

Supplemental Digital Content is available in the text.
Keywords: chromatin, endothelial cells, endothelium, homeostasis, transcription factors
Abstract
Rationale:
The ETS (E-26 transformation-specific) transcription factor ERG (ETS-related gene) is essential for endothelial homeostasis, driving expression of lineage genes and repressing proinflammatory genes. Loss of ERG expression is associated with diseases including atherosclerosis. ERG’s homeostatic function is lineage-specific, because aberrant ERG expression in cancer is oncogenic. The molecular basis for ERG lineage-specific activity is unknown. Transcriptional regulation of lineage specificity is linked to enhancer clusters (super-enhancers).
Objective:
To investigate whether ERG regulates endothelial-specific gene expression via super-enhancers.
Methods and Results:
Chromatin immunoprecipitation with high-throughput sequencing in human umbilical vein endothelial cells showed that ERG binds 93% of super-enhancers ranked according to H3K27ac, a mark of active chromatin. These were associated with endothelial genes such as DLL4 (Delta-like protein 4), CLDN5 (claudin-5), VWF (von Willebrand factor), and CDH5 (VE-cadherin). Comparison between human umbilical vein endothelial cell and prostate cancer TMPRSS2 (transmembrane protease, serine-2):ERG fusion-positive human prostate epithelial cancer cell line (VCaP) cells revealed distinctive lineage-specific transcriptome and super-enhancer profiles. At a subset of endothelial super-enhancers (including DLL4 and CLDN5), loss of ERG results in significant reduction in gene expression which correlates with decreased enrichment of H3K27ac and MED (Mediator complex subunit)-1, and reduced recruitment of acetyltransferase p300. At these super-enhancers, co-occupancy of GATA2 (GATA-binding protein 2) and AP-1 (activator protein 1) is significantly lower compared with super-enhancers that remained constant following ERG inhibition. These data suggest distinct mechanisms of super-enhancer regulation in endothelial cells and highlight the unique role of ERG in controlling a core subset of super-enhancers. Most disease-associated single nucleotide polymorphisms from genome-wide association studies lie within noncoding regions and perturb transcription factor recognition sequences in relevant cell types. Analysis of genome-wide association studies data shows significant enrichment of risk variants for cardiovascular disease and other diseases, at ERG endothelial enhancers and super-enhancers.
Conclusions:
The transcription factor ERG promotes endothelial homeostasis via regulation of lineage-specific enhancers and super-enhancers. Enrichment of cardiovascular disease-associated single nucleotide polymorphisms at ERG super-enhancers suggests that ERG-dependent transcription modulates disease risk.
Maintenance of endothelial homeostasis is essential for vascular health. Disruption of endothelial homeostasis, as observed in atherosclerosis and in chronic inflammatory diseases, leads to profound changes in the phenotype of endothelial cells (ECs) with upregulation of proinflammatory pathways and loss of anti-inflammatory pathways. Loss of endothelial lineage identity is associated with endothelial-to-mesenchymal transition, a process implicated in multiple diseases.1 The transcriptional mechanisms regulating EC lineage identity and maintenance of endothelial homeostasis are also areas of immense interest for vascular regenerative therapies but remain poorly understood.
Meet the First Author, see p 1278
The ETS (E-26 transformation-specific) transcription factor (TF) ERG (ETS-related gene) is a critical regulator of endothelial homeostasis (reviewed in Shah et al2). In the endothelium, ERG expression appears around developmental day E8.5 and is maintained into adulthood.3 ERG is required for endothelial lineage specification,4 vascular development, and angiogenesis5; endothelial-specific deletion of Erg in mouse results in embryonic lethality due to vascular defects.6,7 ERG drives expression of lineage-specific genes such as CDH5 (VE-cadherin), DLL4 (Delta-like protein 4), CLDN5 (claudin-5), and VWF (von Willebrand factor) and controls processes including survival, permeability, and cytoskeletal dynamics (reviewed in Shah et al2). Molecular pathways through which ERG promotes vascular stability and angiogenesis include Wnt/β-catenin signaling6 and angiopoietin-1–dependent Notch signaling.8 ERG maintains vascular homeostasis also by repressing expression of proinflammatory genes such as ICAM1 and IL89,10 and by protecting from endothelial-to-mesenchymal transition.11 In line with its homeostatic role, ERG’s expression is lost in vascular diseases such as the activated endothelium overlying human atherosclerotic plaques.10
However, aberrant expression of ERG in non-EC can be detrimental. ERG overexpression as the result of chromosomal translocations in prostate cancer correlates with malignancy and invasiveness, poor prognosis and shorter survival times.12 In these circumstances, ERG acts as an oncogene. Since the first report of gene fusions between ERG and the regulatory region of the androgen-dependent TMPRSS-2 (transmembrane protease, serine-2) gene,13 the molecular mechanisms through which ERG aberrant expression is oncogenic have been investigated in detail (reviewed in Adamo and Ladomery14). The striking difference between the homeostatic versus oncogenic roles of ERG has important implications for the possible therapeutic potential of this pathway in pathologies associated with cardiovascular disease (CVD).
The mechanistic basis for the difference in ERG’s lineage-specific activity may lie in the chromatin landscape. Genetic regulatory elements, called enhancers, play a key role in mediating the transcriptional regulation of lineage-specific gene expression.15 Enhancer activation by TFs is cooperative and hierarchical.15–17 Members of the ETS, AP-1 (activator protein 1), and GATA families have been shown to functionally interact at enhancers associated with lineage-specific genes.16 Recently, clusters of enhancer elements variably termed super-enhancers, stretch enhancers or enhancer clusters, have been described in numerous cell types, including EC.18–21 These regulatory elements are associated with an extremely high abundance of TFs, H3K27ac-modified nucleosomes and MED1 (Mediator complex subunit 1) and drive cell type-specific gene expression.18,19
Furthermore, super-enhancers are enriched in single nucleotide polymorphisms (SNPs) associated with specific diseases, in a cell type-specific manner.18 The majority of SNPs identified by genome-wide association studies (GWASs) associated with human disease traits are localized to noncoding regions of the genome,22 including promoters and enhancers, and frequently perturb TF recognition sequences. Thus, SNPs associated with CVD risk may affect TF-binding sites within super-enhancers in EC and other cell types relevant to CVD.
In this study, we show that ERG regulates a subset of endothelial super-enhancers, and that CVD-associated SNPs are enriched at ERG enhancers and super-enhancers. The association of ERG-bound loci with CVD risk variants provides candidate SNPs for future studies on the epigenomic pathways underlying cardiovascular pathologies.
Methods
All supporting data are available within the article and in the Online Data Supplement. Sequencing data generated in this study (chromatin immunoprecipitation with deep sequencing [ChIP-seq] data sets for ERG, H3K27ac, and MED1 in human umbilical vein endothelial cells [HUVEC]) have been made publicly available at the NCBI Gene Expression Omnibus and can be retrieved through accession number GSE124893.
An expanded Methods section is available in the Online Data Supplement.
Results
Integrated Genomic Analysis Reveals an ERG-Regulated Transcriptional Program for the Vascular Endothelium
ChIP-seq analysis in HUVEC identified 40 821 genomic ERG-binding sites associated with 14 786 genes. Selected ERG peaks in the promoters of known ERG target genes CDH5 and ICAM1 were validated by ChIP-quantitative polymerase chain reaction (qPCR; Online Figure IA). As expected, the canonical ERG motif (C/a/g)(A/C)GGAA(G/A)23 was the most represented at ERG sites in HUVEC (Online Figure IB). Globally, analysis of the distribution of ERG-binding sites relative to annotated transcription start sites (TSSs) revealed that most ERG-bound regions were intragenic and intergenic sites located distally from the promoter (±2 kb from TSS; Figure 1A).
Figure 1.
