

Abstract
To determine the efficacy, safety and tolerability of nebicapone, a new catechol‐O‐methyltransferase inhibitor for the treatment of motor fluctuations in Parkinson's disease (PD), we conducted a multicenter, randomized, 8‐week double‐blind, placebo‐ and active‐controlled, parallel‐group study comparing nebicapone 50 mg, 100 mg, or 150 mg, entacapone 200 mg (active control) or placebo administered concomitantly with levodopa/carbidopa or levodopa/benserazide. Two hundred and fifty‐two PD patients with motor fluctuations treated with levodopa/carbidopa or levodopa/benserazide (4–8 daily doses) were enrolled and 250 patients were eligible for intention‐to‐treat (ITT) analysis on the basis of having at least one efficacy assessment. The primary endpoint was 8‐week change from baseline in absolute “Off” time duration noted in self‐scoring diaries. At 8 weeks of treatment the mean daily “Off” time decreased significantly compared to placebo for nebicapone 150 mg (−106 min; 95%CI: −192; −21) and entacapone 200 mg (−81 min; 95%CI: −142; −19). The decrease in “Off” time with nebicapone 50 mg or 100 mg did not reach statistical significance. Treatment‐emergent adverse events were reported by 32% to 49% of patients in any treatment group, with no observed dose relationship in the nebicapone groups. Clinically relevant elevations in aspartate transaminase (AST) and/or alanine transaminase (ALT) were observed in 4 of 46 patients with the nebicapone 150 mg dose. The results of this study show that nebicapone 150 mg is efficacious for the treatment of motor fluctuations in PD patients. However, the risk of increasing liver transaminases and its clinically relevance deserves further evaluation.
Keywords: Clinical trial, COMT inhibitors, Nebicapone, Parkinson’s disease
Introduction
Although the number of available efficacious drugs for the treatment of Parkinson's disease (PD) has continuously increased in the last 50 years, levodopa remains the most efficacious antiparkinsonian drug and almost all patients at any disease stage will benefit from its administration [1, 2]. Unfortunately, its long‐term use is associated with the development of unavoidable motor complications, which occur in up to 80% of patients [3, 4]. Several pharmacologic options to minimize the “wearing‐off’’ phenomenon can be used, including adjustment of levodopa treatment, the use of dopamine agonists, monoamine oxidase type B inhibitors, or catechol‐O‐methyl‐transferase (COMT) inhibitors in combination with levodopa/dopa decarboxylase inhibitor (DDCI) [5, 6].
There are two COMT inhibitors used in clinical practice at present: tolcapone and entacapone. They are both efficacious in reducing ”Off” time and increasing ”On” time duration, and in improving motor scores in fluctuators, compared to placebo [7]. However, due to known cases of fatal hepatic toxicity in patients taking tolcapone, its use is now restricted to patients who failed other treatments and requires close monitoring of liver function [8].
Nebicapone is a new reversible and mainly peripherally acting COMT inhibitor currently being developed for use as an adjunct to levodopa/DDCI in the treatment of PD [9, 10, 11, 12, 13, 14]. A previous study in 16 PD patients showed that multiple doses of 75 and 150 mg nebicapone inhibited COMT activity, increased the levodopa bioavailability, and were efficacious and well tolerated when administered concomitantly with standard release 100/25 mg levodopa/carbidopa [15]. The dose of 75 mg nebicapone was shown to be noninferior and the dose of 150 mg nebicapone was shown to be superior to 200 mg entacapone in increasing the plasma level of levodopa [15]. On the basis of these results, a 5‐arm, phase II study was conducted to evaluate the efficacy, safety and tolerability of three different doses of nebicapone compared with entacapone and placebo when administered concomitantly with levodopa/DDCI in PD patients with motor fluctuations.
Methods
Study Participants
Study participants were enrolled at 40 sites in Europe and South America. Eligibility criteria included: age between 30 and 80 years, diagnosis of PD according to the UK Parkinson's Disease Society Brain Bank Clinical Diagnostic Criteria[16], modified Hoehn and Yahr staging <5 during the “Off” time, treatment with levodopa plus DDCI for at least 1 year with good clinical response, treatment with 4–8 daily doses of standard levodopa/DDCI (bedtime dose of a slow‐release formulation was permitted), a stable regimen of levodopa/DDCI and other antiparkinsonian drugs for at least 4 weeks before screening, and signs of “wearing‐off” phenomenon (end‐of‐dose deterioration) with an average total daily “Off” time while awake of at least 1.5 h excluding the early morning prefirst dose “Off” period. Individuals were ineligible to participate if they had a dyskinesia disability score >3 in the Unified Parkinson's Disease Rating Scale (UPDRS item 33), a major depressive episode within the 6 months before screening, being treated with entacapone, tolcapone, neuroleptics, antidepressants (except serotonin‐specific reuptake inhibitors or imipraminics [desipramine, imipramine, clomipramine, and amitriptyline]), monoamine oxidase inhibitors (except selegiline up to 10 mg/day in oral formulation or 1.25 mg/day in buccal absorption formulation or rasagiline up to 1 mg/day), or antiemetics (except domperidone) within the 3 months before screening, and apomorphine within the month before screening, a clinically relevant electrocardiogram abnormality and a history or current evidence of relevant heart disease. An additional exclusion criterion was pregnancy or lactation.
Local independent ethics committees approved the protocol and all patients signed an informed consent before initiating the study.
