

Summary
Background
Tight junction protein degradation is a principal characteristic of the blood–brain barrier (BBB) damage that occurs during brain ischemia.
Aims
We investigated the mechanisms of occludin degradation that underlie permanent middle cerebral artery occlusion (pMCAO) in rats.
Methods and Results
Western blot and Co‐immunoprecipitation data indicated ubiquitination and degradation of occludin in brain after pMCAO, which was consistent with ZO‐1 degradation in penumbra regions as observed at 24 h after pMCAO. We further investigated candidate protease(s) responsible for the degradation of occludin during pMCAO. The intraventricular administration of γ‐secretase blocker DAPT significantly inhibited the pMCAO‐induced neurovascular damage, whereas ALLM and Batimastat, which are inhibitors of calpain and metalloproteinase proteases, respectively, were less effective. Notably, we found that DAPT significantly inhibited BBB disruption in comparison with vehicle treatment, as assessed by Evans blue excretion. Interestingly, the confocal immunostaining revealed that activation of the E3 ubiquitin ligase Itch is associated with degradation of occludin in brain microvessels following ischemia. Furthermore, our data demonstrate that the inhibition of γ‐secretase signaling and the itch‐mediated ubiquitination of occludin likely underlie the vasoprotective effect of DAPT after pMCAO.
Conclusion
The γ‐secretase blocker DAPT reduces the permeability of the BBB by decreasing the ubiquitination and degradation of occludin during permanent brain ischemia, suggesting that γ‐secretase may represent a novel therapeutic target for preventing neurovascular damage.
Keywords: DAPT, Degradation, Occludin, Permanent brain ischemia, Tight junction, Ubiquitination
Introduction
Ischemic stroke is a significant cause of neurovascular dysfunction and leads to cerebral hypoperfusion and blood–brain barrier (BBB) damage 1, 2. The association between ischemic injury and dysfunction of the endothelium and vascular components of the brain has been accepted for several years based on experimental data and clinical observations. Although neurons are thought to be the major target of ischemic insult, the primary etiology of ischemic stroke involves vascular dysfunction 3, 4. The endothelial cells lining the BBB are held together by tight junctions that provide high electrical resistance, which forms an effective vascular barrier against paracellular permeability 5, 6. Cerebral transient or permanent injury induces alterations in many signaling pathways that underlie BBB disruption and lead to degradation of the extracellular matrix and vasogenic edema 1, 7, 8.
Tight junctions are specialized cell–cell adhesion structures that are critical components of the BBB but have been shown to be vulnerable to various deleterious molecules 9, 10, such as the free radicals, matrix metalloproteinases (MMPs), and platelet‐activating factor 9, 10, 11. Although the inflammatory response mediated by the induction of cytokines is known to contribute to brain edema and increased endothelial permeability 12, 13, 14, the precise mechanisms responsible for the development of BBB damage in ischemia remain largely unknown.
Little is known about the pathological role of γ‐secretase activation in brain ischemia‐induced BBB damage in vivo. The present study demonstrates that permanent brain ischemia promotes BBB breakdown and is associated with the activation of γ‐secretase signaling and the ubiquitination and degradation of occludin. Intraventricular treatment with the γ‐secretase blocker DAPT suppressed the ubiquitination and degradation of occludin and inhibited BBB breakdown. These results highlight the importance of the γ‐secretase cascade in the progression of neurovascular damage that occurs during brain ischemia.
Materials and Methods
Experimental Animals and Surgical Procedures
Male Sprague Dawley (SD) rats weighing 200–230 g (8 weeks old) were obtained from Zhejiang Medical Animal Centre (Hangzhou, China). Rats were housed under climate‐controlled conditions with a 12‐h light/dark cycle and provided with standard food and water. Animals were acclimated to their environment for at least 1 week before initiating the experimental protocols. All experimental protocols and animal handling procedures were performed in accordance with the National Institutes of Health (NIH, USA) guidelines for the care and use of laboratory animals and were approved by The Committees for Animal Experiments at Zhejiang University in China. Intracerebroventricular injections were performed using a Hamilton syringe attached to a 27‐gauge needle at 30 min prior to the permanent middle cerebral artery occlusion (pMCAO) surgery. The location of the right lateral ventricle injection was 0.8 mm caudal to the bregma, 1.6 mm lateral to the midline, and at a 4.0 mm depth from the skull surface. For inhibitor treatments, healthy adult SD rats were divided into the following four groups: Vehicle, ALLM (10 mmol/L), Batimastat/BB‐94 (5 mmol/L), and DAPT (50 μmol/L). Each animal was given 10 μL of either artificial cerebrospinal fluid vehicle (aCSF) or the corresponding inhibitor (dissolved in aCSF) over a 10‐min period.
Permanent Middle Cerebral Artery Occlusion Model
Preparation for the pMCAO model in rats was carried out as previously described 15. Animal procedures were approved by the Committee on Animal Experiments at Zhejiang University. The rectal temperature was monitored throughout the surgery, and the body temperature was maintained at 37 ± 0.5°C with a heating blanket. Rats were decapitated at 24 h after ischemia, and the brains were dissected for further analysis.