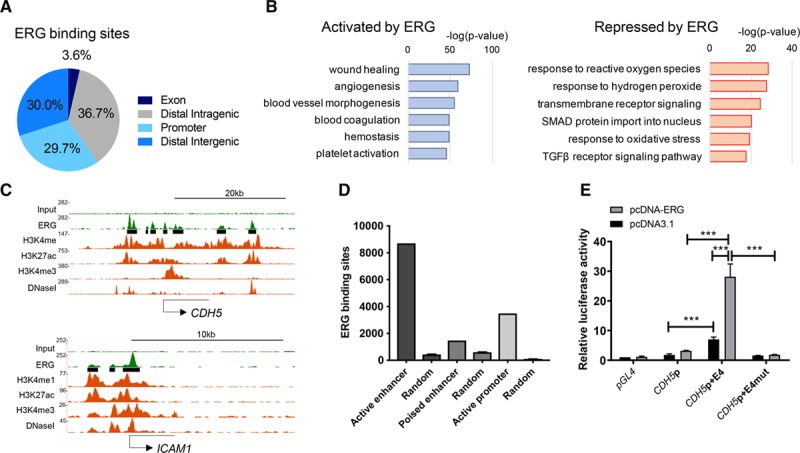
Characterization of ERG (ETS-related gene)-bound enhancers in human umbilical vein endothelial cells (HUVEC). A, Percentage genomic distribution of ERG chromatin immunoprecipitation with deep sequencing (ChIP-seq) peaks. B, Gene ontology analysis showing pathways associated with differentially regulated ERG-bound endothelial target genes (genes activated in blue and genes repressed in red). Significance shown as –log (P value). C, ChIP-seq binding profiles for ERG, H3K4me1, H3K27ac, H3K4me3, and DNase I hypersensitivity in HUVEC; loci of ERG-activated CDH5 (VE-cadherin; top) and ERG-repressed ICAM1 (bottom). The x axis represents the genomic position; the y axis the ChIP-seq signal in reads per million per base pair (rpm/bp). ERG-binding sites are shown as black bars. D, Number of ERG-binding sites associated with specific epigenomic features vs size-matched random regions. Active enhancers are defined by combined H3K4me1 and H3K27ac, poised enhancers by H3K4me1 and H3K27me3, and active promoters by H3K4me3 and H3K27ac. E, Luciferase reporter plasmids (pGL4) containing the CDH5 promoter with or without region E4 or a mutant enhancer (E4mut) were cotransfected into HUVEC along with an ERG cDNA expression plasmid (pcDNA-ERG) or empty vector control, pcDNA3.1. Values are represented as the fold change in relative luciferase activity over the empty vector alone. Values are mean±SEM, n=3. A 2-way ANOVA showed significance (P<0.001) and a post hoc test using Tukey multiple comparisons test shows pairwise differences between specific groups, ***P<0.001. TGF indicates transforming growth factor.
Integrated analysis of global expression profiling24 with ERG ChIP-seq showed that ERG binds 85% (1232/1454) of its activated targets and 80% (939/1180) of its repressed targets (Online Figure IC). Gene ontology pathway analysis revealed that directly activated ERG targets clustered in functions related to angiogenesis, blood vessel morphogenesis, and hemostasis, whereas repressed genes associated with TGF (transforming growth factor)-β/SMAD signaling and stress pathways (Figure 1B), in line with its known roles.
ERG-Bound Enhancers Drive Endothelial Gene Expression
We next examined the relationship between ERG binding and chromatin states in HUVEC using data from the Encyclopedia of DNA Elements Consortium.25 Ninety-seven percent of ERG peaks mapped to regions of DNase I hypersensitivity, a marker of accessible open chromatin.26 Analysis of known ERG target genes showed ERG genomic loci overlapping histone marks of active promoters (H3K4me3 and H3K27ac) and enhancers (H3K4me1 and H3K27ac) at sites of DNase I hypersensitivity (Figure 1C; Online Figure ID). Globally, ERG binding in HUVEC was greatest at active enhancers (Figure 1D).
ERG-bound enhancers were identified in known ERG targets, including CDH5. Four ERG-bound active enhancers, named E1 to E4, were selected based on H3K27ac and H3K4me1 enrichment in a 23 kb region along the CDH5 locus either side of the TSS (Online Figure IIA). The E1, E2, and E4 enhancers, but not the repeat DNA-containing E3 enhancer, were individually cloned into luciferase reporter vectors containing the CDH5 promoter, previously characterized as transactivated by ERG.27 In HeLa cells (which do not express endogenous ERG), all enhancers increased ERG-dependent transactivation of the CDH5 promoter, with E4 being the most active (data not shown). This was then validated in HUVEC, where the E4 enhancer increased basal CDH5 promoter activity >4-fold (Figure 1E); moreover, this region was responsive to ERG transactivation, which further increased luciferase activity by 9-fold compared with CDH5 promoter alone (Figure 1E). Mutation of the 9 AGGAA putative ERG-binding motifs in region E4 (Online Figure IIB and IIC) completely abolished enhancer activity and the response to ERG (Figure 1E).
These findings support a key role for ERG-mediated transactivation of gene expression through EC enhancers.
ERG Binds to HUVEC Super-Enhancers
Multiple ERG-bound enhancers were found in close proximity with each other in ERG-activated genes, such as CDH5 (see Figure 1C). Clusters of enhancers, known as super-enhancers can be distinguished from isolated typical enhancers by enrichment in lineage-specific TF, coactivators such as MED1 and the histone modification mark H3K27ac.18,19,28 Super-enhancers preferentially associate with genes that define cell-lineage identity.18,19 To define endothelial super-enhancers, we identified enhancer regions by co-occupancy of H3K27ac and H3K4me1 in HUVEC.25 Enhancers within 12.5 kb of each other were then concatenated to define a single entity, and ranked by increasing H3K27ac enrichment, as described.28 The analysis identified 917 super-enhancers (Figure 2A; Online Table I) that mapped to 822 genes, including ERG-activated targets VWF, CDH5, ICAM2, SOX17, DLL4, as well as ERG itself (Figure 2A and 2B). A similar super-enhancer profile was obtained when ranked by MED1 enrichment (Online Figure IIIA).
Figure 2.
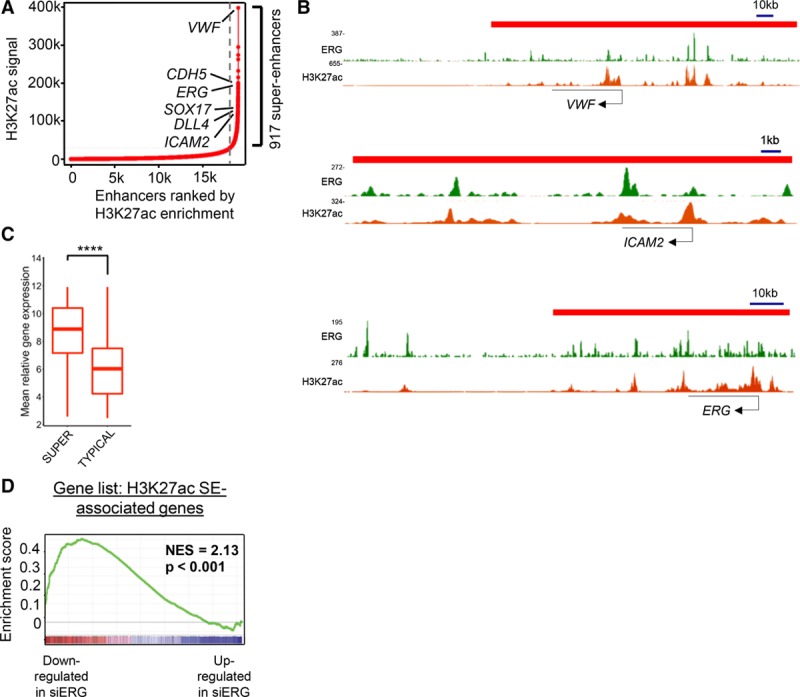
Characterization of human umbilical vein endothelial cell (HUVEC) super-enhancers (SEs) identifies enrichment for ERG (ETS-related gene) and ERG-target genes. A, HUVEC enhancer regions identified by H3K27ac and H3K4me1 were ranked by enrichment of H3K27ac chromatin immunoprecipitation with deep sequencing (ChIP-seq) signal in rpm/bp. SE clusters are shown to the right of the gray dashed line (see also Online Table V). B, HUVEC SE regions indicated as red bars above density plots of ERG ChIP-seq signal alongside H3K27ac at distal enhancers of VWF (von Willebrand factor), ICAM2, and ERG loci. C, Transcriptome profiling of HUVEC shows significantly higher gene expression in SE than in typical enhancers. ***P<0.0001, Wilcoxon rank-sum test. D, Gene set enrichment analysis of the top 500 SE-associated genes compared with the ranked gene list from transcriptome profiling of ERG-deficient HUVEC. Normalized enrichment score (NES)=2.13; P<0.001.
Genes associated with endothelial super-enhancers were found to have significantly higher mean expression levels compared to those associated with typical enhancers (Figure 2C). Gene set enrichment analysis showed significant enrichment of ERG driven genes with the top 500 ranked super-enhancer genes (Figure 2D). These data suggest that ERG regulates endothelial gene expression via super-enhancers.
