

Abstract
Caspases form a family of proteinases required for the initiation and execution phases of apoptosis. Distinct proapoptotic stimuli lead to activation of the initiator caspases-8 and -9, which in turn activate the common executioner caspases-3 and -7 by proteolytic cleavage. Whereas crystal structures of several active caspases have been reported, no three-dimensional structure of an uncleaved caspase zymogen is available so far. We have determined the 2.9-Å crystal structure of recombinant human C285A procaspase-7 and have elucidated the activation mechanism of caspases. The overall fold of the homodimeric procaspase-7 resembles that of the active tetrameric caspase-7. Each monomer is organized in two structured subdomains connected by partially flexible linkers, which asymmetrically occupy and block the central cavity, a typical feature of active caspases. This blockage is incompatible with a functional substrate binding site/active site. After proteolytic cleavage within the flexible linkers, the newly formed chain termini leave the cavity and fold outward to form stable structures. These conformational changes are associated with the formation of an intact active-site cleft. Therefore, this mechanism represents a formerly unknown type of proteinase zymogen activation.
Programmed cell death (apoptosis) is associated with the hierarchical activation of a number of cysteine proteinases with cleavage preference after Asp residues, comprising the caspase family C14 of clan CD (1). Based on their position within this proteolytic cascade, caspases are subdivided into initiator caspases (including caspases-8 and -9) and executioner caspases (including caspases-3, -6, and -7), as well as a third group of caspases (caspases-1, -4, and -5) involved in cytokine activation. In apoptosis, the upstream caspases triggered by cofactor-mediated trans-activation activate the downstream executioner caspases by limited proteolysis (2–4). These executioners, in turn, cleave distinct intracellular proteins involved in promoting the apoptotic phenotype.
Because of the potentially hazardous action of activated caspases in the host cell, effective mechanisms must keep them in a latent form before activation. Both the initiator and the executioner caspases are initially synthesized as single-chain molecules, most of which require proteolytic cleavage in their C-terminal half (physiologically at a distinct Asp–Xxx scissile bond) to become proteolytically active (5). Procaspase-7, in particular, is expressed as a 303-aa residue polypeptide chain. Upon activation in vivo, a short N-terminal peptide is removed, and, more importantly from the perspective of generating catalytic activity, an Ile-Gln-Ala-Asp-↓-Ser-Gly site is cleaved, giving rise to a 175-residue large chain and a 105-residue small chain, comprising the active caspase-7. The only known exception to this mode of activation is caspase-9, whose zymogen does not require proteolytic processing for activity (6, 7).
The first crystal structures of active caspase-1 (8, 9), and later the homologous structures of caspases -3, -7, and -8 (see refs. 10–14), revealed that the active caspases comprise a heterotetramer consisting of two closely associated large and two small subunits, aligned together by means of the central small subunits about a 2-fold axis. The large subunits harbor the catalytic residues His-237 and Cys-285 (using the caspase-1 numbering convention, refs. 8 and 9), whereas the small subunits contribute mainly to formation of the substrate binding region. The preference for substrate cleavage after Asp residues is explained by the side chains of Arg-179 and Arg-341 lining the S1 specificity pocket (with P1, P2, P3, etc. designating the peptide substrate residues N-terminal of the scissile peptide bond and S1, S2, S3, etc. designating the opposite subsites in the enzyme).
In contrast, no structure of an uncleaved caspase zymogen has been reported so far, thus limiting our understanding of the procaspase inactivity and the mechanism of procaspase activation. Our recent structure of cleaved caspase-9 (15) has allowed us to postulate a zymogen conformation, in that the asymmetric dimers have captured two conformations, a catalytically competent and an incompetent one. We have postulated that the zymogen form of caspase-9 is restrained by dislocation of the catalytic and the substrate binding sites. To test our hypothesis and disclose the general mechanism(s) responsible for caspase activation, we have crystallized the C285A variant of human procaspase-7 and have solved its crystal structure. Analysis of this executioner zymogen structure and its comparison with the structures of active caspase-7 unveil the structural basis of the procaspase inactivity and suggest the conformational changes leading to procaspase activation.