Study Design
A double‐blind, randomized, parallel‐group design was used. The study design is summarized in Figure 1. After a screening visit, patients began a 1‐ to 2‐week, single‐blind, placebo run‐in period (Period 1). Eligible patients who satisfactorily completed the run‐in period were randomized to an 8‐week double‐blind treatment period (Period 2) with 1 of 5 different study treatments (50 mg, 100 mg, or 150 mg nebicapone, 200 mg entacapone, placebo) in addition to their usual levodopa/DDCI treatment. Participants were randomized according to a block‐balanced computer‐generated (using SAS® software, version 9.1.3) randomization scheme that was stratified by study site. At randomization, each patient was allocated to 1 of the 5 treatment groups (50 mg, 100 mg, or 150 mg nebicapone, 200 mg entacapone, placebo) in the ratio of 1:1:1:1:1. The lowest randomization number available in the center was assigned. This randomization number identified the patient from randomization until study completion.
Figure 1.
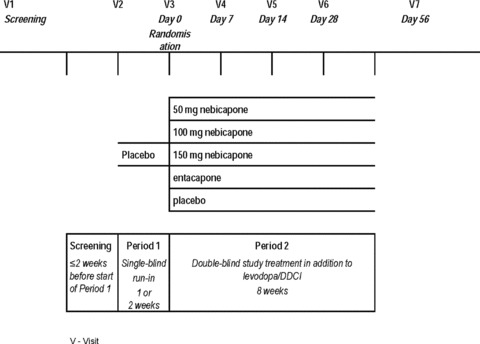
Study design.
Study treatments were administered orally as encapsulated tablets that were identical in appearance and were to be taken concomitantly with each levodopa/DDCI dose.
During Period 1 and Period 2, patients were administered the investigational product in addition to their usual levodopa/DDCI treatment (4–8 daily doses of levodopa/DDCI). The levodopa/DDCI dosage regimen should be kept stable during the whole study. However, during Weeks 1 to 4 of Period 2, the investigator could decrease the total daily dose of levodopa (while maintaining the number of daily intakes) in case of unacceptable adverse events (AEs), e.g., increased intensity or incidence of dyskinesia. If such levodopa daily dosage reduction was considered excessive, the dose could be increased again up to the baseline level. Concomitant anti‐Parkinsonian medication other than levodopa/DDCI remained unchanged during the whole study.
Outcome Measures
The patient (alone or with the family's or caregiver's assistance) was instructed to record the “On/Off” times in his or her diary chart [17] on the 3 days preceding Visits V3, V4, V5, V6, and V7. For each 30 min period during the day, the patients were to rate their mobility as 1 of the following: “Off”—poor mobility or complete immobility (able to move only slowly, or not at all); “On” with troublesome dyskinesia – limited mobility (able to move around despite presence of troublesome dyskinesia); “On” with nontroublesome dyskinesia—good mobility (able to move around relatively well despite presence of dyskinesia); “On” without dyskinesia—excellent mobility (able to move around well); asleep.
The prespecified primary efficacy endpoint was the 8‐week change from baseline in absolute “Off” time in the intent‐to‐treat (ITT) population (all randomized patients with at least one administration of investigational product in Period 2 and at least one assessment of “On/Off” times in the patient's diary). “On/Off” times of the diaries completed before randomization (i.e., at the end of Period 1) were considered as the baseline values.
The secondary efficacy endpoints included: changes in waking hours spent in “On” time without troublesome dyskinesia, changes in the UPDRS, the Modified Abnormal Involuntary Movement Scale (Modified AIMS) [18], an investigator's and patient's global assessment of changes. The UPDRS I scale was applied at screening, V2, V3, and V7. The other UPDRS scales and modified AIMS were applied at all visits. Dyskinesia was assessed by the investigator using the Modified AIMS during the “best‐On” period of the patient. The investigator first observed the patient sitting quietly at rest and then again while the patient counted backwards. Evaluation was to be performed at the same time of day, after the same levodopa intake, by the same investigator each time.
Safety was assessed in all study visits and included evaluation and assessment of treatment‐emergent AEs (TEAEs) and vital signs. It also included the review of clinical laboratory tests (including complete blood cell count, serum chemistry and urine analysis) performed by a central laboratory at visits V1, V4, V6, and V7, 12‐lead ECG, physical/neurological examination and complications of therapy. Clinically relevant laboratory abnormalities were listed as adverse events.
Statistical Analysis
With a sample size of 50 evaluable patients per group (total of 250 evaluable patients), the study was powered (two‐sided; α= 0.05; β= 0.30) to detect a difference of 1.4 h/day in the primary efficacy endpoint. These calculations assumed a standard deviation of 2.2 h, which was derived from the results of a previous pilot study with nebicapone [15].
