

Multiple acyl‐CoA dehydrogenase deficiency (MADD) is an inherited autosomal recessive disorder of fatty acid and amino acid metabolism caused by a defect of electron transfer flavoprotein (ETF) or ETF dehydrogenase (ETFDH) 1. MADD is roughly classified into two forms: an early‐onset form and a late‐onset form. Although the molecular mechanism of MADD is still unclear, riboflavin supplementation has been known to strikingly remit symptoms and signs in a group of MADD patients, especially those with the late‐onset form. Herein, we present a late‐onset case harboring two new mutations with dramatic response to riboflavin.
A 55‐year‐old man was admitted to the hospital because of osphyalgia and muscle weakness. He noticed a sign of mild osphyalgia 10 years ago, which had waxed and waned for years without disturbing his mild physical activities. Two years ago, at the age of 53, signs of dorsalgia and muscle weakness appeared and, as well as osphyalgia, worsened progressively. At the same time, anorexia and intermittent nausea happened as well. He also felt difficulty in walking, swallowing and speaking, and lost his flesh gradually. The other accompanied signs included fatigue in the extensor muscles of neck, and disability in holding his head erect in a longer time. There were neither muscle cramps nor episodes of myoglobinuria during his excises. His past medical history and life history were unremarkable except his disgust with meat. General physical examinations revealed no abnormal findings. On neurological examinations, there was moderate weakness of neck muscles and proximal muscles of the extremities. Moderate atrophy of limbs, trunk, especially sternocleidomastoid was present. Gower's sign was negative. No tremor, ataxia, nor abnormal movements were present. Deep tendon reflexes decreased throughout without clonus. Sensation was normal.
The following enzymes in the serum were abnormally high: creafine kinase 571 U/L (normal 24–200), aspartate aminotransferase 96 U/L (normal 0–40), lactic dehydrogenase 600 U/L (normal 114–240). His proximal muscles electromyography demonstrated myopathic changes. Motor and sensory nerve conduction studies were normal.
The patient received an open muscle biopsy in biceps brachii under local anesthesia. The histochemical stains of all the muscular sections represented similar outcomes: Muscular fibers were abnormally variable in size with moderate to severe vacuoles, which were most prominent in type‐I fibers; oil red O (ORO) stain revealed predominant lipid accumulation in type‐I fibers (Figure 1). On modified Gomori trichrome (MGT) stain, no atypical ragged red fibers (RRFs) were found under the microscopy. The cytochrome oxidase (COX) stains showed lesion fibers deeply stained without enzyme activity missing. Mazarine deposits distributed unequally with mussy grid structure between myofibers were presented on NADH stains. Other stainings including SDH and PAS did not show any specific results.
Figure 1.
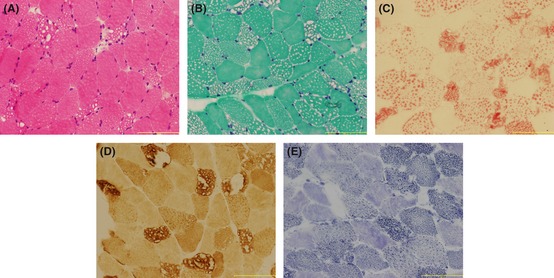
Muscular pathological finding (bar = 100 μm). (A) H&E stain: muscular fibers were abnormally variable in size with moderate to severe vacoules mainly in type‐I fibers (B) MGT stain: similar representation with H&E stain without ragged red fibers (C) ORO stain: predominant lipid accumulation also mainly in type‐I fibers. (D) COX stain: lesion fibers deeply stained without enzyme activity missing (E) NADH stain: mazarine deposits distribute unequally with mussy grid structure between myofibers
Before riboflavin therapy, urine and blood samples were collected for organic acid and acylcarnitine analyses 2, respectively. Abnormal urinary excretion of organic acids, considered pathognomonic for the condition, including glutarate, ethylmalonic acid (EMA) and 2‐hydroxyglutaric acid (2‐HG), was observed. Meanwhile, dry blood acycarnitine analysis with tandem mass spectrometry represented a combined significant elevation of some acylcarnitines with specific carboxylase ranging from medium to long chains (C8, C10, and C12). Considering the biochemical findings, the patient was regarded as sufferring MADD. Furthermore, the DNA sequencing results certified the diagnosis.
The genomic DNAs were isolated from peripheral blood lymphocytes 3. The patient was identified to carry compound heterozygous mutations in ETFDH gene (Figure 2). A missense mutation, c.1522C>A(p.P508T), and a frameshift mutation, c.1583_1584insA(p.N528KfsX3), which produced a premature termination codon (PTC) were found. Seventy‐five unrelated Chinese individuals were screened and none of the mutations appeared in 150 control alleles. Homology search in different species verified all the missense mutations were highly conserved. The SIFT (sorting intolerant from tolerant)‐calculated probabilities of the mutations were 0.00, which indicated both mutations were pernicious to the function of the ETFDH protein. Both mutations located in the 4Fe4S domain (p.P508T & p.N528KfsX3), referenced at the 3D structure of ETFDH. No mutation was identified in ETFA or ETFB gene.
Figure 2.
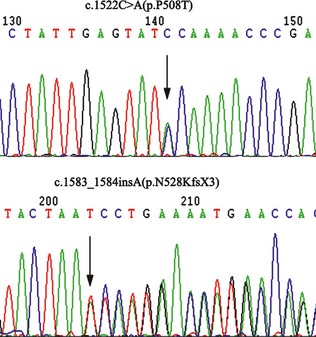
Analysis of the mutations located in electron transfer flavoprotein dehydrogenase gene, showing a missense a mutation, c. 1522C>A(p.P508T), and a frameshift mutation, c.1583_1584insA(p.N528KfsX3)
ELISA analysis of the ETFDH protein in the muscles before treatment revealed that the quantity of the mutant protein in the patient was nearly half of the normal.