Evaluation of BBB Damage
The loss of BBB integrity was assessed by examining the extent of Evans blue solution leakage from the microvessels in the rat brains following intravenous injection 1. Evans blue solution (2% in saline, 4 mL/kg) was intravenously administered via the tail vein at 22 h after the onset of pMCAO. To clear the blood and intravascular Evans blue solution that remained in the vascular system, rats were transcardially perfused with saline under pentobarbital anesthesia at 24 h after pMCAO. After decapitation, the forebrains, excluding the cerebellum, were immediately removed and divided into the ipsilateral and contralateral hemispheres. Each hemisphere was weighed and homogenized in 50% trichloroacetic acid solution for the Evans blue‐injected rats. Following centrifugation at 10,000 g for 10 min, the supernatant was diluted with ethanol (1:3), and the absorbance of Evans blue was measured at 620 nm using a spectrophotometer. The amount of Evans blue in the tissue was quantified using a standard curve derived from known amounts of the dye. Data were expressed as micrograms of Evans blue per gram of tissue.
Protein Extraction, Co‐immunoprecipitation, and Western Blotting
After decapitation, the brains were removed and rinsed once in cold 0.32 M sucrose. Coronal sections (2 mm thick) were prepared using a brain slicer, and the ipsilateral hemispheres including the cortex and striatum were dissected and stored at −80°C. Frozen brain tissue was homogenized in a buffer containing 50 mM Tris‐HCl (pH 7.4), 0.5% Triton X‐100, 4 mM EGTA, 10 mM EDTA, 1 mM Na3VO4, 30 mM sodium pyrophosphate, 50 mM NaF, 50 μg/mL leupeptin, 25 μg/mL pepstatin A, 50 μg/mL trypsin inhibitor, and 1 mM dithiothreitol (DTT). The soluble cytosolic fractions were obtained after a 10‐min centrifugation at 15,000 g. The protein concentration in each fraction was determined using Bradford's method. Membrane fractionation of the brain slices was performed as previously described 16. For immunoprecipitations, 100 μg of cytosolic protein isolated from the contralateral and ipsilateral sides of brain after pMCAO was immunoprecipitated with an anti‐occludin polyclonal antibody (Invitrogen, San Diego, CA, USA), according to a previously described protocol 10. For electrophoresis and Western blotting analysis, samples containing equivalent amounts of protein were run on 10–15% acrylamide denaturing gels (SDS‐PAGE). Proteins were then transferred to an Immobilon PVDF transfer membrane for 1 h at 50 V. The membranes were blocked in 20 mM Tris‐HCl (pH 7.4), 150 mM NaCl, and 0.1% Tween 20 (TBS‐T) containing 5% fat‐free milk powder for 1 h at room temperature and were then incubated with antibodies targeting the following molecules: ZO‐1 and occludin (Invitrogen); calcineurin 17; spectrin, MMP‐9 and HES‐1 (Millipore, Billerica, MA, USA); Notch1 (Cell Signaling Technology Inc., Beverly, MA, USA); MMP2 (BioVision, Mountain View, CA, USA); Jagged‐1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and β‐actin (Sigma Chemical, St. Louis, MO, USA). All primary antibody incubations were performed overnight at 4°C. After rinsing, the membranes were incubated for 60 min at room temperature with the corresponding horseradish peroxidase (HRP)‐conjugated secondary antibody diluted in TBS‐T. Immunoreactive proteins were visualized using an enhanced chemiluminescence detection system (Amersham Life Science, Buckinghamshire, UK).
γ‐Secretase Activity Assay
In vitro γ‐secretase assays were performed as previously described 18. Briefly, solubilized membrane preparations were incubated with a fluorogenic peptide probe (8 μM), cleavage of the peptide by γ‐secretase allowing for the release of a fluorescent signal. Fluorescence was measured using a Spectrafluor plate reader (Thermo Electron) with excitation wave length at 355 nm and emission wave length at 440 nm. Results from 3 independent replicates.
Immunohistochemistry
Rats were anesthetized at the time of sacrifice and were transcardially perfused with 4% paraformaldehyde in phosphate‐buffered saline (PBS) as previously described 19. The whole brains were immediately removed and postfixed overnight at 4°C. The brains were then blocked, and the blocks containing the ventral hippocampus were selected and transversely cut into 35‐μm‐thick serial sections using a vibratome. Sections were incubated at room temperature in PBS with 0.01% Triton‐X100 for 30 min and for 1 h in 3% bovine serum albumin (BSA) in PBS. For immunolabeling, the sections were incubated with antibodies targeting occludin (Invitrogen), CD31 (Abcam, Cambridge, MA, USA) or Itch (Abnova) overnight at 4°C. After washing, the sections were incubated with Alexa 488 anti‐rabbit IgG (Molecular Probes, Eugene, OR, USA) in TNB buffer (1:400). Immunofluorescence was visualized using a Zeiss LSM 510 confocal microscope.
Statistical Analysis
Data are represented as the mean ± SEM. Statistical significance was determined using a one‐way analysis of variance (ANOVA) followed by a Dunnett's test for multi‐group comparisons. A P‐value of P < 0.05 was used to determine statistically significant differences.