Materials and Methods
Protein Purification and Analysis.
Full-length recombinant human procaspase-7 (residues 1–303, equivalent to Met-96 to Gln-402 in the caspase-1 numbering system), with the active site Cys-285 replaced by an Ala residue to avoid autoactivation, was expressed in Escherichia coli and purified as described for the close homologue procaspase-3 (16). Samples for SDS/PAGE were heated at 90°C in SDS buffer containing 50 mM DTT, loaded onto an 18% gel, and stained with Coomassie brilliant blue R250. For N-terminal sequence determination, samples were subjected to automated sequencing on a PROCISE protein sequencer (Applied Biosystems). To show the integrity of the crystalline zymogen, procaspase-7 samples were digested with granzyme B at a 1:1,000 enzyme to substrate ratio in 50 mM Tris⋅HCl, pH 8.0/100 mM NaCl and analyzed by SDS/PAGE. Dynamic light-scattering experiments were performed with a DynaPro light-scattering instrument (Protein Solutions, Lakewood, NJ), by using a 10 μM protein concentration and a 0.02-μm filter.
Crystallization and Data Collection.
Using the sitting drop vapor diffusion method, crystals were grown by mixing 3 μl of a 8 mg/ml procaspase-7 solution with 0.4 μl of a 100 mM ammonium sulfate solution and 1 μl of the reservoir buffer (50 mM Mes, pH 5.6/100 mM ammonium sulfate/10 mM MgCl2/20% PEG 8000), and equilibration against the reservoir buffer at 20°C. These crystals belong to the trigonal space group P3221 and contain two zymogen monomers per asymmetric unit [corresponding to a (vol/vol) solvent content of 60%]. For data collection, the crystals were transferred to a cryobuffer [mother liquor containing 20% (vol/vol) glycerol]. Diffraction data from a single crystal were collected on the BW6 synchrotron beam line at Deutsches Elektronen Synchrotron(Hamburg, Germany) in 0.5° rotation frames by using a MAR-CCD detector (MAR Research, Hamburg). The diffraction images were evaluated, merged, and scaled with MOSFLM (17) and reduced with programs from the CCP4 program suite (18).
Modeling and Refinement.
Orientation and position of the molecules were determined by molecular replacement with AMORE (19), using a truncated model derived from the crystal structure of active caspase-7 in complex with N-acetyl-Asp-Glu-Val-Asp aldehyde (13) and x-ray data from 15.0- to 3.5-Å resolution. The best solutions corresponded to a correlation coefficient of 61% and an R factor of 39%. A 2.9-Å electron density calculated from the correctly positioned caspase-7 model immediately showed extra density in the central cavity region at the dimer interface. The polypeptide chain was manually rebuilt and extended to account for the new density, and the modified model was subjected to several rounds of crystallographic refinement with CNSSOLVE (21). Density calculations and visual inspections as well as manual corrections of the protein models and electron density maps were made by using the graphics program MAIN (22). The target parameters of Engh and Huber (23) were used, and initially strong noncrystallographic restraints were applied. For the final stages of the refinement, individual constrained B factors were also refined, and solvent molecules were included in the model at stereochemically reasonable sites with electron density and difference density peaks above 1.0 and 2.5 σ. In the final model, segments Met-96 to Val-148, Thr-288 to Arg-318/Tyr-319, Ser-339 to Gly-346, and S381A to H381I cannot be traced and are thus disordered, and a number of side chains of flanking residues are also undefined by electron density. Therefore, the position of more than one-third of all nonhydrogen atoms remains undefined in the crystals. The final model corresponding to an Rcryst/Rfree of 26.8/28.5% includes 4,297 defined protein atoms and 25 solvent molecules; 84.2% and 13.7% of all defined residues fall into the allowed and additionally allowed regions of the Ramachandran plot (24). Crystal data and refinement statistics are given in Table 1.
Table 1.