Change from baseline in absolute “Off” time until the end of the 8‐week treatment period was compared between the placebo and the nebicapone groups by an analysis of covariance (ANCOVA) with fixed effects treatment, region, treatment‐by‐region interaction and with the baseline value of absolute “Off” time as covariate. Last observation carried forward (LOCF) was used for patients who did not complete Period 2. An adjustment for the multiple treatment comparisons of nebicapone versus placebo was performed using the Dunnett's procedure. A supportive analysis was made for the primary variable including the use of concomitant anti‐Parkinsonian medication in the ANCOVA model. A secondary analysis using these procedures was also done comparing the entacapone 200 mg group to placebo. Further secondary analyses were made descriptively for absolute “Off” time, percentage “On” time without troublesome dyskinesia, and total scores of the modified AIMS at each visit over the 8‐week treatment period. Except for the modified AIMS, a repeated‐measures ANCOVA was performed for all these variables. Percentage “On” time without troublesome dyskinesia and total scores of modified AIMS were also analyzed by an ANCOVA at the end of the 8‐week treatment period. For UPDRS, descriptive statistics were presented for total scores and treatment comparisons were made by the Kruskal–Wallis test. Total daily dose of levodopa was summarized using descriptive statistics and was compared between treatment groups by the Kruskal–Wallis test. Treatment comparisons for investigator's and patients’ global assessment of change were made by a Cochran–Mantel–Haenszel (CMH) test.
For safety, we compared treatment groups for the occurrence of TEAEs. Clinical laboratory variables and vital signs variables were summarized for each treatment group by calculating summary statistics on the actual values and on the change from baseline.
Results
A total of 298 patients were enrolled in the study, 254 were randomized and 252 received study medication. Of these, 53 participants were randomized to receive nebicapone 50 mg, 53 to nebicapone 100 mg, 46 to nebicapone 150 mg, 50 to entacapone and 50 to placebo (Figure 2). Of the 298 patients enrolled, 44 prematurely discontinued from the study before randomization (16 during screening and 28 during Period 1). The most common primary reasons for discontinuation before randomization were ineligibility (15 patients), withdrawal of consent (11 patients) and AEs (6 patients). Two of the patients randomized to the placebo group also discontinued the study before receiving the investigational treatment (one withdrew consent and the other was lost to follow‐up). An additional 27 patients discontinued the study after the start of the double‐blind phase (Period 2). The most common reasons for discontinuation were AEs (12 patients) and withdrawal of consent [placebo: n = 2 (4.0%), nebicapone 50 mg: n = 2 (3.8%), nebicapone 100 mg: n = 5 (9.4%), nebicapone 150 mg: n = 2 (4.3%), and entacapone 200 mg: n = 1 (2.0%)]. From the 252 study participants 2 patients were excluded from the ITT efficacy analysis because there were no assessments of “On” and “Off” time in their diaries during Period 2. All 252 study participants were included in the safety analysis.
Figure 2.
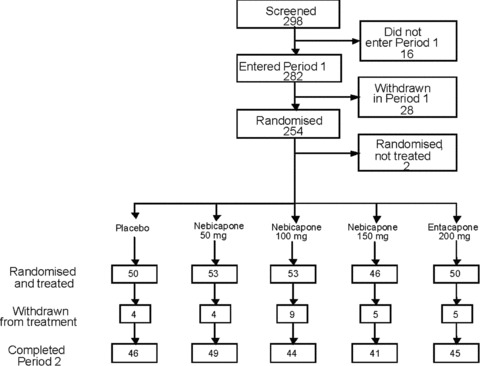
Flow‐chart of participation in the study.
The baseline demographic and clinical characteristics of the study participants randomized to the different treatment arms are presented in Table 1. There were no marked differences between the treatment groups.
Table 1.
Baseline characteristics of study participants (ITT population)
| Characteristic | Placebo (N = 49) | Nebicapone 50 mg (N = 53) | Nebicapone 100 mg (N = 52) | Nebicapone 150 mg (N = 46) | Entacapone 200 mg (N = 50) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sex – N (%) patients | ||||||||||
| Male | 32 | (65.3) | 29 | (54.7) | 31 | (59.6) | 26 | (56.5) | 27 | (54.0) |
| Female | 17 | (34.7) | 24 | (45.3) | 21 | (40.4) | 20 | (43.5) | 23 | (46.0) |
| Age, years | ||||||||||
| Mean (SD) | 64.1 | (10.1) | 65.4 | (8.1) | 62.9 | (10.0) | 64.5 | (8.8) | 65.3 | (8.6) |
| Race – N (%) patients | ||||||||||
| Caucasian | 49 | (100.0) | 52 | (98.1) | 52 | (100.0) | 46 | (100.0) | 49 | (98.0) |
| African | 0 | 1 | (1.9) | 0 | 0 | 1 | (2.0) | |||
| Disease characteristics – Mean duration in years (SD) | ||||||||||
| PD | 8.3 | (3.98) | 8.6 | (5.42) | 7.9 | (4.15) | 8.2 | (4.72) | 8.2 | (4.37) |
| Dyskinesias | 3.6 | (2.58) | 3.6 | (2.85) | 3.7 | (2.94) | 4.8 | (3.69) | 3.2 | (2.29) |
| Motor fluctuations | 3.3 | (2.35) | 3.1 | (3.46) | 2.8 | (2.79) | 2.7 | (2.69) | 2.7 | (2.18) |
| Levodopa treatment | 6.5 | (4.23) | 6.3 | (4.88) | 5.7 | (4.09) | 6.7 | (4.62) | 6.6 | (4.33) |
| Levodopa daily dose, mg | ||||||||||
| Mean (SD) | 712 | (298) | 671 | (278) | 622 | (231) | 633 | (224) | 709 | (307) |
| Hoehn & Yahr score | ||||||||||
| Mean (SD) | 2.6 | (0.6) | 2.5 | (0.4) | 2.6 | (0.6) | 2.6 | (0.7) | 2.7 | (0.6) |
| UPDRS Part III score | ||||||||||
| Mean (SD) | 30.2 | (13.0) | 27.6 | (11.0) | 26.5 | (14.2) | 30.1 | (15.2) | 29.8 | (13.3) |
| Concomitant anti‐Parkinsonian medication in at least 10% of patients in any treatment group – N (%) patients | ||||||||||
| Amantadine | 14 | (28.6) | 12 | (22.6) | 12 | (23.1) | 11 | (23.9) | 13 | (26.0) |
| Bromocriptine | 2 | (4.1) | 3 | (5.7) | 4 | (7.7) | 1 | (2.2) | 6 | (12.0) |
| Pramipexole | 8 | (16.3) | 7 | (13.2) | 10 | (19.2) | 9 | (19.6) | 4 | (8.0) |
| Ropinirole | 7 | (14.3) | 6 | (11.3) | 4 | (7.7) | 5 | (10.9) | 6 | (12.0) |
| Selegiline | 9 | (18.4) | 7 | (13.2) | 12 | (23.1) | 2 | (4.3) | 6 | (12.0) |
| Trihexiphenidyl | 2 | (4.1) | 6 | (11.3) | 5 | (9.6) | 2 | (4.3) | 2 | (4.0) |
N, number of patients; SD, Standard deviation; PD, Parkinson's disease; UPDRS, Unified Parkinson's Disease Rating Scale; ITT, Intention‐to‐treat.