Results
Changes in Tight Junction Proteins and Neurovascular Damage Following Permanent MCAO
The STAIR endorses the testing of agents in permanent models of ischemia to mimic better the typical human stroke without reperfusion 20. To verify the temporal profile of neurovascular damage following pMCAO, we examined the appearance of a 150‐kDa breakdown product of spectrin and a 48‐kDa form of calcineurin by immunoblotting. A significant increase in the breakdown products of spectrin and calcineurin was observed at 2 h after pMCAO and persisted until 24 h after pMCAO (Figure 1A). MMP‐2 and ‐9 have been shown to be upregulated in focal cerebral ischemia and are involved in the breakdown of the BBB 21. Here, we confirmed that pMCAO induced the significant activation of MMP‐2 and MMP‐9 in both the core and penumbra regions (Figure 1B). In parallel with the observed ZO‐1 breakdown, we found evidence for the degradation of occludin in both the core and penumbra regions at 24 h after pMCAO (Figure 1C,D). In addition, we observed a dramatic increase in the appearance of a high molecular weight band for occludin that was between 90 and 118 kDa in size (Figure 1D).
Figure 1.
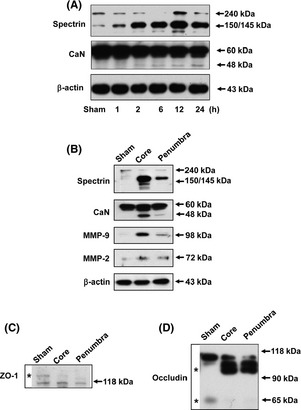
Permanent brain ischemia induces neurovascular damage in rats. (A) Representative immunoblotting image showing spectrin and calcineurin expression following permanent middle cerebral artery occlusion (pMCAO) at the indicated times. (B) Representative immunoblots from the core and penumbra brain regions of pMCAO‐injured rats, as assayed using the indicated antibodies indicative of neurovascular damage. (C) The change in ZO‐1 expression in the core and penumbra brain regions of pMCAO rats. The asterisk indicates the 220‐kDa band of ZO‐1. (D) The changes in occludin expression in the core and penumbra brain regions of pMCAO rats. The asterisk identifies the high molecular weight band and the 65‐kDa band of occludin in sham and pMCAO rats.
The Inhibitory Effect of DAPT on Neuronal Damage Following Permanent MCAO
We next investigated whether inhibition of γ‐secretase 22, metalloproteases or proteases would provide neuroprotection against pMCAO injury. We found that the γ‐secretase inhibitor DAPT ameliorated pMCAO‐induced neuronal damage, as demonstrated by the decrease in the amount of spectrin and calcineurin breakdown products (Figure 2A,B). However, ALLM and Batimastat/BB‐94, broad‐spectrum inhibitors of calpain and metalloproteinase proteases, respectively, did not significantly reduce pMCAO‐induced brain damage in comparison with vehicle‐treated animals (Figure 2A,B).
Figure 2.
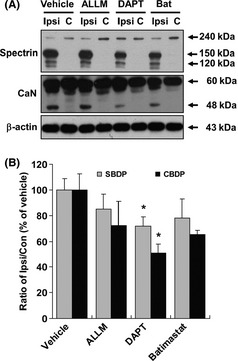
The inhibitory effect of DAPT on the neuronal damage following permanent MCAO. (A) Representative image of an immunoblot using antibodies against spectrin and calcineurin after DAPT, ALLM, and Batimastat treatment in pMCAO rats. Con, contralateral hemisphere; Ipsi, ipsilateral hemisphere. (B) The effect of DAPT, ALLM, and Batimastat treatment on spectrin and calcineurin expression was quantitatively analyzed according to the ratio of the expression in the ipsilateral/contralateral brain. Immunoblotting with an anti‐β‐actin antibody shows equal protein loading in each lane. Data are expressed as the percentage of the values observed in the vehicle‐treated animals (mean ± SEM, n = 5). *P < 0.05 versus vehicle‐treated rats. SBDP, spectrin breakdown products; CBDP, calcineurin breakdown products.
DAPT Attenuates BBB Disruption Following Permanent MCAO
The maintenance of BBB function is critical for a positive clinical outcome after brain ischemic injury 23. Here, we examined the protective effect of DAPT on BBB disruption during pMCAO by quantifying the amount of Evans blue solution that penetrated the brain tissue after injury. In the ipsilateral hemisphere, we observed that the amount of Evans blue solution that extravasated from blood vessels into the brain parenchyma was significantly increased at 24 h after pMCAO (Figure 3). In comparison with vehicle treatment, DAPT treatment (10 and 50 μmol/L) significantly reduced the Evans blue leakage at 24 h after pMCAO in the ipsilateral hemisphere (Figure 3).
Figure 3.

DAPT attenuates the blood–brain barrier (BBB) disruption produced by permanent MCAO. (A) Extensive Evans blue dye extravasation (blue areas) observed in the ipsilateral hemisphere of rat brains after pMCAO. (B) The effect of DAPT treatment on BBB permeability at 24 h after pMCAO. Extravasation of Evans blue was expressed as μg/mg brain tissue. The data in (B) and (C) are shown as the mean ± SEM (n = 5 in each group). **P < 0.01 versus the contralateral side of the same brain. # P < 0.05 versus vehicle‐treated rats. Con, contralateral hemisphere; Ipsi, ipsilateral hemisphere.
