

Summary
Aim
Conclusions on the association between polymorphisms in the vascular endothelial growth factor (VEGF) gene promoter and risk of Alzheimer's disease (AD) are ambiguous, and sufficient evaluation of the association is lacking. Therefore, we performed a meta‐analysis of observational studies to explore the association between polymorphisms in the VEGF gene promoter and AD risk.
Methods
Bibliographical searches were performed in the MEDLINE, EMBASE, and China National Knowledge Infrastructure (CNKI) databases without any language limitations. Three investigators independently assessed abstracts for relevant studies and independently reviewed all eligible studies. A meta‐analysis was conducted using a fixed‐ or random‐effects model. Odds ratios (ORs) and their 95% confidence intervals (CIs) were used to assess the strength of association. All statistical analyses were performed using Stata 11.0 software.
Results
The meta‐analysis of 2787 AD cases and 2841 controls from eight published case‐control studies on the ‐2578C/A polymorphism and 1422 AD cases and 1063 controls from four studies on the ‐1154G/A polymorphism did not show any significant associations. However, in a subgroup analysis stratified by the presence of APOE є4, associations were observed with APOE ε4 (‐) for ‐2578C/A (A vs. C: OR = 1.22, 95% CI = 1.04–1.43, P = 0.014; A/A vs. C/C: OR = 1.59, 95% CI = 1.11–2.27, P = 0.011 and A/A vs. A/C + C/C: OR = 1.46, 95% CI = 1.08–1.99, P = 0.015) and ‐1154G/A (A vs. G: OR = 0.74, 95% CI = 0.62–0.89, P = 0.001; A/A vs. G/G: OR = 0.57, 95% CI = 0.37–0.87, P = 0.009; A/G vs. G/G: OR = 0.69, 95% CI = 0.53–0.89, P = 0.004 and A/A + A/G vs. G/G: OR = 0.66, 95% CI = 0.52–0.85, P = 0.001).
Conclusion
This meta‐analysis showed the risk role of the ‐2578 polymorphism and the protective role of the ‐1154 polymorphism when the APOE є4 status was negative, suggesting that the two polymorphisms in the VEGF promoter may have opposing effects on AD risk in an APOE є4‐independent manner.
Keywords: Alzheimer's disease, Meta‐analysis, Polymorphism, Promoter, Vascular endothelial growth factor
Introduction
Alzheimer's disease (AD), also called senile dementia of the Alzheimer type or primary degenerative dementia of the Alzheimer's type, is a degenerative disease of the central nervous system characterized by progressive cognitive impairment and memory damage. According to data from the World Alzheimer Report, the number of people with AD is forecast to nearly double every 20 years, from 36 million in 2010 to 115 million in 2050, and the costs associated with AD are US$604 billion, approximately 1% of global GDP 1. Therefore, it is particularly urgent to gain insight into the pathogenic factors of AD to discover different strategies for preventive and effective treatment.
The first case of AD was identified in 1901 in a 50‐year‐old woman by German psychiatrist Alois Alzheimer 2; however, its pathological cause, like many other mental diseases, is still unclear. Recently, it was reported that vascular endothelial growth factor (VEGF), which was first purified from the conditioned medium of bovine pituitary follicular stellate cells 3, 4 and is one of the major regulators of angiogenesis 5, may be associated with AD pathogenesis. VEGF has been reported to play a complex role in AD development. Lower concentrations of VEGF have been found in the serum of patients with AD, and decreased serum VEGF levels have been associated with AD in a dose‐dependent manner 6. Furthermore, diminished VEGF immunoreactivity was observed in particularly lesion‐prone regions of AD brains compared with control brains 7. However, other evidence showed that intrathecal levels of VEGF were significantly increased in patients with AD 8, and microvessels isolated from AD brains expressed significantly higher levels of VEGF than microvessels obtained from control brains 9. Moreover, enhanced VEGF immunoreactivity is present in the neocortex of AD brains compared with control brains 10. These findings suggest that VEGF may exert dual effects on the pathological mechanism of AD.
VEGF, the gene encoding the VEGF protein, is located on chromosome 6p21.3, one of the common susceptibility loci associated with AD risk 11, and it consists of eight exons 12. Studies have reported that genetic variants in the promoter of the VEGF gene are associated with significantly different VEGF promoter activities and responsiveness, which affects the VEGF expression level 13.
Furthermore, increased or decreased VEGF expression levels are present in AD patients compared with controls 6, 8, 9. Therefore, VEGF promoter polymorphisms may be associated with the differential expression levels of VEGF linked to AD risk. Recently, the association between two polymorphisms (‐1154G/A and ‐2578C/A) of the VEGF promoter and AD risk has been hotly debated in many studies. However, the conclusions of these studies are ambiguous, and sufficient evaluation of the association is still lacking. Therefore, we performed a meta‐analysis of the existing epidemiologic studies using a comprehensive search strategy to determine whether there is an association between VEGF polymorphisms and AD risk.