Remarkably, the vast majority of super-enhancers (93%) were bound by ERG, compared with only 34% of typical enhancers (Figure 3A). In keeping with higher TF occupancy at super-enhancers,19 the canonical ERG motif was significantly more bound by ERG in super-enhancers compared with typical enhancers (35% versus 25%, respectively; Online Figure IIIB). Furthermore, within the 917 super-enhancers, H3K27ac and ERG-binding signal significantly correlated (Figure 3B). We thus tested whether ERG itself could be used to identify super-enhancers in EC. Using ERG enrichment at active enhancers as the ranking parameter, we identified 1125 super-enhancers in HUVEC (Figure 3C), associated with a similar gene set as the H3K27ac super-enhancers (see Figure 2A). Indeed, gene set enrichment analysis demonstrated a strong positive correlation between super-enhancers defined by ERG and those by H3K27ac (Figure 3D). Moreover, functional clustering of H3K27ac super-enhancers and ERG super-enhancers revealed shared pathways essential to EC identity and function (Figure 3E).
Figure 3.
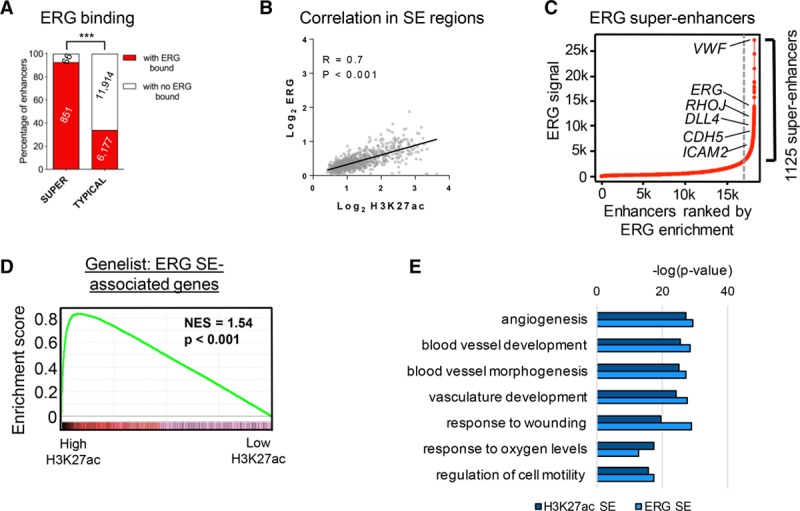
ERG (ETS-related gene) defines super-enhancers (SEs) in human umbilical vein endothelial cells (HUVEC). A, ERG binding at SE and typical enhancer regions. SE have significantly more ERG bound than typical enhancers; ***P<0.0001, Fisher exact test. B, Scatterplot of the correlation between ERG and H3K27ac occupancy (log2 transformed) in the 917 SE regions. Each point is the mean chromatin immunoprecipitation with deep sequencing signal across each SE region; P<0.001. C, SE ranked by ERG enrichment on identified enhancer regions. Known ERG-target genes proximal to SE are indicated (see also Online Table VI). D, Gene set enrichment analysis of the top 500 genes associated with ERG-identified SE compared with the ranked gene list from 917 H3K27ac-enriched SE. Normalized enrichment score (NES)=1.54; P<0.001. E, Gene ontology analysis was performed on the genes associated with SE identified by enrichment for H3K27ac or ERG. The top 7 biological processes ranked by significance (−log (P value)) are depicted.
Thus, ERG binding identifies super-enhancers in differentiated ECs and supports the prominent role of ERG as a lineage-determining TF for the vascular endothelium.
Differential Super-Enhancer Binding Underlies the Lineage-Specific Activity of ERG
At odds with its homeostatic role in EC, aberrant ERG expression due to chromosomal translocations, such as the TMPRSS-2:ERG gene fusions in prostate cells, is a hallmark of cancer (reviewed in Adamo and Ladomery14). To exploit ERG’s therapeutic potential in the vasculature, it is crucial to understand the molecular basis of its lineage-specific role. Therefore, we compared ERG-bound gene targets in HUVEC with those from human prostate epithelial cancer cell line (VCaP) prostate cancer cells carrying the TMPRSS-2:ERG gene fusion. Two publicly available ERG ChIP-seq data sets in VCaP cells29,30 were found to correlate highly (Online Figure IVA); we used the data in Chng et al29 for further analysis. Integration of this ChIP-seq data with transcriptome profiling of ERG-depleted VCaP cells31 identified 584 genes bound and activated by ERG and 589 genes bound and repressed by ERG (Online Figure IVB). Gene ontology pathway analysis showed regulation of genes involved in DNA replication, cell proliferation, cytoskeletal remodeling, and apoptosis (Figure 4A). We next examined the overlap between genes directly bound by ERG in VCaP cells versus HUVEC (Online Figure IVC). Interestingly, only 249 ERG-bound target genes (8%) were found in common between HUVEC and VCaP cells (Figure 4B; Online Table II); of these only 58% were activated or repressed concurrently in both cell types (Figure 4C). Interestingly, even pathways controlled by ERG in both HUVEC and VCaP cells (such as cell migration, adhesion, and Notch signaling) are regulated in a lineage-specific manner (Online Figure IVD). Selected ERG transcriptional targets were validated in HUVEC and VCaP cells by RT-qPCR (Online Figure IVE).
Figure 4.
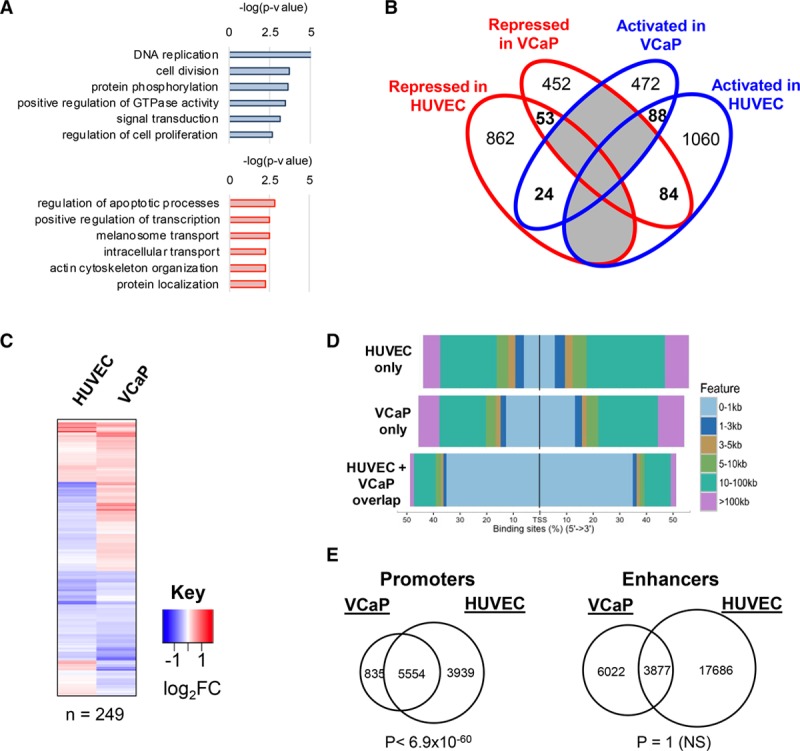
Chromatin immunoprecipitation with deep sequencing analysis identifies a lineage-specific program for ERG (ETS-related gene): ERG expression and binding profile in prostate cancer cells. A, Gene ontology pathway analysis of differentially regulated ERG-bound target genes in human prostate epithelial cancer cell line (VCaP) prostate cancer cells (activated genes in blue and repressed genes in red). Significance of pathway enrichment as −log (P value). B, Venn diagram comparing ERG bound, differentially regulated genes in human umbilical vein endothelial cell (HUVEC) and VCaP cells. Only 249 genes (in bold) are common to the 2 cell types. None of the fractions overlap significantly between the 2-way comparisons using a hypergeometric distribution test. C, Heatmap of expression levels of the shared 249 bound and regulated putative ERG target genes following ERG inhibition in HUVEC and in VCaP cells. D, Genomic view of percentage distribution of ERG peaks relative to transcription start site (TSS) in regions bound by ERG that are shared between HUVEC and VCaP cells, HUVEC only, or VCaP cells only. E, Overlap of ERG-binding sites at H3K4me3-enriched and RefSeq assigned TSS promoter regions (left), and H3K27ac/H3K4me1-enriched enhancers (right), in HUVEC and VCaP cells. Significance reported by a hypergeometric test P value. NS indicates not significant.