Crystal and refinement data of human procaspase-7
| Space group | P3221 |
|---|---|
| Cell constants, Å | a = b = 90.21, c = 183.02 |
| Resolution limits, Å | 59.76–2.90 |
| Total number of observations | 180,534 |
| Number of independent reflections | 19,487 |
| Completeness (%) overall/outer shell (3.06–2.9) | 99.1 (99.1) |
| Rmerge (%) (overall/outer shell)* | 6.8 (38.6) |
| Refinement | |
| Resolution range, Å | 59.76–2.90 |
| Rcryst (%) (outer shell)† | 26.8 (31.3) |
| Rfree (%) (outer shell)‡ | 28.5 (38.6) |
| Reflections used for refinement working/ test set | 18,540/947 |
| Number of defined | |
| Nonhydrogen protein atoms | 4,297 |
| Solvent molecules | 25 |
| Average B factor of, Å2 | 74.88 |
| rms deviations | |
| Bond lengths, Å | 0.011 |
| Bond angles, ° | 1.33 |
Rmerge = ∑hkl∑i|Ii(hkl) − 〈Ihkl〉|/∑hkl∑i|Ii(hkl)|, where 〈Ihkl〉 represents the average intensity for multiple observations of Ii(hkl) for symmetry-related reflections.
Rcryst = ∑hkl∥Fobs| − |Fcalc∥/∑hkl|Fobs|.
Rfree = R factor calculated with 5% of reflections that were not used in the refinement.
Results and Discussion
Protein Characterization and Crystallization.
A construct of a human procaspase-7 Cys285Ala mutant was expressed in E. coli, purified, and crystallized. In SDS/PAGE (Fig. 1), the crystallized procaspase-7, identical to the original sample, migrated as a protein with an apparent molecular mass of 35 kDa (Fig. 1). N-terminal sequencing revealed a major sequence starting at Met-96 (the natural N-terminal residue of procaspase-7) and a minor one starting at Ser-142. The redissolved crystalline procaspase-7 sample could be cleaved by granzyme B, yielding the expected large and small subunits. These results thus show that the crystals contain intact caspase-7 zymogen in a fully activatable form.
Figure 1.
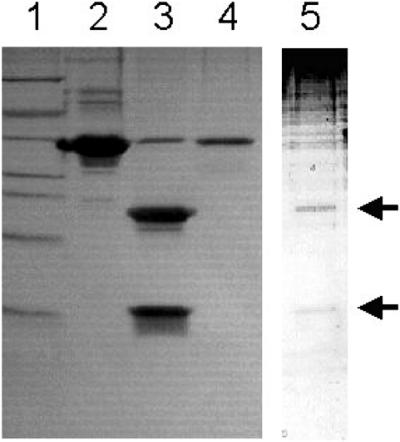
Integrity of crystallized human procaspase-7. Lane 1, molecular weight markers; lane 2, ≈8 μg of recombinant procaspase-7 used for crystallization trials; lane 3, recombinant procaspase-7 after treatment with granzyme B; lane 4, dissolved crystal of procaspase-7; lane 5, dissolved crystal of procaspase-7 treated with granzyme B (enhanced to visualize the weaker bands of lower molecular weights).
The cell constants of these trigonal procaspase-7 crystals (Table 1) are identical to within 2% to those of the complexes of active caspase-7 formed with N-acetyl-Asp-Glu-Val-Asp aldehyde (13), or with a fragment of the physiological inhibitor, X-linked inhibitor of apoptosis protein (XIAP) (20, 25), respectively.
Overall Structure.
The procaspase-7 crystals contain two molecules per asymmetric unit, which form a compact homodimer (Fig. 2A, which also specifies the reference orientation). The procaspase-7 homodimer resembles the heterotetramer of active caspase-7; 387 topologically equivalent Cα atoms (2.0-Å threshold) can be superimposed with an rms deviation of 0.6 Å. As in the active form, both procaspase-7 monomers are arranged side by side but with opposite orientation around a noncrystallographic 2-fold axis (in Fig. 2). In contrast to caspase-7 and the other active caspase structures, however, the (local) homodimer symmetry is significantly broken in procaspase-7, in that the ellipsoidal central cavity of approximate dimensions 10 Å × 25 Å, present on the front side between the monomers of active caspases, is filled with some disrupted electron density, which cannot account for two identically placed polypeptide chains simultaneously (see below).