Materials and Methods
Study Selection
This meta‐analysis was performed according to the methodology advocated by the MOOSE guideline 14. We performed a systematic electronic search using the following terms in the PubMed database (from January 1965 to December 2012): “(Alzheimer* or AD) and (vascular endothelial growth factor or VEGF) and (polymorphism* or genotype* or variant*)”. We subsequently repeated this search in EMBASE (from January 1974 to December 2012), China National Knowledge Infrastructure (CNKI: http://www.cnki.net), and Google Scholar (http://scholar.google.com/). We searched for additional publications in personal reference lists from original research articles and review articles. We also hand‐searched relevant journals and e‐mailed the authors to obtain relevant articles that did not have an abstract or full‐text available in the journals or databases.
Studies included in the meta‐analysis had to meet the following inclusion criteria: (1) case‐control studies; (2) reported outcomes included the number or frequency of AD; (3) published as a full‐text article; and (4) polymorphisms should include at least the ‐2578C/A or ‐1154G/A polymorphism. Studies were excluded based on the following criteria: (1) study design based on family or sibling pairs; (2) no detailed genotype frequency; and (3) insufficient information for data extraction. When multiple publications from the same patient population resource or overlapping data sets were available, only the most recent or largest sample size study was included in the meta‐analysis.
Data Extraction
The citation (titles and abstracts) search and data extraction were carried out independently by three reviewers, and disagreements were resolved by consensus. The following information was collected in a predefined data collection form: the first author's name, year of publication, country of origin, ethnicity, AD diagnosis method, source of controls, proportion of men in cases and controls, total number of cases and controls, mean (range) age of cases and controls, and numbers of cases and controls with different genotypes. The quality of each study selected for inclusion in the meta‐analysis was assessed by the Newcastle‐Ottawa Quality Assessment Scale for case‐control studies 15. For non‐English and non‐Chinese articles, data were abstracted by a single reviewer with the help of translation software.
Statistical Analysis
We used crude odds ratios (ORs) and their 95% confidence intervals (CIs) to assess the strength of association between VEGF polymorphisms and AD risk. First, allelic comparison for each polymorphism (C vs. A for the ‐2578C/A polymorphism and G vs. A for the ‐1154G/A polymorphism) was used to detect overall differences. Second, the additive genetic model (homozygote comparison and heterozygote comparison), recessive genetic model, and dominant genetic model were assessed for each polymorphism. We also performed subgroup analyses according to the presence of the APOE ε4 polymorphism and ethnicity.
The deviation of frequencies of VEGF polymorphisms from the expectation under Hardy–Weinberg equilibrium (HWE) was assessed using the chi‐squared test in controls. A sensitivity analysis for the overall effect was performed using influence analyses to evaluate whether one or more studies markedly affected the results 16. Statistical heterogeneity between studies was tested with the I2 statistic, and I2 values of 25%, 50%, and 75% corresponded to cutoff points for mild, moderate, and extensive statistical inconsistencies, respectively 17. When there was a lack of heterogeneity among studies, the pooled OR estimate was merged using the fixed‐effects model 18. Otherwise, the random‐effects model was applied 19. Publication bias was investigated with Egger's regression asymmetry test and funnel plot 20, 21. All statistical analyses were performed using Stata version 11.0 (College Station, TX, USA).
Results
Study Characteristics
A total of 125 articles were identified in the literature search of PubMed, EMBASE, and CNKI and other searching methods using different combinations of key words (Figure 1). Eight case‐control studies (2787 cases and 2841 controls) for the ‐2578C/A polymorphism 22, 23, 24, 25, 26, 27, 28, 29 and four case‐control studies (1422 cases and 1063 controls) for the G (‐1154)A polymorphism 22, 27, 28, 29 were used to evaluate the association with AD risk (Table 1, Figure 2). The distribution of genotypes among controls was consistent with HWE in all studies. The mean ages of the cases and controls were 74.5 and 74.3 years for the C(‐2578)A polymorphism and 74.6 and 70.5 years for the G(‐1154)A polymorphism. The mean ages at onset of AD of the cases were 70.8 years for the C(‐2578)A polymorphism and 69.8 years for the G(‐1154)A polymorphism. Fewer male participants were observed for all studies (percentage of males ranged from 31.1% to 48.0%), except for one study conducted only in males 26. Age and gender distribution between cases and controls was matched for each study. The control population consisted of study participants who were in good health and without cognitive impairment (MMSE>26), as well as older matched participants who had no family history of dementia. For the C(‐2578)A polymorphism, six studies were conducted in Caucasians 22, 23, 24, 25, 26, 28, one in Asians 27, and one in Africans 29. The criteria for AD diagnosis for five studies were the National Institute of Neurological Disorders and Stroke–Alzheimer Diseases and Related Disorders Association criteria (NINCDS/ADRDA criteria), and three other studies used the third/fourth Diagnostic and Statistical Manual of Mental Disorders criteria (DSM‐III‐R criteria). For the G(‐1154)A polymorphism, there were two studies in Caucasians, one in Africans, and one in Asians. The criteria for AD diagnosis for all studies were NINCDS/ADRDA criteria. Minor allele frequencies (MAFs) were lower in Asians than other ethnicities for the C(‐2578)A polymorphism (Table 2).
Figure 1.

Flow chart of article selection in our meta‐analysis.
Table 1.