Efficacy
Figure 3 displays the mean “On”/”Off” times in the different treatment groups and Table 2 presents the results of the ANCOVA of the primary variable. There was a dose‐dependent reduction in absolute “Off” time in the nebicapone treatment groups. After 8 weeks of treatment, the largest improvement was achieved in the nebicapone 150 mg group with a least squares mean (LS mean) change of −106 minutes compared to placebo (P < 0.05); the differences in the other nebicapone groups were not statistically significant. The reduction in absolute “Off” time from baseline in the entacapone 200 mg group was also statistically significant compared to placebo (−81 min; P < 0.05). The significant decrease in mean absolute “Off” time was also observed for nebicapone 150 mg (−112 min; 95%CI: −189; −34; P < 0.01) and entacapone 200 mg (−65 min; 95%CI: −125; −5; P < 0.05) when the use of concomitant anti‐Parkinsonian medication other than levodopa/DDCI was included in the ANCOVA model.
Figure 3.
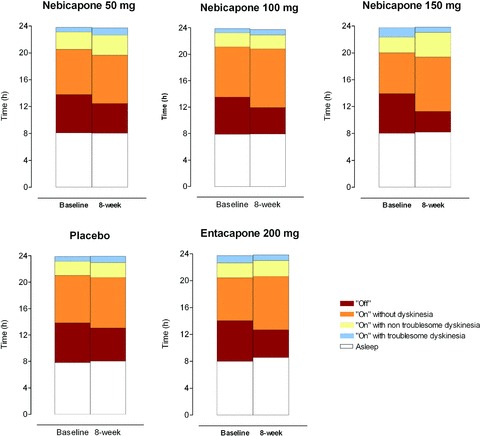
Mean “On”/”Off” times at the baseline and over the 8‐week treatment period in the different treatment groups (ITT population).
Table 2.
Change in “Off” times after 8 weeks of treatment (ITT population)
| Variable | Placebo (N = 49) | Nebicapone 50 mg (N = 53) | Nebicapone 100 mg (N = 52) | Nebicapone 150 mg (N = 46) | Entacapone 200 mg (N = 50) |
|---|---|---|---|---|---|
| Change in “Off” time (minutes) | |||||
| LSMean ± SEM | −35 ± 24.2 | −58 ± 23.1 | −75 ± 23.8 | −142 ± 26.7 | −123 ± 21.9 |
| LSMean change to placebo | − | −23 | −39 | −106 | −81 |
| 95%CI | − | −102; 56 | −120; 41 | −192; −21 | −142; −19 |
| Difference vs. placebo | − | n.s. | n.s. | P < 0.05 | P < 0.05 |
N, number of patients; LSMean, Least square mean; SEM, Standard error of the mean; 95%CI, 95% confidence interval; n.s., not statistically significant; ITT, Intention‐to‐treat.
The ANCOVA for change from baseline in percentage of time “On” without troublesome dyskinesia over the 8 weeks of treatment compared to placebo showed a 0.3% increase for nebicapone 50 mg (95%CI: –7%; 8%), 4% increase for nebicapone 100 mg (95%CI: –4%; 11%), 14% increase for nebicapone 150 mg (95%CI: 5%; 22%) and 8% increase for entacapone 200 mg (95%CI: –0%; 16%). The increase of time “On” without troublesome dyskinesia attained statistical significance only for the nebicapone 150 mg group (P < 0.01).
As observed in Table 3, changes in UPDRS scores were statistically significant for the nebicapone 150 mg group compared to placebo for all scores except UPDRS I (mentation, behavior and mood).
Table 3.