Comparison of ChIP-seq data sets between HUVEC and VCaP cells showed that only 23% of ERG-bound sites are in common between the HUVEC and VCaP genomes (Online Figure IVF). Interestingly, the majority (70%) of these shared sites are located close (±1 kb) to the TSS (Figure 4D). In contrast, the proportion of ERG-binding sites unique to HUVEC or VCaP cells are preferentially located at sites distal to the TSS (Figure 4D). Mapping of ERG genomic binding with histone modification marks confirmed a significant overlap of ERG binding to promoters in HUVEC and VCaP cells (Figure 4E, left). However, no significant overlap was observed between ERG binding at enhancers in HUVEC and VCaP cells, with only 18% of ERG-binding sites found at enhancers in VCaP cells (Figure 4E, right). We next asked whether ERG binding in VCaP cells was also associated with super-enhancers. Using ranked enrichment of H3K27ac from ChIP-seq in VCaP cells,32 we identified 208 super-enhancers (Figure 5A). Genes associated with super-enhancers in VCaP had significantly higher average expression levels compared to those associated with isolated typical enhancers (Online Figure VA), as expected. The vast majority of VCaP super-enhancers were bound by ERG (91%), compared with 48% of typical enhancers (Figure 5B). Interestingly, gene set enrichment analysis showed no significant relationship between super-enhancer–associated genes in VCaP cells and HUVEC (Online Figure VB). Gene ontology analysis revealed different pathways regulated by super-enhancer–associated genes in HUVEC compared with VCaP cells (Figure 5C). Finally, super-enhancers occupied by ERG showed cell-lineage specificity: endothelial ERG-regulated genes (such as CDH5) are associated with super-enhancers in HUVEC but not VCaP cells, and vice versa for VCaP cell genes (such as TMPRSS2; Figure 5D; Online Figure VC).
Figure 5.
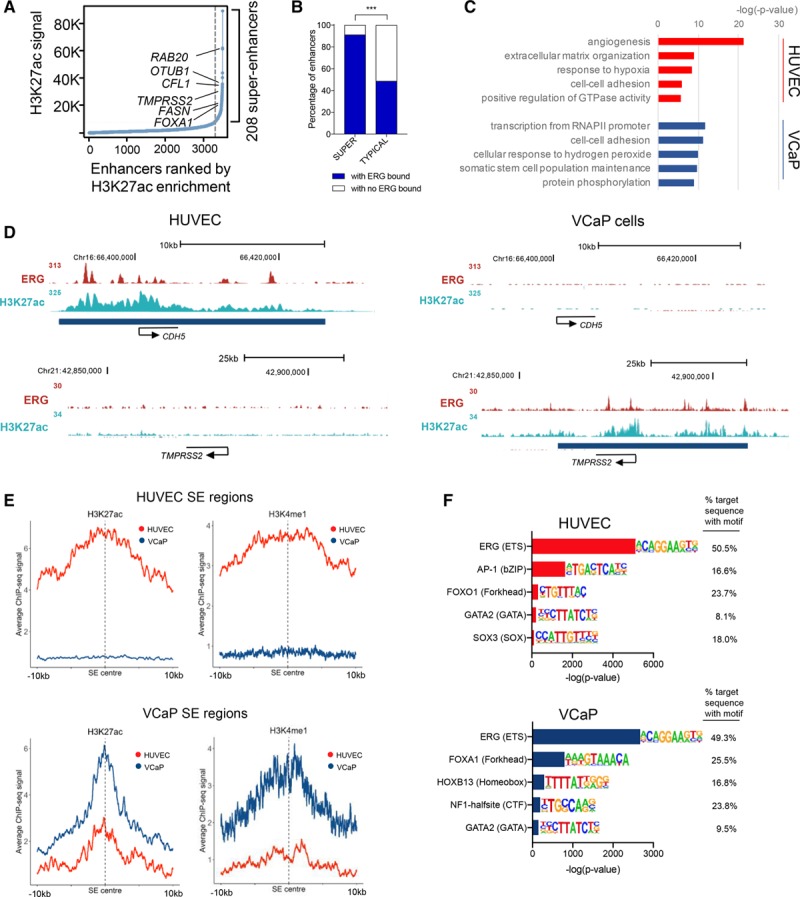
ERG (ETS-related gene) associates with lineage-specific super-enhancers (SEs). A, Identification of human prostate epithelial cancer cell line (VCaP) SE by H3K27ac chromatin immunoprecipitation with deep sequencing (ChIP-seq) signal in rpm/bp (see also Online Table VII). B, ERG binding at SE and typical enhancers in VCaP cells. SE have significantly more ERG binding than typical enhancers; ***P<0.0001, Fisher exact test. C, Gene ontology analysis on the SE-associated genes in human umbilical vein endothelial cell (HUVEC; red) and VCaP cells (blue). The top 5 biological processes in both cells are ranked by significance (−log (P value)). D, ChIP-seq binding profiles for ERG and H3K27ac occupancy in HUVEC (left) and VCaP cells (right). H3K27ac-defined SE regions are depicted as blue bars below tracks. E, Aggregate plots of active histone modifications H3K27ac and H3K4me1 histone modifications from HUVEC and VCaP cells in HUVEC SE (top) and VCaP SE (bottom). Plots centered on the SE center. The average size of VCaP SE is smaller than those identified in HUVEC. F, Motif analysis at ERG-binding sites (±200 bp) in HUVEC and VCaP. Top 5 most enriched transcription factor families are shown with the most highly occurring member of each family represented. Motifs for the binding of critical lineage transcription factors coincide with the ERG motif in both HUVEC and VCaP cells.
The difference between super-enhancer profiles in HUVEC versus VCaP cells suggests that the chromatin landscape is unique to the particular cell type. Analysis of histone modifications associated with active (H3K27ac and H3K4me1) or repressed (H3K27me3) chromatin in HUVEC and VCaP super-enhancer regions supports this hypothesis. Genomic regions corresponding to HUVEC super-enhancers were enriched in active histone marks in HUVEC but not VCaP cells (Figure 5E, top). Conversely, genomic regions corresponding to VCaP super-enhancers were enriched in active marks in VCaP cells but not HUVEC (Figure 5E, bottom). The reverse pattern was observed for the repressive mark H3K27me3 (Online Figure VD).
To further define the lineage-specific machinery of super-enhancers, we focused on TFs that may collaborate with ERG to regulate gene transcription in a cell type-specific manner. We searched for TF DNA motifs located ±200 bp from the ERG-binding sites in HUVEC and VCaP cells. Interestingly, different TF motifs are enriched in ERG-binding sites in the 2 cell types. In HUVEC, these include AP-1, FOXO-1 (forkhead box O1), GATA-2 (GATA-binding protein 2), and SOX-3 (SRY-box 3), TFs known to be important in endothelial gene expression16,33 (Figure 5F, top). In VCaP cells, the enriched motifs included TFs FOXA-1 (forkhead box A1) and HOXB-13 (homeobox B13), TFs previously described to play a role in prostate cancer gene expression34 (Figure 5F, bottom).
These data indicate that ERG’s lineage-specific transcriptional activity is associated with binding to cell type-specific super-enhancers and suggests cooperativity with distinct lineage-specific factors.
ERG Controls the Gene Expression Profile in HUVEC by Regulating the Enhancer and Super-Enhancer Landscape
We investigated whether ERG is required for H3K27 acetylation at endothelial enhancers by performing H3K27ac ChIP-seq analysis in HUVEC treated with control or ERG-siRNA (Online Figure VIA and VIB). In control HUVEC, 56 347 H3K27ac-bound regions were identified, which significantly correlated with those reported in the Encyclopedia of DNA Elements data25 (Online Figure VIC). In ERG-deficient cells, H3K27ac was modulated globally, with a decrease in H3K27ac at 5277 regions (loss) and an increase in 1648 regions (gain; Figure 6A and 6B). Changes in H3K27ac enrichment were validated by ChIP-qPCR across regions associated with selected endothelial ERG target genes (Figure 6C). Globally, in ERG-deficient cells changes in H3K27ac also correlated with the expression profile: expression of genes associated with loss of H3K27ac was significantly downregulated while expression of genes associated with gain of H3K27ac was significantly upregulated (Figure 6D; Online Figure VID). Genomic Regions Enrichment of Annotations Tool analysis of ERG-depleted HUVEC showed that loss of H3K27ac was associated with enrichment of Notch and VEGF (vascular endothelial growth factor) receptor signaling, pathways positively controlled by ERG8,35 (Figure 6E). In contrast, gain of H3K27ac was associated with TGF-β-SMAD signaling, a pathway repressed by ERG11 (Figure 6E).
Figure 6.