Study characteristics from included studies in the meta‐analysis
| First author | Year | Country | Ethnicity | Cases | Controls | Criteria for AD diagnosis | Source of controls | SNP(s) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Na | Ageb | Agec | Genderd | N | Ageb | Genderd | |||||||
| Del Bo 22 | 2005 | Italy | Caucasian | 249 | 77.5 | 74.6 | 37.4 | 347 | 72.2 | 44.6 | NINCDS/ADRDA | Ethnic‐, sex‐, and age‐matched Individuals, MMSE criteria | C(‐2578)AG(‐1154)A |
| Chapuis 23 | 2006 | France | Caucasian | 601 | 72.4 | 69.5 | 39.5 | 631 | 72.5 | 36.0 | NINCDS/ADRDA; DSM‐III‐R | Altruistic volunteers from retirement home or electoral rolls | C(‐2578)A |
| Chiappelli 24 | 2006 | Italy | Caucasian | 317 | 76.2 | – | 32.8 | 320 | 73.0 | 35.0 | NINCDS/ADRDA; DSM‐III‐R | Nondemented controls from the same geographical area, MMSE criteria | C(‐2578)A |
| Mateo 25 | 2006 | Spain | Caucasian | 362 | 75.8 | 72.3 | 30.9 | 428 | 80.5 | 31.1 | NINCDS/ADRDA | Unrelated individuals randomly selected from a nursing home, MMSE criteria | C(‐2578)A |
| Giedraitis 26 | 2009 | Sweden | Caucasian | 85 | – | 80.2 | 100 | 399 | 81.8 | 100 | NINCDS/ADRDA; DSM‐IV‐R |
Healthy controls selected from the Uppsala Longitudinal Study of Adult Men, MMSE criteria |
C(‐2578)A |
| Yuan 27 | 2009 | China | Asian | 279 | 68.7 | 65.4 | 48.0 | 317 | 66.6 | 40.1 | NINCDS/ADRDA | Individuals underwent standard health examinations, MMSE criteria | C(‐2578)AG(‐1154)A |
| Landgren 28 | 2010 | Sweden | Caucasian | 801 | 76.0 | – | 36.6 | 286 | 72.0 | 40.9 | NINCDS/ADRDA | Individuals recruited by advertisement, or spouses or unrelated friends of the patients, MMSE criteria | C(‐2578)AG(‐1154)A |
| Smach 29 | 2010 | Tunisia | Africa | 93 | 73.0 | 70.0 | 47.3 | 113 | 72.0 | 46.0 | NINCDS/ADRDA | Individuals from the same geographical area, with no known personal a family history of dementia | C(‐2578)AG(‐1154)A |
DSM, the Diagnostic and Statistical Manual of Mental Disorders; NINCDS‐ADRDA, the National Institute of Neurological Disorders and Stoke–Alzheimer Diseases and Related Disorders Association; MMSE, mini‐mental state examination; SNP, Single‐nucleotide Polymorphism.
Number.
Age at survey.
Age at onset of Alzheimer's disease.
Percentage of male.
Figure 2.
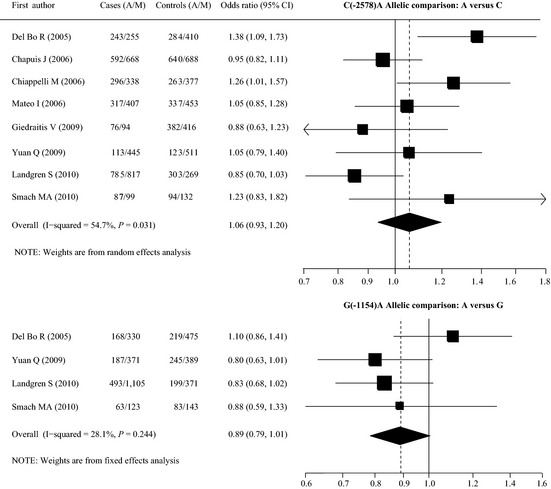
Two polymorphisms of the VEGF gene and the risk of Alzheimer's disease. Note: M: C for the ‐2578C/A polymorphism, G for the ‐1154G/A polymorphism; CI: confidence interval.
Table 2.