Figure 2.

Crystal structure of human procaspase-7. (A) Stereo ribbon plot of the homodimeric procaspase-7, shown in the usual reference orientation. The asymmetric unit comprises two monomers, each consisting of a structured large (light blue and dark blue) and a small subdomain (gray and gray–blue). The local pseudo 2-fold axis is perpendicular to the plane of the figure. Flexible segments not defined by electron density are colored in yellow (see text). The secondary structure elements are assigned as for caspase-1 (8, 9). Segments of caspase-7 (13) exhibiting a conformation different from procaspase-7 are superimposed in red. The picture was made by using molscript (30). (B) Structure-based sequence alignment of selected segments of procaspases-1, -3, -7, -8, and -9. Strictly conserved residues are boxed, the active-site residues Cys-285 and His-237 and the P1 residue Asp-297 (except for caspase-9, where the cleavage site is further downstream) are given with red letters, and residues and elements primarily involved in procaspase activation are highlighted in green.
Each procaspase-7 monomer consists of a large N-terminal and a small C-terminal subdomain, corresponding approximately to the large and small subunits in the active caspase, and a connecting flexible linker. Because of the lack of contiguous electron density accounting for both linkers, the covalent connectivities between the large and the small subdomains are not unequivocally defined in the crystal structure. The chosen zymogen connectivity shown in Fig. 2A, which emphasizes the close association of the respective subdomains instead of monomer interdigitation, seems to be favored, however, mainly because of the identical connectivity observed in the related single-chain cysteine proteinase gingipain R (26).
Similar to the active caspases, each monomer essentially comprises a slightly twisted six-stranded central β-sheet with a few additional hairpins, flanked by five helices on either side (Fig. 2). Up to and including strand 4, both zymogen chains fold virtually identical as in the large subunits of active caspase-7. Following the catalytic residue Cys-285, the zymogen chains run straight on before becoming completely flexible at the strictly conserved Gly-287. In contrast, in active caspase-7 the equivalent C-terminal strands 4 are bent at Cys-285 and Arg-286 and extend toward bulk solvent, where they cross over the N-terminal segments from the opposite small subunits and pack against the hF-s8 loops to form structured four-chain bundles (Fig. 2).
After extending out of the molecular surface as loops, both zymogen linkers insert, in an asymmetrical manner, into the central cavity, before the chains turn back and enter strands 7 (Fig. 2). Instead, in the activated caspase-7, the equivalent N termini of the small subunits enter strands 7 from the above-mentioned four-chain bundle. In the zymogen, both strands 7 slightly bend and run through the exposed disordered s7-hE surface loops, whereas in the active caspases both chains abruptly extend in an elbow-like manner into the central cavity before passing over to the substrate binding sites. Finally, the large hF-s8 loops at both homodimer poles are disordered in procaspase-7, but they are ordered in caspase-7, where their stalks pack together with the chain termini to form four-strand bundles (Fig. 2A).
Dimer Interface.
In procaspase-7, the central cavity left free in the active caspase homodimers (except caspase-9) is filled with two lateral electron density strings and some disrupted central density (Fig. 3A). Both polypeptide chains of the small subdomains can be traced backward beyond Asp-326 of strand 7 for 6–8 residues. In strong contrast to the active caspase-7, both chains (in N-terminal direction) turn back through the Glu-324–Ala–Asp-326 loops toward the molecular center to nestle with their preceding segments Lys-320–Ile–Pro–Val-323 against the walls of the cavity (Fig. 3B). The chain of the right-hand subdomain can be traced backward, with less confidence, even beyond Lys-320 to about Arg-318 (Fig. 3A), whereas the left-side chain does not seem to be fixed until Lys-320. Therefore, in the crystals primarily the C-terminal part of the interdomain linker from the right-hand zymogen monomer seems to slot into the central region, whereas the corresponding linker segment from the left-side monomer is arranged more outside of the central cavity, presumably without adopting a distinct structure (Fig. 3). We believe that this asymmetry reflects an inherent property of the procaspase dimer and that the dimers are essentially selected and packed asymmetrically during crystal growth. Despite this asymmetry, both cavity halves are almost equally occupied by the blocking segments Lys-320–Ile–Pro–Val-323. Their Val-323 isopropyl side chains, in particular, point toward the opposite monomers, preventing insertion of the elbow loops associated with activity (see below).