Change in UPDRS score from baseline after 8 weeks of treatment (ITT population)
| UPDRS Section | Median (min, max) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Timepoint | Placebo (N = 49) | Nebicapone 50 mg (N = 53) | Nebicapone 100 mg (N = 52) | Nebicapone 150 mg (N = 46) | Entacapone 200 mg (N = 50) | ||||||
| UPDRS I | Baseline | 1.0 | (0, 6) | 1.0 | (0, 8) | 1.0 | (0, 6) | 1.0 | (0, 6) | 2.0 | (0, 5) |
| Change at V7 | 0.0 | (−5, 2) | 0.0 | (−4, 3) | 0.0 | (−4, 3) | 0.0 | (−3, 2) | 0.0 | (−2, 3) | |
| UPDRS II at “Off” stage | Baseline | 18.0 | (7, 32) | 19.0 | (6, 35) | 18.0 | (7, 38) | 18.5 | (5, 37) | 17.5 | (7, 34) |
| Change at V7 | −1.0 | (−12, 8) | −3.0 | (−15, 5)* | −4.0 | (−15, 7)* | −4.0 | (−23, 2)** | −2.0 | (−14, 8) | |
| UPDRS II & III at “On” stage | Baseline | 36.0 | (9, 80) | 39.0 | (11, 81) | 33.0 | (4, 95) | 36.5 | (6, 80) | 36.5 | (11, 89) |
| Change at V7 | −3.0 | (−27, 15) | −6.0 | (−30, 12) | −5.0 | (−39, 14) | −11.0 | (−44, 4)** | −6.5 | (−29, 28) | |
| UPDRS III at “On” stage | Baseline | 27.0 | (8, 62) | 28.0 | (7, 62) | 25.5 | (4, 67) | 27.5 | (6, 61) | 29.0 | (10, 68) |
| Change at V7 | −2.0 | (−24, 11) | −5.0 | (−20, 11) | −4.0 | (−22, 10) | −9.0 | (−31, 4)* | −4.0 | (−23, 17) | |
Statistically significant treatment differences vs. placebo: *P < 0.05; **P < 0.01.
N, number of patients; UPDRS, Unified Parkinson's Disease Rating Scale; ITT, Intention‐to‐treat; min, minimum; max, maximum.
The treatment groups were comparable with regard to UPDRS II (activities in daily living) total score at “Off” stage at baseline; decreases from baseline were more pronounced in the active treatment groups than in the placebo group. In general, the UPDRS II total scores decreased over time, with values stabilizing from visitV6 to visitV7. Improvements in single items reflected total scores, with no notable difference between different single items.
The treatment groups were comparable with regard to total score for combined UPDRS II plus UPDRS III(motor examination) at “On” stage at baseline. Decreases from baseline to Visit V7 were more pronounced in the active treatment groups than in the placebo group. Compared to placebo, the largest decrease was observed in the nebicapone 150 mg group (P < 0.01). Change in total score showed a similar pattern over time in all treatment groups, with a decrease until visit V6 and a slight increase from visit V6 to visit V7. With the exception of a slight deterioration in “swallowing” (UPDRS II) in the nebicapone 50 mg group, scores for single items generally reflected the improvements observed for the total score. Results in UPDRS III at “On” stage were generally similar to those for the combined UPDRS II plus III total score at “On” stage.
Only small changes were observed in the modified AIMS test in any treatment group. The ANCOVA of change showed no differences between active treatment groups and placebo.
All active treatment groups showed statistically significant improvement to placebo after 8 weeks of treatment in the investigator's and patients’ global assessments of change. Results were best in the nebicapone 150 mg group, followed by the entacapone group.
Mean values for levodopa dosage at baseline were broadly comparable in all treatment groups (Table 1). Only a small number of patients had a change in levodopa dosage at any visit. The greatest number of patients with a change was observed in the nebicapone 50 mg group. Up to visit V7, changes in levodopa dosage were reported in 4 (8.2%) patients with placebo, 14 (26.9%) patients with nebicapone 50 mg, 6 (11.5%) patients with nebicapone 100 mg, 4 (8.7%) patients with nebicapone 150 mg and 6 (12.8%) patients with entacapone 200 mg. In 3 patients of the placebo group, 1 patient of the nebicapone 50 mg group and 1 patients of the nebicapone 150 mg group the change in levodopa/DDCI dose was an increase, corresponding to a protocol violation. The levodopa/DDCI dose was decreased in the remaining patients who reported a change.
Safety
The incidence of TEAEs in the different treatment groups is presented in Table 4. The overall frequency of TEAEs varied between 32% and 49% of patients across the treatment groups.
Table 4.
TEAEs occurring in at least 5% of patients of any treatment group (Safety population)
| Preferred term | Number (%) of patients | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Placebo (N = 50) | Nebicapone 50 mg (N = 53) | Nebicapone 100 mg (N = 53) | Nebicapone 150 mg (N = 46) | Entacapone 200 mg (N = 50) | ||||||
| Any TEAE | 17 | (34.0) | 17 | (32.1) | 26 | (49.1) | 18 | (39.1) | 18 | (36.0) |
| Diarrhoea | 1 | (2.0) | 0 | 4 | (7.5) | 1 | (2.2) | 1 | (2.0) | |
| Headache | 0 | 0 | 3 | (5.7) | 1 | (2.2) | 1 | (2.0) | ||
| Nausea | 1 | (2.0) | 1 | (1.9) | 3 | (5.7) | 0 | 0 | ||
| Urine colour abnormal | 1 | (2.0) | 2 | (3.8) | 1 | (1.9) | 3 | (6.5) | 5 | (10.0) |
| Transaminase increase | 0 | 0 | 0 | 4 | (8.7) | 0 | ||||
TEAE, treatment‐emergent adverse event; N, number of patients.