Permanent Brain Ischemia Induces Ubiquitination and Degradation of Occludin
Representative immunoblots are presented in Figure 4A and show a significant increase in the high molecular weight isoform of occludin in rats at 6 h after pMCAO as compared to the sham rats. In contrast, we observed degradation of occludin, as evidenced by the loss of its 65‐kDa form, in a time‐dependent manner after pMCAO as compared to sham treatment (Figure 4A). Moreover, our data showing that ubiquitination of occludin occurs in the brain lysates of pMCAO rats was further supported by immunoprecipitation experiments (Figure 4B). Concomitantly, we observed high molecular weight bands of approximately 90–118 kDa in the brain lysates of rats following pMCAO after probing with an anti‐ubiquitin antibody, whereas DAPT treatment significantly decreased the ubiquitination of occludin (Figure 4C). As shown in Figure 4D, the bands larger than the 90‐kDa marker are consistent in size with polyubiquitin ladders. The steady‐state levels of 65‐kDa occludin were reduced by 3‐fold in comparison with the levels detected on the ipsilateral side, suggesting that the steady‐state levels of occludin are inversely proportional to its degree of ubiquitination. Furthermore, DAPT pretreatment significantly decreased the ubiquitination and degradation of occludin at 24 h after pMCAO (Figure 4D,E).
Figure 4.

DAPT attenuates the ubiquitination and degradation of occludin during permanent MCAO. (A) Temporal changes in occludin degradation after pMCAO at the indicated epitopes. (B) Occludin is ubiquitinated in the rat brain following pMCAO. The high molecular weight occludin isoform, which is suggestive of polyubiquitinated occludin, was observed in the rat brain, as detected by the immunoprecipitation of occludin followed by immunoblotting with an anti‐ubiquitin antibody. (C) DAPT pretreatment significantly decreased the ubiquitination of occludin. The asterisk indicates the high molecular weight band. (D) DAPT treatment suppressed both the ubiquitination and degradation of occludin during permanent MCAO. The asterisk indicates the high molecular weight band (a) and the 65‐kDa band for occludin (b) in vehicle‐ and DAPT‐treated rats following pMCAO. (E) Changes in the expression of band (a) and band (b) were analyzed quantitatively as ratio of the values observed in the ipsilateral/contralateral brain. Data are expressed as the percentage of the values observed in vehicle‐treated animals (mean ± SEM, n = 5). *P < 0.05 versus vehicle‐treated rats. Con, contralateral hemisphere; Ipsi, ipsilateral hemisphere.
The Neurovascular Protective Effect of DAPT is Associated with its Inhibitory Effect on γ‐Secretase Activation in pMCAO Rats
As shown in Figure 5A, we observed that the γ‐secretase activity reached its maximum at 6 h and remained elevated for at least 24 h after pMCAO. Consistently, permanent ischemia induced a significant decrease in Notch1 protein expression after 6 h in pMCAO rats (55.9 ± 9.2% vs. sham, P < 0.05; Figure 5B). Parallelled with its inhibitory effect on BBB breakdown and occludin degradation, DAPT treatment (50 μmol/L) significantly blocked Notch1 degradation after pMCAO (Figure 5C). Hairy enhancer of split‐1 (HES‐1) is a transcriptional target that is downstream of activated Notch1 24. DAPT treatment (50 μmol/L) significantly suppressed the pMCAO‐induced elevation of HES‐1 (Figure 5C). However, no significant differences in Jagged‐1 expression were found between each group throughout the experiments (Figure 5C).
Figure 5.
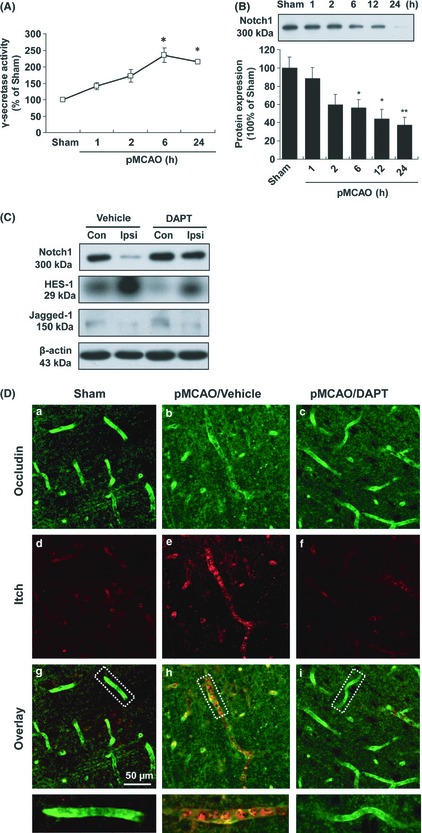
The neurovascular protective effect of DAPT is associated with its inhibitory effect on γ‐secretase and Itch activation in pMCAO rats. (A) The changes in γ‐secretase activity in the rat brain following pMCAO. Data are presented as mean ± SEM (n = 3); *P < 0.05 versus sham‐operated rats. (B) Representative immunoblots probed with an anti‐Notch1 antibody. A 300‐kDa Notch1 band was observed in the brain extracts from sham and pMCAO rats. Data are expressed as the percentage of the value observed in sham‐operated animals (mean ± SEM, n = 4). *P < 0.05 versus sham‐operated rats. (C) Representative immunoblots from brain lysates of vehicle‐ and DAPT‐treated pMCAO rats when probed with anti‐Notch1, ‐HES‐1 and ‐Jagged‐1 antibodies. Con, contralateral hemisphere; Ipsi, ipsilateral hemisphere. (D) Fluorescent staining of occludin and Itch in the cerebral microvessels of pMCAO rats. Anti‐occludin (a, b, and c) and ‐Itch (d, e, and f) staining was performed 24 h after pMCAO with or without DAPT treatment. Scale bar = 50 μm.