Figure 3.

Occupancy of the central cavity by the blocking segments. (A) Section of the dimer interface region, superimposed with the well contoured lateral electron density stretches accounting for the blocking segments Lys-320–Asp326, and the disrupted central density (green). The final electron density is contoured at 1.0 σ. The main-chain segments are colored as in Fig. 2. The side chains of some selected residues are shown as stick models. (B) Close-up stereo view around the right-side blocking loop. The electron density for residues Ile-321 to Asp-326 contoured at 1.0 σ is superimposed.
As stated previously, the polypeptide chains bend abruptly in the active caspases at Ser-332 to run through the short Ser-332–Ser-339 elbow hairpin loops before forming part of the substrate binding regions (Fig. 4). Both exposed Val-334–Pro–Gly–Tyr-337 tight turns extend far into the space of the central cavity. In procaspase-7, this site is occupied by the blocking segment as well as additionally blocked by the preceding linker segment from the opposite monomer. The elbow conformation associated with the active caspase state would not be compatible with the presence of an opposite zymogen partner, owing to strong clashes of residues Val-334 and Pro-335 with Val-323 and Arg-318, respectively (Fig. 3). Also notable is that the side chains, making up the compact cavity-lining Tyr-331–Arg-286–Pro-335 stack in the active executioner caspases, are disordered in procaspase-7.
Figure 4.

The substrate binding region in procaspase-7. Procaspase-7 segments are shown in blue, gray, and yellow (flexible), whereas deviating caspase-7 segments are shown in red. Residues that form the substrate binding site in active caspases (e.g., Trp-340 and Arg-341) as well as Val-334 and Pro-335 of the elbow loop are shown for procaspase-7 and active caspase-7 as stick models. The catalytic His-237 and the position of Cys-285 are indicated (orange).
Substrate Binding Region.
In the active caspase-7, the extrusion of the Ser-332–Ser-339 elbow loops is closely linked with the structuring of Ser-339–Trp–Arg-341 (Fig. 4), which serve as S1–S3 binding templates for the antiparallel alignment of polypeptide substrates from P1 to P3. In contrast, in procaspase-7 the equivalent chain segments are, owing to lack of the interspersed elbow loops, shifted by five residues, so that both Val-334–Pro–Gly-336 segments are placed approximately at the site occupied by the Ser-339–Trp–Arg-341 alignment segments in the active enzyme. Trp-340 and Arg-341 located in exposed surface loops, and Gln-381D of the hF-s8 loops forming the S4 subsite in the active caspase-7, are fully disordered in the zymogen. It is notable that the zymogen conformation of the Val-334–Pro–Gly-336 segments stabilized through the hydrophobic core formed by the side chains of Val-334, Val-377, Trp-348, and Phe-380 completely differs from that of the Ser-339–Trp–Arg-341 alignment segment in the active enzyme, and that the S1, S2, and S4 subsites are incompletely or not at all formed, so that peptide substrates cannot be properly bound. The active Cys-285 residues occupy part of the space that will become the S1 pocket upon activation cleavage, so that the oxyanion holes formed by the amido nitrogens of Cys-285 and Gly-238 are slightly disrupted. Therefore, the zymogen conformation is incompatible with productive substrate binding and proteolytic cleavage.
Mechanism of Procaspase Activation.
The current structure not only explains the inactivity of procaspase-7 but also hints at the activation mechanism (Fig. 5). In the zymogen homodimer, one of the two intact linkers will slot into the groove of the central cavity, whereas the other is placed more outside such that both C-terminal blocking segments can nestle, assisted by the linker tension, against opposite walls of this cavity. These blocking segments, although probably not too tightly fixed, physically prevent entry of the elbow loops from the respective opposite monomer, thereby impairing formation of a functional substrate binding and active site. Both activation cleavage sites, even the one from the more buried linker, are quite exposed on the dimer surface and are therefore accessible to proteolytic attack, in particular by upstream caspases.