Review of the most frequent individual TEAEs with regard to time of onset in Period 2 showed that nearly all cases of diarrhea were reported between Day 28 and Day 56 of treatment, no relationship could be identified between duration of study treatment and onset of headache, nausea was noted mainly between Day 0 and Day 13, and cases of abnormal urine color were generally noted between Day 0 and Day 13.
The vast majority of TEAEs were classified by the investigator as of mild or moderate intensity. One patient in the nebicapone 50 mg group experienced severe dyskinesia. Severe TEAEs observed in the nebicapone 100 mg group were two cases of pain and one case each of diarrhea, dyskinesia, anxiety, insomnia and hallucination. Severe TEAEs reported in the nebicapone 150 mg group were one case each of metatarsalgia and hypertension, and one case of prostate cancer. Severe TEAEs reported in the entacapone 200 mg group were one case each of diarrhea, dyskinesia and hallucination. One patient in the placebo group reported severe myalgia.
Transaminase elevations were reported as a TEAE in 1 patient with nebicapone 50 mg and 5 patients with nebicapone 150 mg. In 4 of the 5 patients in the nebicapone 150 mg group these were clinically relevant elevations in aspartate transaminase (AST) and/or alanine transaminase (ALT). Clinically relevant transaminase elevations were predefined as values of AST ≥117 IU/L in men and ≥111 IU/L in women (normal range: 11–39 U/L in men and 11–37 U/L in women) and/or values of ALT ≥135 IU/L in men and ≥129 IU/L in women (normal range: 8–45 U/L in men and 8–43 U/L in women). No changes in serum bilirubin were reported and hepatitis viral serology showed negative results. Transaminase abnormal values rapidly decreased to normal after cessation of the investigational product.
TEAEs leading to withdrawal occurred in 12 patients: 2 (4.0%) placebo, 4 (7.5%) nebicapone 100 mg, 3 (6.5%) nebicapone 150 mg, and 3 (6.0%) entacapone patients. The only TEAE leading to withdrawal that occurred in more than 1 patient was an increase in ALT reported by 2 (4.3%) patients in the nebicapone 150 mg group; increases in ALT or AST levels leading to discontinuation were not reported by patients in any other nebicapone group.
Discussion
This double‐blind, randomized, parallel‐group study investigated the efficacy and safety of three doses of a new COMT inhibitor nebicapone as compared to placebo and entacapone in fluctuating PD patients. Following 8 weeks of concomitant administration with levodopa/DDCI, the highest dose of nebicapone (150 mg) was associated with the greatest decrease in “Off” time. This nebicapone dose reduced “Off” time at 8‐weeks of treatment by 106 min compared to placebo. These results replicated the effect found in a previous pilot study, after one week of treatment with nebicapone 150 mg (103 min) [15]. The 200 mg entacapone dose, used in this study as an active control arm, also showed a significant decrease in “Off” time, thus confirming the integrity of the trial design. Globally, the study does not support the results of the prior pilot study suggesting that nebicapone 75 mg was not inferior and nebicapone 150 mg was superior to entacapone in increasing plasma level of levodopa [15]. In the present study only nebicapone 150 mg and entacapone 200 mg (standard dose) significantly modified “On” and “Off” times. The lower doses of nebicapone (50 mg and 100 mg) showed to decrease “Off” time, but not significantly different from placebo. In most efficacy variables the effect size of entacapone 200 mg was smaller than that of nebicapone 150 mg, but the overall results show no superiority of nebicapone 150 mg over entacapone 200 mg.
When comparing these results with data available from other COMT inhibitor trials, the magnitude of the “Off” time reductions with nebicapone (150 mg) appear closer to the effects reported with tolcapone than to entacapone. In a Cochrane Collaboration review, a meta‐analysis of four entacapone versus placebo studies estimated an “Off” time reduction of 41 min (95% CI: 13 min, 68 min) [19]. Although it was not possible to perform a similar meta‐analysis for the tolcapone trials the descriptive analysis of the data suggested that tolcapone produced slightly larger significant reductions in “Off” time, levodopa dose and improvements in “On” time [19].
The overall frequency of TEAEs varied between 32% and 49% of patients across the treatment groups, but no dose relationship was observed in the nebicapone groups. The highest incidence was in the nebicapone 100 mg group. TEAEs reported by at least 5% of patients were diarrhea, headache, nausea, and urine discoloration. These TEAEs had been reported in other clinical trials with nebicapone and are known adverse effects of other COMT inhibitors such as entacapone and tolcapone [20]. The discoloration is presumed to be due to the presence of the COMT inhibitor and its metabolites in urine [21].
Clinically relevant elevations in AST and/or ALT were observed in 4 of 46 patients with the highest nebicapone dose (150 mg). No clinically relevant elevations in AST or ALT occurred in any of the other treatment groups, including nebicapone 50 mg and nebicapone 100 mg. No cases of hepatitis or other serious liver failure cases have been reported with entacapone [22]. Abnormal liver enzyme levels were reported by 1–3% of patients in tolcapone phase II/III trials [23, 24, 25]. Later in the tolcapone postmarketing period, the occurrence of several cases of acute hepatotoxicity with three fatalities that were attributed to tolcapone, led to tolcapone being suspended in the EU (late 1998), and labelling was tightened in the US [26]. A recent reanalysis of tolcapone safety data concluded that, since the labeling restrictions in 1998, there have been more than 40,000 patient‐years of tolcapone treatment worldwide, with only three reports of severe, but reversible, liver injury and no reports of hepatic fatality [8]. The cause of hepatotoxicity with tolcapone has not been clearly established, although current hypothesis suggests a possible interference with mitochondrial respiration in hepatocytes, by uncoupling oxidative phosphorylation [27].