DAPT Attenuates the Itch Activation and Degradation of Occludin in Brain Microvessels During Permanent Brain Ischemia
There is limited information on the role of ubiquitin ligases in the ubiquitination and degradation of tight junction proteins in a pathophysiological context 20, 25. We next examine whether the degradation of occludin coincides with changes in the E3 ubiquitin ligase Itch in the brain vascular endothelium. Consistent with the immunoblot data, an immunohistochemical analysis of the control sections revealed strong immunoreactivity for occludin in a continuous, interendothelial staining pattern that colocalized with the endothelial marker CD31 (Supplementary Figure S1A, B). In the penumbra of the rat forebrain after pMCAO, immunoreactivity for Itch was predominantly observed in the brain microvessel endothelium of the ipsilateral side at 24 h after injury (Figure 5D‐e) and occurred in parallel with decreased occludin immunoreactivity (Figure 5D‐b). Moreover, DAPT treatment (50 μmol/L) partially blocked pMCAO‐induced Itch immunoreactivity in the brain microvessels, paralleled with inhibitory effect on occludin degradation (Figure 5D‐f,i).
Discussion
Recent studies suggest that damage to tight junction proteins may play a role in the pathological process of neurovascular damage during brain ischemia; however, the underlying cellular and molecular mechanisms remain elusive 14, 26. The current study demonstrates the following critical findings: (1) permanent brain ischemia leading to BBB breakdown is associated with the ubiquitination and degradation of occludin, (2) intraventricular treatment with the γ‐secretase blocker DAPT effectively suppresses the ubiquitination and degradation of occludin and inhibits BBB breakdown, (3) the inhibition of Notch1/HES‐1 signaling and Itch overactivation may contribute to the neurovascular protective effect of DAPT following pMCAO.
Tight junctions form a virtually impermeable barrier between endothelial cells by preventing the free passage of molecules and ions; they also join together the cytoskeletons of adjacent cells 5, 6. Experimental evidence confirms that occludin, a four‐pass integral transmembrane protein containing an intracellular amino and carboxy terminus, is a functional component of the paracellular barrier. The overall hydrophilicity of occludin predicts two extracellular loops bounded by NH2‐terminal and COOH‐terminal cytoplasmic domains 6, 27. Currently, there is not sufficient information to describe the function of occludin in maintaining BBB integrity during brain ischemia.
The ubiquitin‐proteasome system degrades intracellular proteins including membrane‐surface receptors 28, 29. In the present study, the ubiquitination and degradation of occludin in the rat brain following the onset of pMCAO was observed. Notably, our data indicated that the ubiquitination of occludin was significantly elevated in the brain lysates of pMCAO rats. The observed accumulation of ubiquitinated occludin protein in the pMCAO rats suggests that an ubiquitin‐dependent mechanism is responsible for recognizing defective tight junction proteins in the brain. We further demonstrated that a greater extent of ischemic injury corresponded to a greater extent of ubiquitination and degradation of occludin in the compensative phases of ischemia. However, occludin appeared to lose its adaptive response and its ability to relieve the burden of excessive injury in the late phase of ischemia, as we observed that the degradation of occludin occurred at 6 h after pMCAO.
To determine the specific protease(s) involved in the proteolysis of occludin, the rats subjected to pMCAO were intraventricularly treated with different protease inhibitors. Notably, we observed the γ‐secretase inhibitor DAPT significantly blocked the pMCAO‐induced neurovascular damage, whereas ALLM and Batimastat, which are inhibitors of calpain and metalloproteinase proteases, respectively, were ineffective. It has been reported that synthetic MMP inhibitors restore the integrity of the BBB in the early stages but are ineffective in the later stages after there has been intense damage to the blood vessels in the brain 21. Moreover, Liebetrau and colleagues found that one potential calpain inhibitor failed to attenuate the brain ischemic damage in rats following 3 h of ischemia and 24 h of reperfusion 30. Thus, we postulate that ALLM and Batimastat may provide modest neuroprotection in a transient rat MCAO model when administered before the ischemic insult, but not when administered following severe injury in the pMCAO model.