Figure 5.
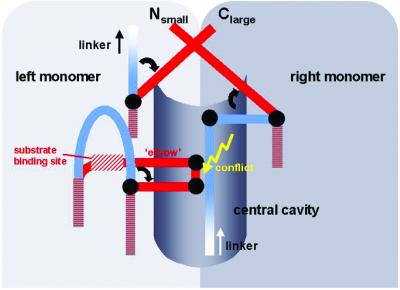
Schematic representation of the procaspase activation mechanism. In the zymogen, both blocking segments and part of the linker occupy the central cavity, preventing intrusion of the elbow loop from the opposite monomer. Upon activation cleavage, the newly formed N and C termini turn away from the cavity crossing over each other to form stable structures. This allows the elbow loop to expand into the now empty cavity, enabling the substrate alignment segment to shift and adopt its active conformation. As a consequence, the substrate binding subsites and the active sites become functional.
Upon activation cleavage in both linkers, the newly formed N and C termini loose their tension toward the molecular center and can leave their blocking positions in the cavity. Equally important, they gain the ability to turn outward and to form the tight four-strand bundles at both dimer poles, which would confer further stability to this open active conformation. Evacuation of the central cavity, in turn, will allow the substrate alignment segments to glide toward the central cavities and to fill them with their elbow loops. In addition, these exposed loops may assist in pushing the blocking segments out of the cavity. Stabilized by aromatic stack formation, the substrate alignment segments and the hF-s8 loops would be locked in their correct place, so that the S1, S2, and S4 subsites will be sculpted, the active Cys-285 will be positioned properly, and the oxyanion holes will be formed, rendering the substrate binding sites and the active sites functional.
This activation mechanism predicts that the linkers do not have to be cleaved precisely at the Asp-297–Xaa bond, as long as cleavage occurs within the linker region so that the newly formed termini are able to fold outward and to form the stable four-strand bundles. This is in agreement with activation cleavage experiments performed with proteinases of different specificity such as cathepsin G or subtilisin Carlsberg (27). Furthermore, this activation mechanism requires the presence of a dimer, in that both the cavity blockage in the zymogen (giving rise to inactivity) as well as the formation of a functional binding site and catalytic site in the active enzyme depend on intermonomer interactions.
With the important exception of caspase-9, most caspases are likely to be dimers under normal physiological concentrations. Our preliminary dynamic light-scattering data confirm this also for procaspase-7 down to low μM concentrations. In addition, crosslinking and gel-filtration experiments indicate a dimer-like behavior for the other major executioner, procaspase-3, as well (M.R., unpublished results). Of course, in vivo local cell conditions as well as accessory proteins could influence these monomer–dimer equilibria.
General Implications for Procaspase Activation.
The activation mechanism outlined above for procaspase-7 holds probably true for most other procaspases. This assumption is supported by a number of conserved residues. Examples of residues or segments located at hinge points and mediating conformational changes between the inactive and the active state are the highly or strictly conserved residues Arg-286, Gly-287, Ser-322, and the Glu-324–Ala–Asp-326-like turn segment, whereas the Ile-321–Pro–Val-like blocking segment, or residues involved in four-strand bundle package and stack formation, represent examples of critical residues/segments involved in the stabilization of one state or the other (Fig. 2).
Each of the caspases will possess some distinctive features, however, which certainly will require the elucidation of additional zymogen structures in the future. We note, for instance, that in strand 8 of caspase-1, an (inserted) Arg-391 residue bulges out of the sheet into the central cavity, with its extending side chain placed at the site, which in procaspase-7 is occupied by the blocking segment (Fig. 2B). A procaspase-7-like placement of the blocking segment would require a conformational change in the Arg-391 side chain, possibly brought about by salt bridge formation with Glu-324. We also note that most procaspases possess longer interdomain linkers than procaspase-7, which may influence stability of the respective zymogens. Furthermore, the N-terminal procaspase peptides, which are particularly long in the cytokine activators and apoptosis initiators, may fold back and interact with the catalytic domain, thereby influencing the activation and modulating the activity. Procaspase-1, for instance, seems to attain full activity only after the complete removal of the N-terminal peptides (28).