After diagnosis, the patient received a single riboflavin therapy (100 mg/d) along with a low protein/fat and high carbohydrate diet. With treatment, creatase levels returned to normal, symptoms subsided and exercise intolerance improved within a few weeks. Withdrawal without consulting happened 3 months after beginning therapy. During the 6‐year telephone follow‐up, he remained free of any symptoms and was competent for daily life and light manual labor.
MADD is a rare but treatable cause of proximal myopathy in adult patients in whom the presenting feature was muscle weakness. In the case, muscle biopsy narrowed the differential diagnosis by revealing lipid accumulation in muscle fibers. The diagnosis of MADD was made based on elevated urine organic acid especially 2‐hydroxyglutaric acid. The investigations of DNA sequencing confirmed the diagnosis of MADD. Noteworthily, typical neck flexor and extensor weakness was observed in our patient and has been described as a characteristic clinical feature in MADD patients. So, MADD should be suspected in any patient presenting neck muscle weakness.
To our knowledge, homozygous and compound heterozygous mutations in ETFA, ETFB, or ETFDH are responsible for all MADD except one case 4. By now, more than 80 mutations have been reported all over the world, but the same mutant site is rarely found in different populations. Only in Asian people are hot‐spot mutations of ETFDH gene found: c.770A>G (p.G362R) and c.1227A>C (p.L409F) in northern Chinese patients 2, 5, and c.250G>A (p.A84T) in Taiwanese 6 and southern Chinese patients 7. This indicates that ETFDH mutations may be ethnic‐specific, which is meaningful to molecular epidemiology of the disease. Mutations in ETFA and ETFB tend to cause neonatal‐onset forms, whereas ETFDH mutations often present with late‐onset forms 5, 8, 9, 10. In the present late‐onset patient who responded well to riboflavin, two novel mutations in ETFDH gene were found and proved to be causal mutations rather than neutral polymorphisms. They mildly affect lipid metabolism especially under metabolic stress. Confusingly, the patient has been keeping healthy for 6 years in spite of withdrawal without consulting after the first 3 months therapy. Considering that clinical features may be remarkably variable in patients harboring the same mutation 6, we hypothesize that environment take a modifying effect on the phenotype.
In summary, we report a late‐onset RR‐MADD patient in a Chinese family and present two novel mutations (one missense and one insertion) in ETFDH gene which further expand the spectrum of mutations found in patients with RR‐MADD.
The first two authors contributed equally to this work.
References
- 1. Gordon N. Glutaric aciduria types I and II. Brain Dev 2006;28:136–140. [DOI] [PubMed] [Google Scholar]
- 2. Wen B, Dai T, Li W, et al. Riboflavin‐responsive lipid‐storage myopathy caused by ETFDH gene mutations. J Neurol Neurosurg Psychiatry 2010;81:231–236. [DOI] [PubMed] [Google Scholar]
- 3. Wanders RJ, Vreken P, den Boer ME, Wijburg FA, van Gennip AH, I Jlst L. Disorders of mitochondrial fatty acyl‐CoA beta‐oxidation. J Inherit Metab Dis 1999; 22:442–487. [DOI] [PubMed] [Google Scholar]
- 4. Cotelli MS, Vielmi V, Rimoldi M, et al. Riboflavin‐responsive multiple acyl‐CoA dehydrogenase deficiency with unknown genetic defect. Neurol Sci 2011. DOI: 10.1007/s10072-011-0900-1. [DOI] [PubMed] [Google Scholar]
- 5. Wang Y, Zhao D, Hong D, Wang Z. Hot spot mutations in electron transfer flavoprotein dehydrogenase gene of riboflavin responsive lipid storage myopathy in 20 Chinese families. Chin J Neurol 2011;44:309–313. [Google Scholar]
- 6. Lan MY, Fu MH, Liu YF, et al. High frequency of ETFDH c.250G>A mutation in Taiwanese patients with late‐onset lipid storage myopathy. Clin Genet 2010;78:565–569. [DOI] [PubMed] [Google Scholar]
- 7. Wang ZQ, Chen XJ, Murong SX, Wang N, Wu ZY. Molecular analysis of 51 unrelated pedigrees with late‐onset multiple acyl‐CoA dehydrogenation deficiency (MADD) in southern China confirmed the most common ETFDH mutation and high carrier frequency of c.250G>A. J Mol Med (Berl) 2011;89:569–576. [DOI] [PubMed] [Google Scholar]
- 8. Yotsumoto Y, Hasegawa Y, Fukuda S, et al. Clinical and molecular investigations of Japanese cases of glutaric acidemia type 2. Mol Genet Metab 2008;94:61–67. [DOI] [PubMed] [Google Scholar]
- 9. Olsen RK, Andresen BS, Christensen E, Bross P, Skovby F, Gregersen N. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl‐CoA dehydrogenation deficiency. Hum Mutat 2003;22:12–23. [DOI] [PubMed] [Google Scholar]
- 10. Olsen RK, Olpin SE, Andresen BS, et al. ETFDH mutations as a major cause of riboflavin‐responsive multiple acyl‐CoA dehydrogenation deficiency. Brain 2007;130:2045–2054. [DOI] [PubMed] [Google Scholar]