Several studies with nebicapone in healthy subjects [9, 10, 11, 12, 13, 14, 28] and in PD patients [15], in which more than 180 subjects were exposed to different dosage regimens of nebicapone, did not raise any liver safety concerns. In a placebo‐controlled study (submitted to publication) in which healthy subjects were administered a relatively high daily dose of nebicapone for 7 days, a clinically significant increase in ALT values was observed in 1 subject treated with nebicapone 6 × 100 mg/day and 1 subject treated with 6 × 200 mg/day, for 7 days. However, in both cases the ALT levels were found to be above the upper limit of the normal range at admission to the study and bilirubin values remained within the normal range.
Overall, the previous data do not suggest a potential hepatotoxic effect of nebicapone. However, the results of the current study raise a concern about its liver safety.
In conclusion, the results of this study show that nebicapone 150 mg is efficacious for the treatment of motor fluctuations in PD patients. However, the risk of increasing liver transaminases and its clinically relevance deserves further evaluation.
Disclosures
T. Nunes, J. F. Rocha, and P. Soares‐da‐Silva are employees of BIAL – Portela & Co, SA, S. Mamede do Coronado, Portugal. L. Almeida was employee of BIAL at the time of the study. The other authors have had research and consultancy contracts with several companies involved in drug development in Parkinson's disease, including BIAL.
Conflict of Interest
The authors have no conflict of interest.
Acknowledgments
We would like to thank the staff of SCOPE International AG (Mannheim, Germany) for their committed involvement in the study management. This study was sponsored by BIAL – Portela & Ca SA.
Participant investigators: Austria: Werner Poewe, Innsbruck, Thomas Brücke, Vienna; France: Olivier Rascol, Toulouse, Alain Destée, Lille; Hungary: Béla Clemens, Debrecen, Judit Semjén, Eger, László Bartos, Veszprém, Péter Diószeghy, Nyíregyháza, Levente Kerényi, Székesfehérvár; Poland: Teofan Domżał, Warszawa, Andrzej Bogucki, Łodź, Andrzej Szczudlik, Kraków, Janusz Kapustecki, Częstochowa, Anna Szczepańska‐Szerej, Lublin, Jan Ścigalski, Kraków, Andrzej Potemkowski, Szczecin; Romania: Ovidiu Bajenaru, Bucharest, Cristina Mitu, Bucharest, Ioana Mindruta, Bucharest, Marina Ticmeanu, Bucharest, Emilian Manescu, Campulung Muscel, Gabriel Boeru, Bucharest, Mirela Chiru, Bucharest; Portugal: Joaquim Ferreira, Lisboa, Luis Cunha, Coimbra, Bastos Lima, Porto; Brazil: Egberto Barbosa, São Paulo, Henrique Ferraz, São Paulo, Elizabeth Quagliato, Campinas, Helio Teive, Curitiba, Vitor Tumas, Ribeirão Preto, Carlos Rieder, Porto Alegre, Ailton Melo, Salvador, Francisco Cardoso, Belo Horizonte; Argentina: Rolando Giannaula, Buenos Aires, Federico Micheli, Buenos Aires, Nélida Garreto, Buenos Aires, Manuel Pardal, Buenos Aires, Raúl Dominguez, Buenos Aires, Oscar Gershanik, Buenos Aires; Ukraine: Alla Goloborod’ko, Odessa, Natalia Buchakchiyskaya, Zaporizhzhja, Nataliya Grytsay, Poltava, Yury Golovchenko, Kyiv, Andriy Dubenko, Kharkov, Iryna Karaban, Kyiv; “for the Nebicapone Study Group”.