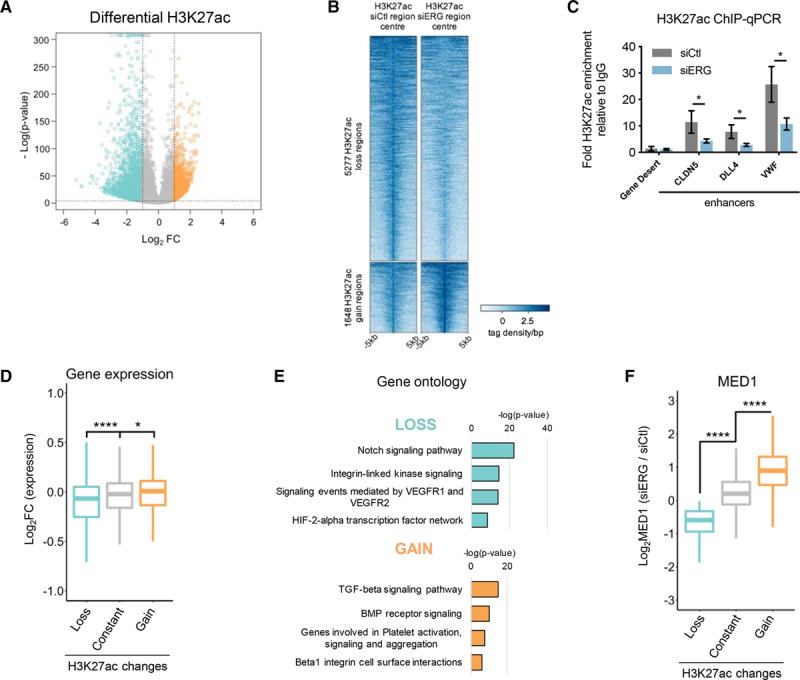
ERG (ETS-related gene) contributes to enhancer activation in human umbilical vein endothelial cell (HUVEC). A, Volcano plot showing log2 fold change (FC) vs –log (P value) of differential H3K27ac enrichment at siCtl H3K27ac regions in response to ERG knockdown. Loss and gain enhancer regions are selected by −1≥log2FC≥1; −log (P value)≥4. B, Heatmap of H3K27ac enrichment over input in all loss and gain regions. Signal ±5 kb from the center of H3K27ac siCtl or siERG regions as tag density/bp. C, Chromatin immunoprecipitation-quantitative polymerase chain reaction (ChIP-qPCR) of H3K27ac at enhancers of CLDN5 (claudin-5), DLL4 (Delta-like protein 4), and VWF (von Willebrand factor) with a negative control gene desert, in siCtl vs siERG-treated HUVEC. Graph represents fold change over IgG, n=3. *P<0.05, paired 2-tailed t test. D, Boxplot representing log2FC from transcriptome profiling data following ERG knockdown in H3K27ac regions changed in response to siERG (as defined in A). P<0.0001, Kruskal-Wallis test and post hoc test using Wilcoxon rank-sum test, ****P<0.0001; *P<0.05. E, Pathway analysis of genes associated with loss and gain of H3K27ac. F, Boxplot showing the log2FC of MED (Mediator complex subunit)-1 occupancy in siERG-treated HUVEC in H3K27ac regions changed in response to siERG. P<0.0001, Kruskal-Wallis test and post hoc test using Wilcoxon rank-sum test, ****P<0.0001. TGF indicates transforming growth factor. BMP indicates bone morphogenetic protein; HIF, hypoxia inducible factor; TGF, transforming growth factor; and VEGFR, vascular endothelial growth factor receptor.
To investigate the effect of ERG depletion on the recruitment of basal transcriptional machinery to enhancers, we performed ChIP-seq for MED1 in control and ERG-deficient HUVEC. Loss or gain of MED1 occupancy in ERG-deficient cells coincided with a decrease or increase in H3K27ac, respectively (Figure 6F). These data indicate that ERG plays a role in modulating endothelial enhancers. MED1 is part of a large complex (Mediator) which interacts with super-enhancers.19 We therefore investigated the role of ERG in the organization of endothelial super-enhancers. H3K27ac ChIP-seq analysis in control HUVEC identified 1015 super-enhancer clusters (Figure 7A; siCtl), in line with the HUVEC super-enhancers profile identified from the Encyclopedia of DNA Elements data (see Figure 2A). ERG depletion by siRNA caused changes in H3K27ac levels leading to a redistribution of endothelial super-enhancers (Figure 7A; siERG). Comparison of H3K27ac super-enhancers in control versus ERG-depleted HUVEC identified a subset of 107 super-enhancers with decreased H3K27ac levels following loss of ERG. Among the ERG-regulated super-enhancers were those associated with key endothelial genes including DLL4, NRARP, and CLDN5 (Figure 7B). The majority of super-enhancers showed no significant changes following ERG-siRNA, and only a few super-enhancers showed increased H3K27ac. MED-1 occupancy was also reduced in the subset of ERG-regulated super-enhancers (decreased super-enhancer) compared to those unchanged (constant super-enhancer; Figure 7C). Importantly, the ERG-dependent decrease in H3K27ac levels correlates with reduced expression of ERG target genes (Online Figure VIIA). Thus, ERG is functionally required to dynamically modulate H3K27ac levels in ECs leading to redistribution of a subset of super-enhancers.
Figure 7.
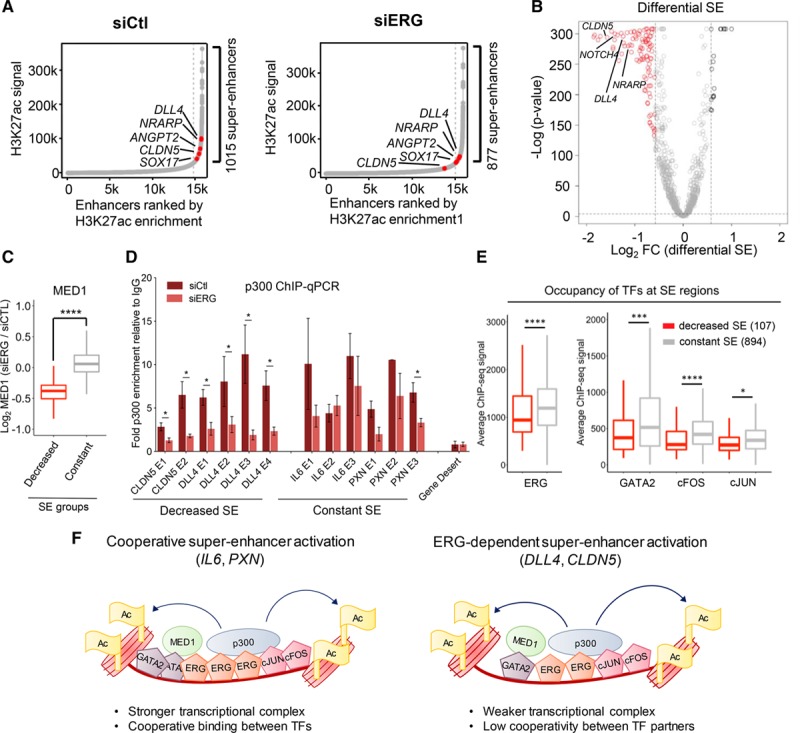
ERG (ETS-related gene) regulates distinct endothelial super-enhancers (SEs). A, SE identification on siCtl H3K27ac using H3K27ac enrichment from siCtl and siERG-treated human umbilical vein endothelial cell (HUVEC). Selected genes relevant to endothelial function are indicated in red (see also Online Table VIII). B, Volcano plot depicting the log2FC vs –log (P value) of differential SEs at 1015 H3K27ac SE identified in siCtl-treated cells. Significantly up or downregulated SEs are selected according to −0.58≥log2FC≥0.58 (−1.5≥FC≥1.5); −log (P value)≥4. C, Boxplot representing the log2FC of MED (Mediator complex subunit)-1 occupancy in siERG-treated HUVEC in decreased SE and constant SE. ****P<0.0001, Wilcoxon rank-sum test. D, Chromatin immunoprecipitation-quantitative polymerase chain reaction (ChIP-qPCR) of p300 enrichment in siCtl or siERG-treated HUVEC at selected SE constituent enhancers (E) associated with decreased SE (CLDN5 [claudin-5], DLL4 [Delta-like protein 4] or constant SE (IL6, PXN). Data are represented as fold change over IgG, n=4. *P<0.05, paired 2-tailed t test. E, Genomic occupancy of ERG and collaborative TFs: GATA-2 (GATA-binding protein 2), cFOS, and cJUN at decreased SE compared with constant SE, measured by the chromatin immunoprecipitation with deep sequencing signal in HUVEC. ****P<0.0001; ***P<0.001; *P<0.05, Wilcoxon rank-sum test. F, Model of ERG-dependent SE assembly in EC. Cooperative SE activation is associated with a strong transcriptional complex (ERG, AP-1 [activator protein 1], GATA-2) at constant SE (left). In a subset of SE, activation of SE is strongly dependent on ERG due to less abundance of transcription factor network partners with reduced cooperativity (right).
