Paroxysmal dyskinesias: Clinical features and classification. Ann Neurol 1995;38:571–579. [DOI] [PubMed] [Google Scholar]
- 3. Bhatia KP. Paroxysmal dyskinesias. Mov Disord 2011, 26:1157–65. [DOI] [PubMed] [Google Scholar]
- 4. Blakeley J, Jankovic J. Secondary paroxysmal dyskinesias. Mov Disord 2002;17:726–734. [DOI] [PubMed] [Google Scholar]
- 5. Lee SW, Donlon S, Caplan JP. Steroid responsive encephalopathy associated with autoimmune thyroiditis (SREAT) or Hashimoto's encephalopathy: A case and review. Psychosomatics 2011;52:99–108. [DOI] [PubMed] [Google Scholar]
- 6. Mijajlovic M, Mirkovic M, Dackovic J, Zidverc‐Trajkovic J, Sternic N. Clinical manifestations, diagnostic criteria and therapy of Hashimoto's encephalopathy: Report of two cases. J Neurol Sci 2010;288:194–196. [DOI] [PubMed] [Google Scholar]
- 7. Oide T, Tokuda T, Yazaki M, et al Anti‐neuronal autoantibody in Hashimoto encephalopathy: Neuropathological, immunohistochemical, and biochemical analysis of two patients. J Neurol Sci 2004;217:7–12. [DOI] [PubMed] [Google Scholar]
- 8. Castillo P, Woodruff B, Caselli R, et al Steroid‐responsive encephalopathy associated with autoimmune thyroiditis. Arch Neurol 2006;63:197–202. [DOI] [PubMed] [Google Scholar]
- 9. Zhao W, Li J, Wang J, et al A case of Hashimoto encephalopathy: Clinical manifestation, imaging, pathology, treatment and prognosis. The Neurologist 2011;17:141–143. [DOI] [PubMed] [Google Scholar]