Furthermore, we found that intraventricular treatment with DAPT suppressed the aggregation and degradation of occludin, in addition to its inhibition of BBB breakdown and neurovascular damage. Li and colleagues recently reported that DAPT protects the brain against damage caused by cerebral ischemia 31. These authors demonstrated that DAPT decreased brain water content and infarct sizes after pMCAO, although they did not identify the molecular mechanisms involved in this neurovascular damage 31. One key remaining question is whether γ‐secretase activation during the early period of brain ischemia eventually leads to the overaccumulation of aggregates of ubiquitinated occludin and occludin degradation. Thus, we examined whether the inhibition of γ‐secretase would be associated with ischemia‐induced BBB damage. DAPT is commonly used as a cell permeable inhibitor for enzymes of the secretase family and may be helpful in studying occludin degradation in pMCAO. Our findings are in agreement with and extend upon previous studies 31, as we found that DAPT treatment attenuates BBB damage that can be detected with Evans blue staining. Additionally, we demonstrated that the decrease in occludin ubiquitination and degradation occurred in parallel with the neurovascular protective effect of DAPT. Furthermore, we also evaluated the pharmacological profile of DAPT in Notch1/HES‐1 signaling, which is a sensitive substrate of γ‐secretase, and our findings support the notion that γ‐secretase may play a role in permanent focal ischemia, as DAPT is only protective when it significantly downregulates the breakdown of Notch1 and the transcriptional activation of HSE‐1.
The ubiquitin‐proteasome system is the major pathway for protein degradation in the cytoplasm of eukaryotic cells 28, 29, 32, yet there have been few studies describing this system in the microvessels of the brain. During pMCAO, we assessed the specific components of the BBB and targets for ubiquitination by examining the changes in expression of the E3 ubiquitin–protein ligase Itch, a member of the HECT domain‐containing ubiquitin–protein ligases. Using yeast two‐hybrid screening identified that the NH2‐terminal region of occludin binds specifically to a multidomain of Itch. We found that immunoreactivity for Itch was predominantly observed in the microvessel endothelium on the ipsilateral side of the brain at 24 h and occurred in parallel with a decrease in occludin immunoreactivity. Thus, our data suggest that the tight junction‐specific protein occludin may be a functional target of the E3 ubiquitin–protein ligase Itch in the microvessels of brain after pMCAO. Based on the inhibitory effect of DAPT on Itch overactivation in brain microvessels, we hypothesize that the E3 ubiquitin‐protein ligase Itch may be a potential downstream target of γ‐secretase activation following pMCAO.
The altered ability of the ubiquitin‐proteasome pathway to effectively degrade misfolded proteins produces protein aggregation and neuronal death in virtually all pathologic conditions 33, 34, 35, 36, 37. Several studies suggest that protein aggregation is a secondary cellular reaction that reflects an imbalance in protein homeostasis between the amount of unfolded protein and the capacity of the protein quality control systems to eliminate these misfolded proteins 35, 37. The accumulation of aggregates of ubiquitinated occludin in the microvasculature of the brain after ischemia may be partially attributed to alterations in the ubiquitin‐proteasome pathway. Our data suggest that unfolded occludin proteins may be too numerous to be completely degraded by proteasomes. Previous data have shown that proteasome peptidase activity declines after brain ischemia/reperfusion 38. In the present study, we did not determine whether aggregated occludin was toxic during brain ischemia, although it is known that protein aggregates in vitro can destabilize artificial phospholipid bilayers, which leads to permeabilization of the membrane 39.
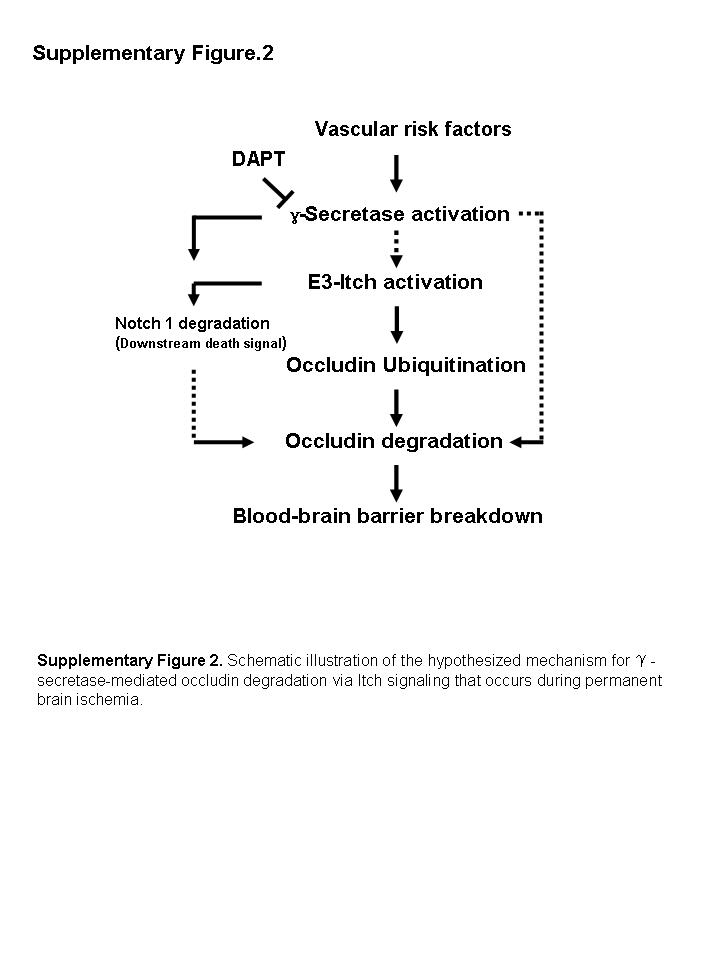
In conclusion, our study demonstrates that inhibition of ubiquitin‐mediated occludin degradation in brain microvessels by γ‐secretase inhibition contributes to an attenuation of the BBB breakdown that occurs during pMCAO. Therefore, our data outline a mechanistic model demonstrating the early activation of γ‐secretase by vascular risk factors, secondary activation of Itch in microvessels, degradation of occludin protein, BBB breakdown and eventual neurovascular damage during the pathological processes of permanent brain ischemia (Supplementary Figure S2). Based on the present results, we hypothesize that modulation of the γ‐secretase pathway and Itch activity may represent an important strategy to prevent or improve the neurovascular complications associated with stroke.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Figure S1. The colocalization of occludin and CD31 in cerebal microvessels of rats.