Cooperative TF Binding and p300 Recruitment in the Regulation of HUVEC Super-Enhancers
ERG has been shown to bind to p30035; thus, we hypothesized that ERG-dependent changes in H3K27ac at super-enhancers might be linked to the recruitment of p300 by ERG. This was tested by ChIP-qPCR for p300 enrichment on selected loci associated with validated ERG targets (CLDN5and DLL4), where H3K27ac was decreased on loss of ERG (decreased super-enhancer). These showed a significant decrease in p300 occupancy following ERG inhibition, suggesting that ERG is required to recruit p300 at these sites (Figure 7D). However, ERG inhibition did not consistently affect p300 recruitment at constant super-enhancers typified by IL6 and PXN (Figure 7D). Online Figure VIIB illustrates the ERG-dependent decrease in H3K27ac and MED1 occupancy observed at the CLDN5 and DLL4 gene loci, compared to loci associated with constant super-enhancers, IL6 and PXN. These findings suggest that ERG regulates a subset of super-enhancers partly through recruitment of the histone acetyltransferase p300.
We speculated that in the super-enhancers that remain constant following loss of ERG, other TFs might compensate for its absence. Previous studies have identified GATA and AP-1 (FOS/JUN) TF families as cooperating with ETS factors in regulating endothelial gene expression16,36; moreover, cJUN has been shown to bind p300.37 Analysis of ChIP-seq data from the Encyclopedia of DNA Elements for GATA-2, cFOS, and cJUN in HUVEC25 showed significant global overlap with ERG-bound sites (Online Figure VIIC). Higher occupancy of GATA-2, cFOS, and cJUN was present at constant super-enhancers compared with decreased super-enhancers (Figure 7E). This global distribution is reflected at representative loci for the 2 groups; CLDN5, DLL4 (decreased super-enhancer) and IL6, PXN (constant super-enhancer; Online Figure VIIB).
These data suggest a model (Figure 7F) in which the majority of super-enhancers are regulated by a cooperative TF network involving ERG, AP-1, and GATA-2 that provide a strong transcriptional complex; thus loss of ERG can be compensated. However, in a subset of super-enhancer–associated lineage genes including CLDN5 and DLL4, AP-1 and GATA-2 are less abundant, and therefore there is low cooperativity and super-enhancer assembly and gene expression are strongly dependent on ERG.
Risk Variants for Cardiovascular and Other Diseases Are Enriched at ERG Super-Enhancers
Several studies have recently shown that disease-associated SNPs identified through genome-wide association studies are preferentially enriched in the super-enhancer regions of disease-relevant cells and can map to TF-binding sites.18,38 Endothelial dysfunction is implicated in many diseases. We examined the enrichment of disease-associated variants at ERG-binding loci, ERG-bound enhancers and ERG super-enhancers, using SNPs reported in the NCBI dbGaP39 and NHGRI-GWAS catalogs.40 We determined enrichment by using a null distribution of background population variants. Analysis at ERG-binding loci identified association with SNPs for immune diseases and CVD (Online Figure VIIIA). At ERG-bound enhancers, the significance of enrichment is greatly amplified, with a similar repertoire of disease traits (Online Figure VIIIB). Interestingly, analysis at ERG super-enhancers identified SNPs for CVD as the most highly associated disease trait (P=1.1×10−14; Figure 8A). ERG super-enhancers were also enriched in diseases for digestive system (P=5.7×10−5) and respiratory tract (P=6.1×10−5; Figure 8A). This enrichment was not identified at size- and chromosome-matched regions randomly shuffled (permuted) across permissive chromatin. Further interrogation of the CVD-associated SNPs revealed strong significant enrichment for traits such as abdominal aortic aneurysm (P=1.7×10−13), coronary artery disease (P=2.6×10−10), myocardial infarction (P=5.1×10−9), and hypertension (P=1.1×10−8), but not heart failure (Figure 8B), suggesting a closer association with diseases where EC contribute most to the pathogenesis.
Figure 8.

ERG (ETS-related gene) super-enhancers (SEs) prioritize cardiovascular disease (CVD) variants. A, Overlap of genome-wide association studies single nucleotide polymorphisms (SNPs) associated with disease trait classes within ERG-defined SE (orange) with chromosome and size-matched random SE (gray). Significance of enrichment was calculated by binomial distribution test with red dashed line indicating P=0.05. Some points overlay. B, Test for enrichment of selected CVD trait-associated SNPs within ERG-defined SE (orange) with chromosome and size-matched random SE (gray), as performed in A. Random region were restricted to open chromatin by excluding placement in repressed chromatin states in human umbilical vein endothelial cell.
These results identify a novel mechanism through which disease-associated noncoding SNPs may cause vascular dysfunction and increased disease risk, and suggest a possible functional link between ERG-dependent transcriptional regulation of endothelial gene expression and the predisposition to CVD.
Discussion
In this study, we characterize the TF ERG as a crucial regulator of enhancers and super-enhancers in HUVEC. Multiple studies have shown that ERG is essential to maintain endothelial homeostasis (reviewed in Shah et al2). Here, we define the ERG-dependent endothelial epigenome and associate this with genetic variants linked to CVD and other diseases, suggesting novel potential strategies for biomarkers and target identification.
Our study identifies ERG as a positive regulator of a core set of putative endothelial super-enhancers in HUVEC. We describe an ERG-dependent subset of super-enhancers, associated with essential endothelial genes such as DLL4, CLDN5, and NRARP. Crucially, ERG-dependent super-enhancers are significantly associated with ERG-activated genes. Analysis of DLL4 and CLDN5 loci shows that recruitment of p300 at these super-enhancers is controlled by ERG; direct interaction between ERG and p30035 suggests a mechanism by which ERG recruits p300 to genomic loci for H3K27 acetylation. However, inhibition of ERG expression in HUVEC did not perturb activity in most super-enhancer regions. This is not surprising because super-enhancers are characterized by the presence of multiple TF-binding sites and a high degree of enrichment of transcriptional coactivators, providing opportunities for cooperative binding and synergistic gene activation.18,19 Transcriptional networks consisting of members of the ETS (including ERG’s closest homolog FLI-1 [friend leukemia integration 1 transcription factor]), AP-1 and GATA families have been shown to bind endothelial enhancers.16,41 Furthermore, ERG and AP-1 have been shown to functionally interact at composite DNA-binding sites in non-EC.42 As suggested from studies on AP-1,43 endothelial enhancer selection may be facilitated by cooperative ERG-AP-1 binding, where ERG is acting as the endothelial-specific TF. A recent study investigated the effect of combined knockdown of ERG and its closest homolog FLI-1 on global H3K27ac levels in HUVEC and found a significant loss of H3K27ac on key endothelial genes,41 supporting the notion that multiple TFs are directing cooperative transcriptional regulation. In our study, we found that the majority of endothelial super-enhancers are co-occupied by ERG, AP-1 members cFOS/cJUN, and GATA-2. Interestingly, we found that the subset of super-enhancers not affected by ERG depletion is highly co-occupied by ERG, AP-1, and GATA-2, while lower levels of all TFs are present at the subset sensitive to ERG depletion. We propose that a cooperative TF network is able to compensate for ERG depletion at most super-enhancers; however, a specific subset of core super-enhancers is strictly dependent on ERG function, highlighting its key role in regulating endothelial gene expression.
ERG may modulate super-enhancer activity through mechanisms other than p300 recruitment. A potential mechanism may be through targeting the activity of the BAF (BRG-1–associated factors) chromatin remodeling complex which disrupts histone-DNA interactions to control access to DNA.44 Recent studies have indicated a role for both AP-1 and ERG in binding to BAF subunits to establish accessible chromatin for enhancer selection and target gene regulation.43,45 The mechanism through which ERG modulates MED-1 recruitment remains unclear. Mediator complex has been implicated in long-range chromatin interactions functionally combining enhancers from many kilobases away. In fact, super-enhancers have been shown to form higher-order 3-dimensional (3D) chromatin structures which are likely to coordinate their activity in an orchestrated manner.38,46 In this study, we followed the current standard convention when annotating super-enhancer–associated regions with ERG-bound loci, namely by their linear distance along the epigenome.28 This methodology does not take into account the complex 3D chromatin structure. Further studies will be required to map super-enhancers using long-range chromatin interactions in the HUVEC genome.