Procaspase-9, besides its marked tendency to dissociate (15), deviates structurally from the other procaspases. It possesses a seven-residue longer s3–s3a surface loop (7), which in the caspase-9 dimer reaches over to the opposite monomer, thereby filling the central cavity together with the bulky side chains of residues Phe-390 and Phe-393 (15). Therefore, in a procaspase-9 dimer, none of the linkers can insert into the central cavity, and the following blocking segments are probably hindered from nestling into their respective docking sites. The caspase-9 structure furthermore indicates that the intrusion of one elbow loop into the central cavity causes a slight asymmetric shift of both s3–s3a loops toward the opposite dimer pole, which might simultaneously hamper the extrusion of the second elbow loop. We note that procaspase-9 possesses an exceptionally long linker, which could project much further out from the molecular surface than in procaspase-7. A further remarkable feature of caspase-9 is Gly-286, whose position in all other known caspases (Fig. 2B) is occupied by a residue with a long charged/polar side chain (Arg/Gln in human caspases), which in the active executioners is sandwiched between the side chains of (Tyr)-331 and Pro-335, together forming a compact stack lining the central cavity. Consequently, the intrusion of the elbow loop from the opposite monomer becomes possible without proteolytic cleavage, in agreement with the significant proteolytic activity observed for the caspase-9 zymogen (6).
The caspase activation mechanism outlined above shares some properties with the mechanism determined for the activation of trypsin-like serine proteinases (29), in that the zymogen structure, inactive mainly because of an incompletely formed substrate binding site, is upon activation cleavage transformed into the active enzyme, characterized by a functional binding site/active site. As in trypsinogen, this transformation is brought about by stabilizing interactions of the newly formed chain termini with the main molecular body. The terminal residues of the newly liberated N- and C-terminal chains are, however, not directly involved in these interactions, making a precise activation cleavage unnecessary.
Acknowledgments
We thank Chris Froelich for granzyme B, Reinhard Mentele for sequencing, Dieter Jenne and Scott Snipas for experimental help, and Marianne Braun, Reiner Kiefersauer, and John Richardson for helpful discussions. The financial support by the SFB469 of the LM University, Munich, and the Fonds der Chemischen Industrie (to W.B.), and by National Institutes of Health Grant NS37878 (to G.S.S.) is kindly acknowledged.
Note Added in Proof.
After this manuscript was submitted, a paper describing a similar procaspase-7 structure was published (31).
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1GQF).
References
- 1.Barrett A J, Rawlings N D. Biol Chem. 2001;382:727–733. doi: 10.1515/BC.2001.088. [DOI] [PubMed] [Google Scholar]
- 2.Salvesen G S, Dixit V M. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 3.Thornberry N A, Lazebnik Y. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 4.Nicholson D W. Cell Death Differ. 1999;6:1028–1042. doi: 10.1038/sj.cdd.4400598. [DOI] [PubMed] [Google Scholar]
- 5.Stennicke H R, Salvesen G S. Biochim Biophys Acta. 2000;1477:299–306. doi: 10.1016/s0167-4838(99)00281-2. [DOI] [PubMed] [Google Scholar]