{kind=link}
Figure S2. Schematic illustration of the hypothesized for γ‐secretase mediated occludin degradation via Itch signaling that occurs during permanent brain ischemia.
{kind=link}
Acknowledgments
This work was funded by National Natural Science Foundations of China (30973521 to F.H); Projects of International Cooperation and Exchanges NSFC (81120108023 to F.H); Zhejiang Provincial Natural Science Foundation of China (R2100281 to F.H; 2010R50049 to YM.L). The Zhejiang Provincial Qianjiang Talent Plan (2012R10036 to FH).
References
- 1. Han F, Shirasaki Y, Fukunaga K. Microsphere embolism‐induced endothelial nitric oxide synthase expression mediates disruption of the blood‐brain barrier in rat brain. J Neurochem 2006;99:97–106. [DOI] [PubMed] [Google Scholar]
- 2. Zeng XN, Xie LL, Liang R, Sun XL, Fan Y, Hu G. AQP4 knockout aggravates ischemia/reperfusion injury in mice. CNS Neurosci Ther 2012;18:388–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cheng T, Liu D, Griffin JH, et al. Activated protein C blocks p53‐mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med 2003;9:338–342. [DOI] [PubMed] [Google Scholar]
- 4. Tao RR, Ji YL, Lu YM, Fukunaga K, Han F. Targeting nitrosative stress for neurovascular protection: new implications in brain diseases. Curr Drug Targets 2012;13:272–284. [DOI] [PubMed] [Google Scholar]
- 5. Chun HB, Scott M, Niessen S, et al. The proteome of mouse brain microvessel membranes and basal lamina. J Cereb Blood Flow Metab 2011;31:2267–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu WY, Wang ZB, Zhang LC, Wei X, Li L. Tight junction in blood‐brain barrier: an overview of structure, regulation, and regulator substances. CNS Neurosci Ther 2012;18:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Song SW, Sun Y, Su BL, et al. Risperidone enhances the vulnerability to stroke in hypertensive rats. CNS Neurosci Ther 2012;18:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zou YX, Zhang XH, Su FY, Liu X. Importance of Riboflavin Kinase in the Pathogenesis of Stroke. CNS Neurosci Ther 2012;18:834–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. del Zoppo GJ. Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience 2009;158:972–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Han F, Tao RR, Zhang GS, et al. Melatonin ameliorates ischemic‐like injury‐evoked nitrosative stress: involvement of HtrA2/PED pathways in endothelial cells. J Pineal Res 2011;50:281–291. [DOI] [PubMed] [Google Scholar]
- 11. Stowe AM, Adair‐Kirk TL, Gonzales ER, et al. Neutrophil elastase and neurovascular injury following focal stroke and reperfusion. Neurobiol Dis 2009;35:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pendyala S, Usatyuk PV, Gorshkova IA, Garcia JG, Natarajan V. Regulation of NADPH oxidase in vascular endothelium: the role of phospholipases, protein kinases, and cytoskeletal proteins. Antioxid Redox Signal 2009;11:841–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harhausen D, Prinz V, Ziegler G, et al. CD93/AA4.1: a novel regulator of inflammation in murine focal cerebral ischemia. J Immunol 2010;184:6407–6417. [DOI] [PubMed] [Google Scholar]
- 14. Lochhead JJ, McCaffrey G, Quigley CE, et al. Oxidative stress increases blood‐brain barrier permeability and induces alterations in occludin during hypoxia‐reoxygenation. J Cereb Blood Flow Metab 2010;30:1625–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boscia F, Gala R, Pignataro G, et al. Permanent focal brain ischemia induces isoform‐dependent changes in the pattern of Na+/Ca2+ exchanger gene expression in the ischemic core, periinfarct area, and intact brain regions. J Cereb Blood Flow Metab 2006;26:502–517. [DOI] [PubMed] [Google Scholar]
- 16. Birukova AA, Zebda N, Fu P, Poroyko V, Cokic I, Birukov KG. Association between adherens junctions and tight junctions via Rap1 promotes barrier protective effects of oxidized phospholipids. J Cell Physiol 2011;226:2052–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fukunaga K, Sato H, Takatsu K, Tominaga A, Miyamoto E. Monoclonal antibody against a multifunctional calmodulin‐dependent protein kinase from rat brain and the tissue distribution of the enzyme. Biomed Res 1986;7:405–413. [Google Scholar]
- 18. Farmery MR, Tjernberg LO, Pursglove SE, Bergman A, Winblad B, Näslund J. Partial purification and characterization of gamma‐secretase from post‐mortem human brain. J Biol Chem 2003;278:24277–24284. [DOI] [PubMed] [Google Scholar]