Aberrant expression of lineage-specific TF in other tissues cause deregulated activation of a transcriptome profile detrimental to the cell15; this is indeed the case with ERG, which acts as an oncogene when overexpressed in cells such as the prostate epithelium.13 We show that ERG-associated super-enhancer profiles are markedly different in HUVEC compared with VCaP cells. Thus, ERG does not co-opt an endothelial genomic profile in VCaP cells but controls fundamentally different pathways in these 2 cell types, partly through selective super-enhancer binding. These findings provide some insight into the molecular basis for ERG’s homeostatic versus oncogenic functions. We postulate that different ERG-dependent gene expression between HUVEC and VCaP cells may be regulated in part by the activity of cell-specific pioneer factors which act to prime chromatin for accessibility at lineage-specific sites.47
Super-enhancer regions are commonly enriched in cell type-specific disease-related SNPs.18,38 Hogan et al16 identified disease trait-associated SNPs for coronary artery disease and hypertension within aortic endothelial enhancers. Our analysis of ERG-bound super-enhancers revealed enrichment for SNPs associated with diseases that have a vascular component, including predisposition to CVDs, such as atherosclerosis and coronary artery disease. These data support the notion that active maintenance of endothelial homeostasis through transcriptional programs is essential protection against a number of diseases, most of all CVD. Interestingly, recent genome-wide association studies meta-analysis revealed a novel risk locus for abdominal aortic aneurysm within the ERG gene itself.48 Further studies will determine the functional role of noncoding variants associated with ERG enhancers, and will provide crucial insight into the contribution of ERG, cooperative TF and cofactor binding in complex disease susceptibility.
In conclusion, this study provides novel evidence on the transcriptional and epigenetic mechanisms which controls lineage-specific gene expression in EC and identifies a possible functional link between regulation of ERG activity and human disease. These associations will provide valuable insights for investigating the role of ERG-dependent regulatory programs in maintaining endothelial homeostasis and protecting against vascular diseases.
Acknowledgments
We thank Dr Joan Ponsà (Imperial College London, United Kingdom) for helpful discussions.
Sources of Funding
This study was funded by grants from the British Heart Foundation (RG/11/17/29256; RG/17/4/32662; FS/15/65/32036; and PG/17/33/32990) and Cancer Research UK.
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- BAF
- BRG-1–associated factors
- ChIP-seq
- chromatin immunoprecipitation with deep sequencing
- CLDN5
- claudin-5
- CVD
- cardiovascular disease
- EC
- endothelial cell
- ERG
- ETS-related gene
- ETS
- E-26 transformation-specific
- HUVEC
- human umbilical vein EC
- MED1
- Mediator complex subunit-1
- SNP
- single nucleotide polymorphism
- TF
- transcription factor
- TGF
- transforming growth factor
- TMPRSS2
- transmembrane protease, serine-2
- TSS
- transcription start site
- VCaP
- human prostate epithelial cancer cell line
- VWF
- von Willebrand factor
Current address for Y. Yang: Department of Cell and Developmental Biology, University College London, London, United Kingdom.
Current address for L. Osuna Almagro: Brain Metastasis Group, Centro Nacional de Investigaciones Oncológicas, Madrid, Spain.
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.118.313788.
Novelty and Significance
What Is Known?
The ETS (E-26 transformation-specific) transcription factor (TF) ERG (ETS-related gene) is essential for endothelial homeostasis, whereas its aberrant overexpression in cancer is oncogenic.
Lineage-specific TFs bind to chromatin regulatory elements, called super-enhancers, which are enriched in single nucleotide polymorphisms (SNPs) associated with specific diseases, including cardiovascular disease (CVD).
Epigenomic mechanisms are emerging as key players in CVD; little is known about the epigenetic regulation of endothelial function and its links to CVD.
What New Information Does This Article Contribute?
The TF ERG drives endothelial lineage genes via super-enhancers.
Comparison of human umbilical vein endothelial cells and prostate cancer.
TMPRSS-2 (transmembrane protease, serine-2):ERG fusion-positive human prostate epithelial cancer cell line (VCaP) cells reveals distinctive lineage-specific transcriptome and super-enhancer profiles.
ERG regulates a core set of endothelial super-enhancers that have reduced TF cooperativity.
CVD-associated SNPs are enriched at ERG super-enhancers.
There is an emerging link between the transcriptional and epigenetic regulation of endothelial gene expression and CVD. Most disease-associated SNPs lie within noncoding regions of the genome and perturb TF recognition sequences. A key question is whether disruption of TF pathways which promote endothelial cell homeostasis modulates disease risk. This is suggested by the presence of SNPs in endothelial cell enhancers linked to CVD. In this study, we focus on the TF ERG which is required for endothelial lineage specification, vascular development, and angiogenesis and plays an essential role in maintaining vascular homeostasis. Profiling global ERG DNA binding reveals that ERG binds to and regulates endothelial super-enhancers. This is lineage-specific since oncogenic ERG in prostate cancer VCaP cells binds different super-enhancers compared with human umbilical vein endothelial cell. ERG binding at endothelial super-enhancers is associated with CVD-associated SNPs, suggesting that perturbation of ERG DNA-binding motifs in super-enhancers may modulate disease risk. In summary, we identify a novel mechanism through which the ERG TF promotes endothelial cell homeostasis via regulation of super-enhancers. Binding of ERG to super-enhancers may have functional consequences for CVD risk. These findings may open new avenues of research focusing on the epigenomic mechanisms underlying CVD pathologies.
References
- 1.Dejana E, Hirschi KK, Simons M. The molecular basis of endothelial cell plasticity. Nat Commun. 2017;8:14361. doi: 10.1038/ncomms14361. doi: 10.1038/ncomms14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shah AV, Birdsey GM, Randi AM. Regulation of endothelial homeostasis, vascular development and angiogenesis by the transcription factor ERG. Vascul Pharmacol. 2016;86:3–13. doi: 10.1016/j.vph.2016.05.003. doi: 10.1016/j.vph.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vlaeminck-Guillem V, Carrere S, Dewitte F, Stehelin D, Desbiens X, Duterque-Coquillaud M. The Ets family member Erg gene is expressed in mesodermal tissues and neural crests at fundamental steps during mouse embryogenesis. Mech Dev. 2000;91:331–335. doi: 10.1016/s0925-4773(99)00272-5. [DOI] [PubMed] [Google Scholar]
- 4.Ginsberg M, James D, Ding BS, et al. Efficient direct reprogramming of mature amniotic cells into endothelial cells by ETS factors and TGFβ suppression. Cell. 2012;151:559–575. doi: 10.1016/j.cell.2012.09.032. doi: 10.1016/j.cell.2012.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu F, Patient R. Genome-wide analysis of the zebrafish ETS family identifies three genes required for hemangioblast differentiation or angiogenesis. Circ Res. 2008;103:1147–1154. doi: 10.1161/CIRCRESAHA.108.179713. doi: 10.1161/CIRCRESAHA.108.179713. [DOI] [PubMed] [Google Scholar]
- 6.Birdsey GM, Shah AV, Dufton N, Reynolds LE, Osuna Almagro L, Yang Y, Aspalter IM, Khan ST, Mason JC, Dejana E, Göttgens B, Hodivala-Dilke K, Gerhardt H, Adams RH, Randi AM. The endothelial transcription factor ERG promotes vascular stability and growth through Wnt/β-catenin signaling. Dev Cell. 2015;32:82–96. doi: 10.1016/j.devcel.2014.11.016. doi: 10.1016/j.devcel.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vijayaraj P, Le Bras A, Mitchell N, Kondo M, Juliao S, Wasserman M, Beeler D, Spokes K, Aird WC, Baldwin HS, Oettgen P. Erg is a crucial regulator of endocardial-mesenchymal transformation during cardiac valve morphogenesis. Development. 2012;139:3973–3985. doi: 10.1242/dev.081596. doi: 10.1242/dev.081596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah AV, Birdsey GM, Peghaire C, Pitulescu ME, Dufton NP, Yang Y, Weinberg I, Osuna Almagro L, Payne L, Mason JC, Gerhardt H, Adams RH, Randi AM. The endothelial transcription factor ERG mediates Angiopoietin-1-dependent control of Notch signalling and vascular stability. Nat Commun. 2017;8:16002. doi: 10.1038/ncomms16002. doi: 10.1038/ncomms16002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan L, Nikolova-Krstevski V, Zhan Y, Kondo M, Bhasin M, Varghese L, Yano K, Carman CV, Aird WC, Oettgen P. Antiinflammatory effects of the ETS factor ERG in endothelial cells are mediated through transcriptional repression of the interleukin-8 gene. Circ Res. 2009;104:1049–1057. doi: 10.1161/CIRCRESAHA.108.190751. doi: 10.1161/CIRCRESAHA.108.190751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sperone A, Dryden NH, Birdsey GM, Madden L, Johns M, Evans PC, Mason JC, Haskard DO, Boyle JJ, Paleolog EM, Randi AM. The transcription factor Erg inhibits vascular inflammation by repressing NF-kappaB activation and proinflammatory gene expression in endothelial cells. Arterioscler Thromb Vasc Biol. 2011;31:142–150. doi: 10.1161/ATVBAHA.110.216473. doi: 10.1161/ATVBAHA.110.216473. [DOI] [PubMed] [Google Scholar]