VEGF two polymorphisms genotype distribution among AD cases and controls in the included studies
| First author | Cases | Controls | P (HWE) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| M/M (APOE ε4+/−) | M/A (APOE ε4+/−) | A/A (APOE ε4+/−) | MAF (%)a | M/M (APOE ε4+/−) | M/A (APOE ε4+/−) | A/A (APOE ε4+/−) | MAF (%)a | ||
| C(‐2578)A | |||||||||
| Del Bo R | 65 (−/−) | 125 (−/−) | 59 (−/−) | 48.8 | 114 (−/−) | 182 (−/−) | 51 (−/−) | 40.9 | 0.114 |
| Chapuis J | 186 (−/−) | 296 (−/−) | 148 (−/−) | 47.0 | 182 (−/−) | 324 (−/−) | 158 (−/−) | 48.2 | 0.557 |
| Chiappelli M | 83 (40/34) | 172 (74/73) | 62 (22/30) | 46.7 | 106 (15/90) | 165 (26/134) | 49 (13/35) | 41.1 | 0.245 |
| Mateo I | 119 (−/−) | 169 (−/−) | 74 (−/−) | 43.8 | 130 (−/−) | 193 (−/−) | 72 (−/−) | 42.7 | 0.980 |
| Giedraitis V | 26 (−/−) | 42 (−/−) | 17 (−/−) | 44.7 | 106 (−/−) | 204 (−/−) | 89 (−/−) | 47.9 | 0.626 |
| Yuan Q | 175 (47/128) | 95 (27/68) | 9 (6/3) | 20.3 | 204 (15/189) | 103 (10/93) | 10 (2/8) | 19.4 | 0.488 |
| Landgren S | 208 (131/61) | 401 (283/118) | 192 (134/74) | 49.0 | 66 (26/57) | 137 (38/99) | 83 (23/43) | 53.0 | 0.514 |
| Smach MA | 25 (10/15) | 49 (24/25) | 19 (11/8) | 46.8 | 35 (6/29) | 62 (10/52) | 16 (3/13) | 41.6 | 0.169 |
| G(‐1154)A | |||||||||
| Del Bo R | 109 (−/−) | 112 (−/−) | 28 (−/−) | 33.7 | 161 (−/−) | 153 (−/−) | 33 (−/−) | 31.6 | 0.114 |
| Yuan Q | 118 (30/88) | 135 (38/97) | 26 (12/14) | 33.5 | 111 (9/102) | 167 (14/153) | 39 (4/35) | 38.6 | 0.488 |
| Landgren S | 389 (254/135) | 327 (237/90) | 83 (56/27) | 30.9 | 121 (38/83) | 129 (39/90) | 35 (10/25) | 34.9 | 0.514 |
| Smach MA | 39 (17/22) | 45 (23/22) | 9 (5/4) | 33.9 | 42 (6/36) | 59 (12/47) | 12 (1/11) | 36.7 | 0.169 |
M: C for C(‐2578)A polymorphism, G for G(‐1154)A polymorphism.
Minor allele frequency.
Quantitative Synthesis
The results of the overall meta‐analysis did not suggest any associations between the two VEGF polymorphisms (C(‐2578)A and G(‐1154)A) and AD susceptibility for all genetic models (allelic comparison: OR = 1.06/0.89, 95% CI = 0.94–1.20/0.79–1.01, P = 0.377/0.067; homozygote comparison: OR = 1.14/0.82, 95% CI = 0.87–1.49/0.62–1.08, P = 0.350/0.161; dominant model: OR = 1.02/0.85, 95% CI = 0.90–1.15/0.72–1.00, P = 0.798/0.053; and recessive model: OR = 1.07/0.89, 95% CI = 0.93–1.23/0.68–1.16, P = 0.328/0.383, respectively) (Table 2). Because of the limited number of studies, we could not stratify by ethnicity (only one study in Asians 27 and one in Africans 29 for both the C(‐2578)A and G(‐1154)A polymorphisms), but after excluding two studies not conducted in Caucasians, the overall association was not changed (Table 3).
Table 3.
Total and stratified analysis of VEGF gene two polymorphisms on AD
| Variables | Na | Cases/Controls | Allelic comparison | Homozygote comparison | Heterozygote comparison | Dominant genetic model | Recessive genetic model | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P b | I2 | OR (95% CI) | P b | I2 | OR (95% CI) | P b | I2 | OR (95% CI) | P b | I2 | OR (95% CI) | P b | I2 | |||
| C(‐2578)A | |||||||||||||||||
| Total | 8 | 2,816/2,841 | 1.06 (0.94, 1.20) | 0.377 | 54.7 | 1.14 (0.87, 1.49) | 0.350 | 57.4 | 1.02 (0.90, 1.15) | 0.798 | 0.0 | 1.04 (0.92, 1.17) | 0.563 | 24.4 | 1.07 (0.93, 1.23) | 0.328 | 49.2 |
| Caucasian | 6 | 2,444/2,411 | 1.04 (0.90, 1.21) | 0.568 | 65.8 | 1.11 (0.82, 1.51) | 0.507 | 67.5 | 1.00 (0.87, 1.15) | 0.963 | 0.0 | 1.02 (0.90, 1.17) | 0.739 | 43.8 | 1.10 (0.86, 1.39) | 0.455 | 60.8 |
| APOE | |||||||||||||||||
| ε4+ | 4 | 809/187 | 1.02 (0.80, 1.28) | 0.900 | 0.0 | 1.03 (0.64, 1.63) | 0.914 | 0.0 | 1.24 (0.85, 1.81) | 0.262 | 0.0 | 1.16 (0.82, 1.66) | 0.396 | 0.0 | 0.86 (0.58, 1.28) | 0.461 | 0.0 |
| ε4‐ | 4 | 637/842 | 1.22 (1.04, 1.43) | 0.014 | 11.8 | 1.61 (1.12, 2.32) | 0.010 | 22.4 | 1.15 (0.91, 1.46) | 0.246 | 0.0 | 1.22 (0.97, 1.53) | 0.091 | 0.0 | 1.48 (1.08, 2.01) | 0.013 | 0.0 |
| G(‐1154)A | |||||||||||||||||
| Total | 4 | 1,420/1,062 | 0.89 (0.79, 1.01) | 0.066 | 28.1 | 0.82 (0.62, 1.08) | 0.160 | 9.4 | 0.85 (0.71, 1.02) | 0.077 | 0.0 | 0.85 (0.72, 1.00) | 0.053 | 19.9 | 0.89 (0.68, 1.16) | 0.383 | 0.0 |
| Caucasian | 2 | 1,048/632 | 0.95 (0.72, 1.25) | 0.716 | 67.2 | 0.93 (0.56, 1.57) | 0.798 | 52.7 | 0.91 (0.67, 1.24) | 0.548 | 47.3 | 0.92 (0.65, 1.30) | 0.635 | 63.0 | 0.96 (0.69, 1.34) | 0.804 | 15.1 |
| APOE | |||||||||||||||||
| ε4+ | 3 | 672/133 | 0.92 (0.70, 1.22) | 0.580 | 0.0 | 0.91 (0.48, 1.71) | 0.762 | 0.0 | 0.86 (0.57, 1.29) | 0.463 | 0.0 | 0.96 (0.40, 2.31) | 0.936 | 75.8 | 0.98 (0.30, 3.24) | 0.977 | 77.7 |
| ε4‐ | 3 | 499/582 | 0.74 (0.62, 0.89) | 0.001 | 0.0 | 0.57 (0.37, 0.87) | 0.009 | 0.0 | 0.69 (0.53, 0.89) | 0.004 | 0.0 | 0.66 (0.52, 0.85) | 0.001 | 0.0 | 0.69 (0.46, 1.03) | 0.068 | 0.0 |
CI, Confidence interval.