- 6.Stennicke H R, Deveraux Q L, Humke E W, Reed J C, Dixit V M, Salvesen G S. J Biol Chem. 1999;274:8359–8362. doi: 10.1074/jbc.274.13.8359. [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez J, Lazebnik Y. Genes Dev. 1999;13:3179–3184. doi: 10.1101/gad.13.24.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker N P, Talanian R V, Brady K D, Dang L C, Bump N J, Ferenz C R, Franklin S, Ghayur T, Hackett M C, Hammill L D. Cell. 1994;78:343–352. doi: 10.1016/0092-8674(94)90303-4. [DOI] [PubMed] [Google Scholar]
- 9.Wilson K P, Black J A, Thomson J A, Kim E E, Griffith J P, Navia M A, Murcko M A, Chambers S P, Aldape R A, Raybuck S A. Nature (London) 1994;370:270–275. doi: 10.1038/370270a0. [DOI] [PubMed] [Google Scholar]
- 10.Rotonda J, Nicholson D W, Fazil K M, Gallant M, Gareau Y, Labelle M, Peterson E P, Rasper D M, Ruel R, Vaillancourt J P, et al. Nat Struct Biol. 1996;3:619–625. doi: 10.1038/nsb0796-619. [DOI] [PubMed] [Google Scholar]
- 11.Blanchard H, Kodandapani L, Mittl P R, Marco S D, Krebs J F, Wu J C, Tomaselli K J, Grutter M G. Struct Fold Des. 1999;7:1125–1133. doi: 10.1016/s0969-2126(99)80179-8. [DOI] [PubMed] [Google Scholar]
- 12.Watt W, Koeplinger K A, Mildner A M, Heinrikson R L, Tomasselli A G, Watenpaugh K D. Struct Fold Des. 1999;7:1135–1143. doi: 10.1016/s0969-2126(99)80180-4. [DOI] [PubMed] [Google Scholar]
- 13.Wei Y, Fox T, Chambers S P, Sintchak J, Coll J T, Golec J M, Swenson L, Wilson K P, Charifson P S. Chem Biol. 2000;7:423–432. doi: 10.1016/s1074-5521(00)00123-x. [DOI] [PubMed] [Google Scholar]
- 14.Mittl P R, DiMarco S, Krebs J F, Bai X, Karanewsky D S, Priestle J P, Tomaselli K J, Grütter M G. J Biol Chem. 1997;272:6539–6547. doi: 10.1074/jbc.272.10.6539. [DOI] [PubMed] [Google Scholar]
- 15.Renatus M, Stennicke H R, Scott F L, Liddington R C, Salvesen G S. Proc Natl Acad Sci USA. 2001;98:14250–14255. doi: 10.1073/pnas.231465798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riedl S J, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik S W, Liddington R C, Salvesen G S. Cell. 2001;104:791–800. doi: 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- 17.Leslie A. In: Crystal Computing V. Moras D, Podjarny A D, Thierry J C, editors. Oxford: Oxford Univ. Press; 1991. pp. 27–38. [Google Scholar]
- 18.Collaborative Computational Project No. 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 19.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 20.Chai J, Shiozaki E, Srinivasula S M, Wu Q, Dataa P, Alnemri E S, Shi Y. Cell. 2001;104:769–780. doi: 10.1016/s0092-8674(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 21.Brünger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 22.Turk D. Ph.D. thesis. Munich: Technische Universität; 1992. [Google Scholar]
- 23.Engh R, Huber R. Acta Crystallogr A. 1991;47:392–400. [Google Scholar]
- 24.Laskowski R, MacArthur M, Hutchinson E, Thorton J. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 25.Huang Y, Park Y C, Rich R L, Segal D, Myszka D G, Wu H. Cell. 2001;104:781–790. [PubMed] [Google Scholar]
- 26.Eichinger A, Beisel H G, Jacob U, Huber R, Medrano F J, Banbula A, Potempa J, Travis J, Bode W. EMBO J. 1999;18:5453–5462. doi: 10.1093/emboj/18.20.5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou Q, Salvesen G S. Biochem J. 1997;324:361–364. doi: 10.1042/bj3240361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamin T T, Ayala J M, Miller D K. J Biol Chem. 1996;271:13273–13282. doi: 10.1074/jbc.271.22.13273. [DOI] [PubMed] [Google Scholar]
- 29.Huber R, Bode W. Acc Chem Res. 1978;11:114–122. [Google Scholar]
- 30.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 31.Chai J, Wu Q, Shiozaki E, Srinivasula S M, Alnemri E S, Shi Y. Cell. 2001;107:399–407. doi: 10.1016/s0092-8674(01)00544-x. [DOI] [PubMed] [Google Scholar]