- 11.Dufton NP, Peghaire CR, Osuna-Almagro L, Raimondi C, Kalna V, Chuahan A, Webb G, Yang Y, Birdsey GM, Lalor P, Mason JC, Adams DH, Randi AM. Dynamic regulation of canonical TGFβ signalling by endothelial transcription factor ERG protects from liver fibrogenesis. Nat Commun. 2017;8:895. doi: 10.1038/s41467-017-01169-0. doi: 10.1038/s41467-017-01169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hägglöf C, Hammarsten P, Strömvall K, Egevad L, Josefsson A, Stattin P, Granfors T, Bergh A. TMPRSS2-ERG expression predicts prostate cancer survival and associates with stromal biomarkers. PLoS One. 2014;9:e86824. doi: 10.1371/journal.pone.0086824. doi: 10.1371/journal.pone.0086824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 14.Adamo P, Ladomery MR. The oncogene ERG: a key factor in prostate cancer. Oncogene. 2016;35:403–414. doi: 10.1038/onc.2015.109. doi: 10.1038/onc.2015.109. [DOI] [PubMed] [Google Scholar]
- 15.Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16:144–154. doi: 10.1038/nrm3949. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hogan NT, Whalen MB, Stolze LK, Hadeli NK, Lam MT, Springstead JR, Glass CK, Romanoski CE. Transcriptional networks specifying homeostatic and inflammatory programs of gene expression in human aortic endothelial cells. Elife. 2017;6:e22536. doi: 10.7554/eLife.22536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaikkonen MU, Spann NJ, Heinz S, Romanoski CE, Allison KA, Stender JD, Chun HB, Tough DF, Prinjha RK, Benner C, Glass CK. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell. 2013;51:310–325. doi: 10.1016/j.molcel.2013.07.010. doi: 10.1016/j.molcel.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaikkonen MU, Niskanen H, Romanoski CE, Kansanen E, Kivelä AM, Laitalainen J, Heinz S, Benner C, Glass CK, Ylä-Herttuala S. Control of VEGF-A transcriptional programs by pausing and genomic compartmentalization. Nucleic Acids Res. 2014;42:12570–12584. doi: 10.1093/nar/gku1036. doi: 10.1093/nar/gku1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown JD, Lin CY, Duan Q, et al. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell. 2014;56:219–231. doi: 10.1016/j.molcel.2014.08.024. doi: 10.1016/j.molcel.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei GH, Badis G, Berger MF, et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 2010;29:2147–2160. doi: 10.1038/emboj.2010.106. doi: 10.1038/emboj.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Birdsey GM, Dryden NH, Shah AV, Hannah R, Hall MD, Haskard DO, Parsons M, Mason JC, Zvelebil M, Gottgens B, Ridley AJ, Randi AM. The transcription factor Erg regulates expression of histone deacetylase 6 and multiple pathways involved in endothelial cell migration and angiogenesis. Blood. 2012;119:894–903. doi: 10.1182/blood-2011-04-350025. doi: 10.1182/blood-2011-04-350025. [DOI] [PubMed] [Google Scholar]
- 25.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Birdsey GM, Dryden NH, Amsellem V, Gebhardt F, Sahnan K, Haskard DO, Dejana E, Mason JC, Randi AM. Transcription factor Erg regulates angiogenesis and endothelial apoptosis through VE-cadherin. Blood. 2008;111:3498–3506. doi: 10.1182/blood-2007-08-105346. doi: 10.1182/blood-2007-08-105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chng KR, Chang CW, Tan SK, Yang C, Hong SZ, Sng NY, Cheung E. A transcriptional repressor co-regulatory network governing androgen response in prostate cancers. EMBO J. 2012;31:2810–2823. doi: 10.1038/emboj.2012.112. doi: 10.1038/emboj.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu J, Yu J, Mani RS, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17:443–454. doi: 10.1016/j.ccr.2010.03.018. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang S, Kollipara RK, Srivastava N, et al. Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer. Proc Natl Acad Sci USA. 2014;111:4251–4256. doi: 10.1073/pnas.1322198111. doi: 10.1073/pnas.1322198111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asangani IA, Dommeti VL, Wang X, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–282. doi: 10.1038/nature13229. doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanki Y, Nakaki R, Shimamura T, Matsunaga T, Yamamizu K, Katayama S, Suehiro JI, Osawa T, Aburatani H, Kodama T, Wada Y, Yamashita JK, Minami T. Dynamically and epigenetically coordinated GATA/ETS/SOX transcription factor expression is indispensable for endothelial cell differentiation. Nucleic Acids Res. 2017;45:4344–4358. doi: 10.1093/nar/gkx159. doi: 10.1093/nar/gkx159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pomerantz MM, Li F, Takeda DY, et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat Genet. 2015;47:1346–1351. doi: 10.1038/ng.3419. doi: 10.1038/ng.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fish JE, Cantu Gutierrez M, Dang LT, et al. Dynamic regulation of VEGF-inducible genes by an ERK/ERG/p300 transcriptional network. Development. 2017;144:2428–2444. doi: 10.1242/dev.146050. doi: 10.1242/dev.146050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Val S, Black BL. Transcriptional control of endothelial cell development. Dev Cell. 2009;16:180–195. doi: 10.1016/j.devcel.2009.01.014. doi: 10.1016/j.devcel.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim BK, Im JY, Han G, Lee WJ, Won KJ, Chung KS, Lee K, Ban HS, Song K, Won M. p300 cooperates with c-Jun and PARP-1 at the p300 binding site to activate RhoB transcription in NSC126188-mediated apoptosis. Biochim Biophys Acta. 2014;1839:364–373. doi: 10.1016/j.bbagrm.2014.03.004. doi: 10.1016/j.bbagrm.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 38.Pasquali L, Gaulton KJ, Rodríguez-Seguí SA, et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet. 2014;46:136–143. doi: 10.1038/ng.2870. doi: 10.1038/ng.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mailman MD, Feolo M, Jin Y, et al. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39:1181–1186. doi: 10.1038/ng1007-1181. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacArthur J, Bowler E, Cerezo M, et al. The new NHGRI-EBI catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45:D896–D901. doi: 10.1093/nar/gkw1133. doi: 10.1093/nar/gkw1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagai N, Ohguchi H, Nakaki R, Matsumura Y, Kanki Y, Sakai J, Aburatani H, Minami T. Downregulation of ERG and FLI1 expression in endothelial cells triggers endothelial-to-mesenchymal transition. PLoS Genet. 2018;14:e1007826. doi: 10.1371/journal.pgen.1007826. doi: 10.1371/journal.pgen.1007826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Madison BJ, Clark KA, Bhachech N, Hollenhorst PC, Graves BJ, Currie SL. Electrostatic repulsion causes anticooperative DNA binding between tumor suppressor ETS transcription factors and JUN-FOS at composite DNA sites. J Biol Chem. 2018;293:18624–18635. doi: 10.1074/jbc.RA118.003352. doi: 10.1074/jbc.RA118.003352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, Greenberg ME. AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol Cell. 2017;68:1067.e12–1082.e12. doi: 10.1016/j.molcel.2017.11.026. doi: 10.1016/j.molcel.2017.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Becker PB, Hörz W. ATP-dependent nucleosome remodeling. Annu Rev Biochem. 2002;71:247–273. doi: 10.1146/annurev.biochem.71.110601.135400. doi: 10.1146/annurev.biochem.71.110601.135400. [DOI] [PubMed] [Google Scholar]
- 45.Sandoval GJ, Pulice JL, Pakula H, et al. Binding of TMPRSS2-ERG to BAF chromatin remodeling complexes mediates prostate oncogenesis. Mol Cell. 2018;71:554–566.e7. doi: 10.1016/j.molcel.2018.06.040. doi: 10.1016/j.molcel.2018.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miguel-Escalada I, Bonàs-Guarch S, Cebola I, et al. Human pancreatic islet 3d chromatin architecture provides insights into the genetics of type 2 diabetes. bioRxiv. 2018:400291. doi: 10.1101/400291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zaret KS, Mango SE. Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr Opin Genet Dev. 2016;37:76–81. doi: 10.1016/j.gde.2015.12.003. doi: 10.1016/j.gde.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones GT, Tromp G, Kuivaniemi H, et al. Meta-analysis of genome-wide association studies for abdominal aortic aneurysm identifies four new disease-specific risk loci. Circ Res. 2017;120:341–353. doi: 10.1161/CIRCRESAHA.116.308765. doi: 10.1161/CIRCRESAHA.116.308765. [DOI] [PMC free article] [PubMed] [Google Scholar]