Number of comparisons.
P‐value of Z‐test for significant test. Bold values are statistically significant.
For the C(‐2578)A polymorphism, when stratified according to the APOE ε4 status, significantly increased associations were found for APOE ε4 (‐) (A vs. C: OR = 1.22, 95% CI = 1.04–1.43, P = 0.014; A/A vs. C/C: OR = 1.59, 95% CI = 1.11–2.27, P = 0.011 and A/A vs. A/C + C/C: OR = 1.46, 95% CI = 1.08–1.99, P = 0.015) (Table 3).
For the G(‐1154)A polymorphism, when stratified according to the APOE ε4 status, significantly decreased associations were found for APOE ε4 (‐) (A vs. G: OR = 0.74, 95% CI = 0.62–0.89, P = 0.001; A/A vs. G/G: OR = 0.57, 95% CI = 0.37–0.87, P = 0.009; A/G vs. G/G: OR = 0.69, 95% CI = 0.53–0.89, P = 0.004 and A/A + A/G vs. G/G: OR = 0.66, 95% CI = 0.52–0.85, P = 0.001) (Table 3).
There was significant heterogeneity between the results of individual studies in the total analysis of the C(‐2578)A polymorphism with allelic comparison (I2 = 54.7%), homozygote comparison (I2 = 57.4%), and the recessive genetic model (I2 = 49.2%), but after stratifying for the presence of APOE ε4, the heterogeneity disappeared (I2 = 0.0% for all genetic models for both APOE ε4 (+) and APOE ε4 (‐), except for homozygote comparison (I2 = 22.4%)) (Table 3). No significant heterogeneity was observed in the total analysis of the G(‐1154)A polymorphism with different models, and I2 ranged from 0.0% to 28.1% (Table 3).
Sensitivity Analysis and Bias Diagnosis
We performed a sensitivity analysis to explore whether modifying the studies included in the meta‐analysis could influence the overall effects. The influence analysis indicated that no single study qualitatively affected the summary risks, as indicated by the sensitivity analysis for the ‐2578C/A polymorphism (data not shown). However, after excluding one study by Del Bo et al. 22 on the ‐1154G/A polymorphism, significant associations were found for allelic comparison (OR = 0.83, 95% CI = 0.72–0.95, P = 0.009), homozygote comparison (OR = 0.71, 95% CI = 0.51–0.98, P = 0.037), heterozygote comparison (OR = 0.78, 95% CI = 0.64–0.96, P = 0.020), and the dominant model (OR = 0.77, 95% CI = 0.63–0.93, P = 0.008). Egger's test and funnel plot were performed to access publication bias. Ultimately, the results did not suggest any evidence of publication bias (all P‐values >0.10) (Table 4, Figure 3).
Table 4.
Publication bias tests (Egger's funnel plot for publication bias test) for VEGF gene two polymorphisms
| Genetic type | Coefficient | Standard error | t | P‐value | 95% CI of intercept |
|---|---|---|---|---|---|
| C(‐2578)A | |||||
| Allelic contrast | 1.712 | 2.011 | 0.85 | 0.427 | −3.208, 6.633 |
| Homozygote comparison | 1.440 | 1.676 | 0.86 | 0.423 | −2.661, 5.542 |
| Heterozygote comparison | 0.780 | 1.288 | 0.61 | 0.567 | −2.372, 3.931 |
| Dominant genetic model | 1.236 | 1.708 | 0.72 | 0.496 | −2.942, 5.414 |
| Recessive genetic model | 1.322 | 1.417 | 0.93 | 0.387 | −2.145, 4.789 |
| G(‐1154)A | |||||
| Allelic contrast | 0.744 | 3.162 | 0.24 | 0.836 | −12.861, 14.348 |
| Homozygote comparison | 0.433 | 2.591 | 0.17 | 0.883 | −10.715, 11.580 |
| Heterozygote comparison | 0.250 | 2.485 | 0.10 | 0.929 | −10.440, 10.940 |
| Dominant genetic model | 0.302 | 2.951 | 0.10 | 0.928 | −12.393, 12.997 |
| Recessive genetic model | 0.460 | 1.954 | 0.24 | 0.836 | −7.948, 8.868 |
Figure 3.