- 19. Han F, Shioda N, Moriguchi S, et al. Spiro[imidazo[1,2‐a]pyridine‐3,2‐indan]‐2(3H)‐one (ZSET1446/ST101) treatment rescues olfactory bulbectomy‐induced memory impairment by activating Ca2+/calmodulin kinase II and protein kinase C in mouse hippocampus. J Pharmacol Exp Ther 2008;326:127–134. [DOI] [PubMed] [Google Scholar]
- 20. Fisher M. Stroke therapy academic industry roundtable. Recommendations for advancing development of acute stroke therapies: Stroke Therapy Academic Industry Roundtable 3. Stroke 2003;34:1539–1546. [DOI] [PubMed] [Google Scholar]
- 21. Rosenberg GA, Yang Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg Focus 2007;22:E4. [DOI] [PubMed] [Google Scholar]
- 22. Ni CY, Murphy MP, Golde TE, Carpenter G. gamma‐Secretase cleavage and nuclear localization of ErbB‐4 receptor tyrosine kinase. Science 2001;294:2179–2181. [DOI] [PubMed] [Google Scholar]
- 23. Kawai N, Stummer W, Ennis SR, Betz AL, Keep RF. Blood‐brain barrier glutamine transport during normoglycemic and hyperglycemic focal cerebral ischemia. J Cereb Blood Flow Metab 1999;19:79–86. [DOI] [PubMed] [Google Scholar]
- 24. Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature 1995;377:355–358. [DOI] [PubMed] [Google Scholar]
- 25. Traweger A, Fang D, Liu YC, et al. The tight junction‐specific protein occludin is a functional target of the E3 ubiquitin‐protein ligase itch. J Biol Chem 2002;277:10201–10208. [DOI] [PubMed] [Google Scholar]
- 26. Stamatovic SM, Keep RF, Wang MM, Jankovic I, Andjelkovic AV. Caveolae‐mediated internalization of occludin and claudin‐5 during CCL2‐induced tight junction remodeling in brain endothelial cells. J Biol Chem 2009;284:19053–19066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF‐mediated disruption of endothelial CLN‐5 promotes blood‐brain barrier breakdown. Proc Natl Acad Sci U S A 2009;106:1977–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rott R, Szargel R, Haskin J, et al. α‐Synuclein fate is determined by USP9X‐regulated monoubiquitination. Proc Natl Acad Sci U S A 2011;108:18666–18671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Altun M, Zhao B, Velasco K, et al. Ubiquitin‐specific protease 19 (USP19) regulates hypoxia‐inducible factor 1α (HIF‐1α) during hypoxia. J Biol Chem 2012;287:1962–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liebetrau M, Martens H, Thomassen N, et al. Calpain inhibitor A‐558693 in experimental focal cerebral ischemia in rats. Neurol Res 2005;27:466–470. [DOI] [PubMed] [Google Scholar]
- 31. Li S, Zhang X, Wang Y, Ji H, Du Y, Liu H. DAPT protects brain against cerebral ischemia by down‐regulating the expression of Notch 1 and Nuclear factor kappa B in rats. Neurol Sci 2012. doi: 10.1007/s10072-012-0948-6. [DOI] [PubMed] [Google Scholar]
- 32. Wakatsuki S, Yumoto N, Komatsu K, Araki T, Sehara‐Fujisawa A. Roles of meltrin‐beta/ADAM19 in progression of Schwann cell differentiation and myelination during sciatic nerve regeneration. J Biol Chem 2009;284:2957–2966. [DOI] [PubMed] [Google Scholar]
- 33. Niwa J, Yamada S, Ishigaki S, et al. Disulfide bond mediates aggregation, toxicity, and ubiquitylation of familial amyotrophic lateral sclerosis‐linked mutant SOD1. J Biol Chem 2007;282:28087–28095. [DOI] [PubMed] [Google Scholar]
- 34. Janué A, Olivé M, Ferrer I. Oxidative stress in desminopathies and myotilinopathies: a link between oxidative damage and abnormal protein aggregation. Brain Pathol 2007;17:377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee JT, Wheeler TC, Li L, Chin LS. Ubiquitination of alpha‐synuclein by Siah‐1 promotes alpha‐synuclein aggregation and apoptotic cell death. Hum Mol Genet 2008;17:906–917. [DOI] [PubMed] [Google Scholar]
- 36. Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol 2009;11:219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Raspe M, Gillis J, Krol H, et al. Mimicking proteasomal release of polyglutamine peptides initiates aggregation and toxicity. J Cell Sci 2009;122:3262–3271. [DOI] [PubMed] [Google Scholar]
- 38. Ge P, Luo Y, Liu CL, Hu B. Protein aggregation and proteasome dysfunction after brain ischemia. Stroke 2007;38:3230–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Canale C, Torrassa S, Rispoli P, et al. Natively folded HypF‐N and its early amyloid aggregates interact with phospholipid monolayers and destabilize supported phospholipid bilayers. Biophys J 2006;91:4575–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The colocalization of occludin and CD31 in cerebal microvessels of rats.
Figure S2. Schematic illustration of the hypothesized for γ‐secretase mediated occludin degradation via Itch signaling that occurs during permanent brain ischemia.