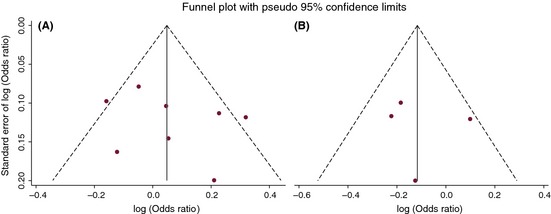
Funnel plot of two VEGF gene polymorphisms and Alzheimer's disease risk. Note: (A) ‐2578C/A allelic comparison, A versus C; (B) ‐1154G/A allelic comparison, A versus G.
Discussion
Our meta‐analysis included 2787 AD cases and 2841 control subjects from eight published case‐control studies on the ‐2578C/A polymorphism and 1422 AD cases and 1063 controls from four studies on the ‐1154G/A polymorphism. The results from the meta‐analysis showed that there was no association overall between the two polymorphisms of the VEGF promoter and AD risk.
However, it might be argued that the negative association between VEGF polymorphisms (‐2578C/A and ‐1154G/A) and AD risk is due to heterogeneity. Although we attempted to overcome this limitation using a random‐effects model to adequately capture the trade‐off between the association estimates in comparison with significant heterogeneity, which were consistent with weighted estimates, the results of this meta‐analysis should be accepted with caution.
APOE є4 plays an important role in the pathogenic mechanism of AD by regulating the formation of Aβ, and APOE є4 is the only established genetic risk factor for AD 30. We found interesting results after stratifying the data by APOE є4 status in the meta‐analysis. The results from the stratification analysis showed a risk association for the ‐2578 variant and a protective association for the ‐1154 variant. The possible reasons for the positive associations of these two VEGF promoter variants and their opposing effects on AD risk in an APOE є4‐independent manner are as follows: (1) the number of analyzed studies is limited after stratification, so the results may yield false‐positives because of bias; or (2) these two VEGF promoter variants may be associated with AD risk in an APOE є4‐independent manner. In fact, several investigations have reported APOE4‐independent genetic effects on AD risk. Lin et al. 31 reported that the rs11833579 polymorphism of the NINJ2 gene was significantly associated with AD in non‐APOE є4 carriers after controlling for the false discovery rate (AOR = 0.38, 95% CI = 0.18–0.82). Cellini et al. 32 also documented that the late‐onset AD association for the rs661057 SNP was confined to APOEε4 noncarriers for both the T/T genotype (P = 0.002; OR, 1.86 [95% CI, 1.22–2.85]) and T allele (P = 0.002; OR, 1.61 [1.18–2.21]). The independent role of the two polymorphisms should warrant further investigation because AD patients without APOE є4 alleles have no other major genetic risk factors that might contribute to them developing the disease. Another explanation for these results could be that these two VEGF promoter variants may have opposing effects on AD risk in the APOE є4 (‐) sample. The risk role of the ‐2578 polymorphism and the protective role of the ‐1154 polymorphism were noted in this meta‐analysis. Evidence has shown that the two polymorphisms at ‐2578 A and ‐1154 A in the VEGF promoter have lower activity and VEGF expression levels than ‐2578 C and ‐1154 G in peripheral blood mononuclear cells 33. Furthermore, previous studies have reported that reduced VEGF levels cause adult‐onset neuron degeneration reminiscent of human neurodegenerative disorder 34 and promote neuron degeneration by limiting neural tissue perfusion and VEGF‐dependent neuroprotection 34. Therefore, it is reasonable to hypothesize that the deficiency in VEGF expression may be associated with neuron dysfunction. Surprisingly, the results from the meta‐analysis were contrary to the protective role and lower activity of VEGF in the pathogenesis of AD. However, an in vitro study may provide a logical explanation for the contrasting results. The findings from an in vitro study showed that treatment with a high dose of VEGF (≥500 ng/mL) decreased neuronal survival and expression of the anti‐apoptotic protein Bcl‐2 while increasing the proapoptotic proteins caspase 3 and phosphorylated p38 MAPK. Additionally, high‐dose VEGF negated the decrease in Aβ evoked by low‐dose VEGF 35. Therefore, taken together with our results, VEGF may exert both beneficial and deleterious effects in the brain.
One limitation of our study was that stratification by age, gender, and ethnicity could not be effectively performed in the meta‐analysis because of unavailable information on age and gender as well as limited information on ethnicity in the included case‐control studies. Age, gender, and ethnicity are the most important confounding factors, and age‐, gender‐, and ethnicity‐dependent genetic effects on AD risk have been reported in many studies 36, 37, 38. In the included articles, adjustment for age and gender was performed for the ‐2578C/A polymorphism in only three articles 22, 23, 25 and in only one article for the ‐1154G/A polymorphism 22. Consequently, it is difficult to systematically determine the role of age and gender in the association between VEGF and AD in the meta‐analysis. Additionally, we could only reanalyze studies with Caucasians to determine whether Caucasians had different risks from the overall effects. We could not combine the remaining two studies (one from Asia and the other from Africa) due to different ethnicities. Therefore, it is necessary to further explore the effect of age, gender, and ethnicity on the association between VEGF and AD in future studies.
Sample size bias often exists in observational studies. However, we had no ability to ascertain whether studies included in our review had an adequate sample size. The genetic power calculation is often used to evaluate the sample size in genetic analyses on associations between polymorphisms and diseases, but no study reported an a priori sample size calculation in the included studies. An inadequate choice of sample size may lead to chance and exaggerate (or dilute) the association between VEGF and AD.
In conclusion, the results from our meta‐analysis provide preliminary evidence that the ‐1154G/A and ‐2578C/A VEGF polymorphisms may have opposing effects on AD susceptibility in an APOE є4‐independent manner when stratified by APOE є4 status. However, whether the two polymorphisms in the promoter affect the pathogenesis of AD alone or together with demographic characteristics is not clear. Therefore, more well‐designed epidemiological studies on the two polymorphisms, as well as age, gender, and ethnicity, which were not systematically covered by the existing studies, will be necessary to validate these findings in further studies.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by funding from the National Nature Science Foundation of China (grant numbers 31171219, 81271213, 81070878, 81271214, and 81261120404), the Natural Science Foundation of Guangdong Province (No. S2012010008222), and the Science and Technology Innovation Fund of Guangdong Medical College (No. STIF 201101).
References
- 1. Alzheimer's disease International . World Alzheimer Report 2009‐2011. 2012. Available at: http://www.alz.co.uk/research/world-report (accessed on 15 November 2012).
- 2. Berchtold NC, Cotman CW. Evolution in the conceptualization of dementia and Alzheimer's disease: Greco‐Roman period to the 1960s. Neurobiol Aging 1998;19:173–189. [DOI] [PubMed] [Google Scholar]
- 3. Ferrara N, Henzel WJ. Pltubry follicuiar cells secrete a VEGF in femoral head of growing rat novel heparin‐binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun 1989;161:851–858. [DOI] [PubMed] [Google Scholar]
- 4. Gospodarowicz D, Abraham JA, Schilling J. Isolation and characterization of a vascular endothelial cell mitogen produced by pituitary‐derived folliculostellate cells. Proc Natl Acad Sci USA 1989;86:7311–7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shibuya M. Vascular endothelial growth factor‐dependent and ‐independent regulation of angiogenesis. BMB Rep 2008;41:278–286. [DOI] [PubMed] [Google Scholar]
- 6. Mateo I, Llorca J, Infante J, et al. Low serum VEGF levels are associated with Alzheimer's disease. Acta Neurol Scand 2007;116:56–58. [DOI] [PubMed] [Google Scholar]
- 7. Provias J, Jeynes B. Neurofibrillary tangles and senile plaques in Alzheimer's brains are associated with reduced capillary expression of vascular endothelial growth factor and endothelial nitric oxide synthase. Curr Neurovasc Res 2008;5:199–205. [DOI] [PubMed] [Google Scholar]
- 8. Tarkowski E, Issa R, Sjogren M, et al. Increased intrathecal levels of the angiogenic factors VEGF and TGFbeta in Alzheimer's disease and vascular dementia. Neurobiol Aging 2002;23:237–243. [DOI] [PubMed] [Google Scholar]
- 9. Thirumangalakudi L, Samany PG, Owoso A, Wiskar B, Grammas P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer's disease. J Alzheimers Dis 2006;10:111–118. [DOI] [PubMed] [Google Scholar]
- 10. Kalaria RN, Cohen DL, Premkumar DR, Nag S, LaManna JC, Lust WD. Vascular endothelial growth factor in Alzheimer's disease and experimental ischemia. Brain Res Mol Brain Res 1998;62:101–105. [DOI] [PubMed] [Google Scholar]
- 11. Blacker D, Bertram L, Saunders AJ, et al. Results of a high‐resolution genome screen of 437 Alzheimer's disease families. Hum Mol Genet 2003;12:23–32. [DOI] [PubMed] [Google Scholar]
- 12. Tischer E, Mitchell R, Hartman T, et al. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem 1991;266:11947–11954. [PubMed] [Google Scholar]
- 13. Stevens A, Soden J, Brenchley PE, Ralph S, Ray DW. Haplotype analysis of the polymorphic human vascular endothelial growth factor gene promoter. Cancer Res 2003;63:812–816. [PubMed] [Google Scholar]
- 14. Stroup DF, Berlin JA, Morton SC, et al. Meta‐analysis of observational studies in epidemiology: a proposal for reporting. Meta‐analysis Of Observational Studies in Epidemiology (MOOSE) group. JAMA 2000;283:2008–2012. [DOI] [PubMed] [Google Scholar]
- 15. Stang A. Critical evaluation of the Newcastle‐Ottawa scale for the assessment of the quality of nonrandomized studies in meta‐analyses. Eur J Epidemiol 2010;25:603–605. [DOI] [PubMed] [Google Scholar]
- 16. Tobias A. Assessing the influence of a single study in the meta‐analysis estimate. Stata Tech Bull 1999;8:15–17. [Google Scholar]
- 17. Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ (Clinical researched) 2003;327:557–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 1959;22:719–748. [PubMed] [Google Scholar]
- 19. DerSimonian R, Kacker R. Random‐effects model for meta‐analysis of clinical trials: an update. Contemp Clin Trials 2007;28:105–114. [DOI] [PubMed] [Google Scholar]
- 20. Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics 1994;50:1088–1101. [PubMed] [Google Scholar]
- 21. Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Del Bo R, Scarlato M, Ghezzi S, et al. Vascular endothelial growth factor gene variability is associated with increased risk for AD. Ann Neurol 2005;57:373–380. [DOI] [PubMed] [Google Scholar]
- 23. Chapuis J, Tian J, Shi J, et al. Association study of the vascular endothelial growth factor gene with the risk of developing Alzheimer's disease. Neurobiol Aging 2006;27:1212–1215. [DOI] [PubMed] [Google Scholar]
- 24. Chiappelli M, Borroni B, Archetti S, et al. VEGF gene and phenotype relation with Alzheimer's disease and mild cognitive impairment. Rejuvenation Res 2006;9:485–493. [DOI] [PubMed] [Google Scholar]
- 25. Mateo I, Llorca J, Infante J, et al. Case‐control study of vascular endothelial growth factor (VEGF) genetic variability in Alzheimer's disease. Neurosci Lett 2006;401:171–173. [DOI] [PubMed] [Google Scholar]
- 26. Giedraitis V, Kilander L, Degerman‐Gunnarsson M, et al. Genetic analysis of Alzheimer's disease in the Uppsala Longitudinal Study of Adult Men. Dement Geriatr Cogn Disord 2009;27:59–68. [DOI] [PubMed] [Google Scholar]
- 27. Yuan Q, Zuo X, Jia J. Association between promoter polymorphisms of vascular endothelial growth factor gene and sporadic Alzheimer's disease among Northern Chinese Han. Neurosci Lett 2009;457:133–136. [DOI] [PubMed] [Google Scholar]
- 28. Landgren S, Palmer MS, Skoog I, et al. No association of VEGF polymorphism with Alzheimer's disease. Neuromolecular Med 2010;12:224–228. [DOI] [PubMed] [Google Scholar]
- 29. Smach MA, Charfeddine B, Othman LB, et al. ‐1154G/A and ‐2578C/A polymorphisms of the vascular endothelial growth factor gene in Tunisian Alzheimer patients in relation to beta‐amyloid (1‐42) and total tau protein. Neurosci Lett 2010;472:139–142. [DOI] [PubMed] [Google Scholar]
- 30. Leoni V. The effect of apolipoprotein E (ApoE) genotype on biomarkers of amyloidogenesis, tau pathology and neurodegeneration in Alzheimer's disease. Clin Chem Lab Med 2011;49:375–383. [DOI] [PubMed] [Google Scholar]
- 31. Lin KP, Chen SY, Lai LC, et al. Genetic polymorphisms of a novel vascular susceptibility gene, Ninjurin2 (NINJ2), are associated with a decreased risk of Alzheimer's disease. PLoS ONE 2011;6:e20573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cellini E, Tedde A, Bagnoli S, et al. Implication of sex and SORL1 variants in italian patients with Alzheimer disease. Arch Neurol 2009;66:1260–1266. [DOI] [PubMed] [Google Scholar]
- 33. Shahbazi M, Fryer AA, Pravica V, et al. Vascular endothelial growth factor gene polymorphisms are associated with acute renal allograft rejection. J Am Soc Nephrol 2002;13:260–264. [DOI] [PubMed] [Google Scholar]
- 34. Oosthuyse B, Moons L, Storkebaum E, et al. Deletion of the hypoxia‐response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet 2001;28:131–138. [DOI] [PubMed] [Google Scholar]
- 35. Sanchez A, Tripathy D, Luo J, Yin X, Martinez J, Grammas P. Neurovascular unit and the effects of dosage in VEGF toxicity: role for oxidative stress and thrombin. J Alzheimers Dis 2013;34:281–291. [DOI] [PubMed] [Google Scholar]
- 36. Combarros O, Rodero L, Infante J, et al. Age‐dependent association between the Q7R polymorphism in the Saitohin gene and sporadic Alzheimer's disease. Dement Geriatr Cogn Disord 2003;16:132–135. [DOI] [PubMed] [Google Scholar]
- 37. Zou F, Gopalraj RK, Lok J, et al. Sex‐dependent association of a common low‐density lipoprotein receptor polymorphism with RNA splicing efficiency in the brain and Alzheimer's disease. Hum Mol Genet 2008;17:929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wollmer MA, Kapaki E, Hersberger M, et al. Ethnicity‐dependent genetic association of ABCA2 with sporadic Alzheimer's disease. Am J Med Genet B Neuropsychiatr Genet 2006;141B:534–536. [DOI] [PubMed] [Google Scholar]