References
- 1. Lewitt PA. Levodopa for the treatment of Parkinson's disease. N Engl J Med 2008;359:2468–2476. [DOI] [PubMed] [Google Scholar]
- 2. Olanow CW, Agid Y, Mizuno Y, et al Levodopa in the treatment of Parkinson's disease: Current controversies. Mov Disord 2004;19:997–1005. [DOI] [PubMed] [Google Scholar]
- 3. Jankovic J. Motor fluctuations and dyskinesias in Parkinson's disease: Clinical manifestations. Mov Disord 2005;20(Suppl 11):S11–S16. [DOI] [PubMed] [Google Scholar]
- 4. Schrag A, Quinn N. Dyskinesias and motor fluctuations in Parkinson's disease. A community‐based study. Brain 2000;123(Pt 11):2297–2305. [DOI] [PubMed] [Google Scholar]
- 5. Goetz CG, Poewe W, Rascol O, Sampaio C. Evidencebased medical review update: Pharmacological and surgical treatments of Parkinson's disease: 2001 to 2004. Mov Disord 2005;20:523–539. [DOI] [PubMed] [Google Scholar]
- 6. Pahwa R, Factor SA, Lyons KE, et al Practice Parameter: Treatment of Parkinson disease with motor fluctuations and dyskinesia (an evidence‐based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2006;66:983–995. [DOI] [PubMed] [Google Scholar]
- 7. Deane K, Spieker S, Clarke CE. Catechol‐O‐methyltransferase inhibitors versus active comparators for levodopa‐induced complications in Parkinson's disease. Cochrane Database Syst Rev 2004. doi: 10.1002/14651858.CD004553.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Olanow CW, Watkins PB. Tolcapone: An efficacy and safety review (2007). Clin Neuropharmacol 2007;30:287–294. [DOI] [PubMed] [Google Scholar]
- 9. Almeida L, Falcao A, Vaz‐da‐Silva M, et al Effect of nebicapone on the pharmacokinetics and pharmacodynamics of warfarin in healthy subjects. Eur J Clin Pharmacol 2008;64:961–966. [DOI] [PubMed] [Google Scholar]
- 10. Almeida L, Soares‐da‐Silva P. Pharmacokinetics and pharmacodynamics of BIA 3‐202, a novel COMT inhibitor, during first administration to humans. Drugs R D 2003;4:207–217. [DOI] [PubMed] [Google Scholar]
- 11. Almeida L, Soares‐da‐Silva P. Pharmacokinetic and pharmacodynamic profiles of BIA 3‐202, a novel catechol‐O‐methyltransferase (COMT) inhibitor, during multiple‐dose administration to healthy subjects. J Clin Pharmacol 2003;43:1350–1360. [DOI] [PubMed] [Google Scholar]
- 12. Almeida L, Vaz‐da‐Silva M, Silveira P, et al Pharmacokinetic‐pharmacodynamic interaction between BIA 3‐202, a novel COMT inhibitor, and levodopa/carbidopa. Clin Neuropharmacol 2004;27:17–24. [DOI] [PubMed] [Google Scholar]
- 13. Silveira P, Vaz‐da‐Silva M, Almeida L, et al Pharmacokinetic‐pharmacodynamic interaction between BIA 3‐202, a novel COMT inhibitor, and levodopa/benserazide. Eur J Clin Pharmacol 2003;59:603–609. [DOI] [PubMed] [Google Scholar]
- 14. Vaz‐da‐Silva M, Loureiro AI, Nunes T, et al Pharmacokinetic‐pharmacodynamic interaction between nebicapone, a novel catechol‐o‐methyltransferase inhibitor, and controlled‐release levodopa/carbidopa 200 mg/50 mg: Randomized, double‐blind, placebo‐controlled, crossover study in healthy subjects. Drugs R D 2008;9:435–446. [DOI] [PubMed] [Google Scholar]
- 15. Ferreira JJ, Almeida L, Cunha L, et al Effects of nebicapone on levodopa pharmacokinetics, catechol‐O‐methyltransferase activity, and motor fluctuations in patients with Parkinson disease. Clin Neuropharmacol 2008;31:2–18. [DOI] [PubMed] [Google Scholar]
- 16. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: A clinico‐pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hauser RA, Friedlander J, Zesiewicz TA, et al A home diary to assess functional status in patients with Parkinson's disease with motor fluctuations and dyskinesia. Clin Neuropharmacol 2000;23:75–81. [DOI] [PubMed] [Google Scholar]
- 18. Guy W. Assessment manual for psychopharmacology. Washington , DC : Department of Health Education and Welfare, 1976. [Google Scholar]
- 19. Deane K, Spieker S, Clarke CE. Catechol‐O‐methyltransferase inhibitors for levodopa‐induced complications in Parkinson's disease. Cochrane Database Syst Rev 2004. doi: 10.1002/14651858.CD004554.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bonifacio MJ, Palma PN, Almeida L, Soares‐da‐Silva P. Catechol‐O‐methyltransferase and its inhibitors in Parkinson's disease. CNS Drug Rev 2007;13:352–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Palma PN, Bonifacio MJ, Almeida L, Soares‐da‐Silva P. Restoring dopamine levels In: Smith HJ, Simons C, Sewell RDE, editors. Protein misfolding in neurodegenerative diseases. Boca Raton , FL , USA : CRC Press, 2008;415–445. [Google Scholar]
- 22. Brooks DJ, Agid Y, Eggert K, Widner H, Ostergaard K, Holopainen A. Treatment of end‐of‐dose wearing‐off in parkinson's disease: Stalevo (levodopa/carbidopa/entacapone) and levodopa/DDCI given in combination with Comtess/Comtan (entacapone) provide equivalent improvements in symptom control superior to that of traditional levodopa/DDCI treatment. Eur Neurol 2005;53:197–202. [DOI] [PubMed] [Google Scholar]
- 23. Baas H, Beiske AG, Ghika J, Jackson M, Oertel WH, Poewe W, Ransmayr G. Catechol‐O‐methyltransferase inhibition with tolcapone reduces the “wearing off” phenomenon and levodopa requirements in fluctuating parkinsonian patients. J Neurol Neurosurg Psychiatry 1997;63:421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rajput AH, Martin W, Saint‐Hilaire MH, Dorflinger E, Pedder S. Tolcapone improves motor function in parkinsonian patients with the “wearing‐off” phenomenon: A double‐blind, placebo‐controlled, multicenter trial. Neurology 1997;49:1066–1071. [DOI] [PubMed] [Google Scholar]
- 25. Waters CH, Kurth M, Bailey P, et al; The Tolcapone Stable Study Group . Tolcapone in stable Parkinson's disease: Efficacy and safety of long‐term treatment. The Tolcapone Stable Study Group. Neurology 1997;49:665–671. [DOI] [PubMed] [Google Scholar]
- 26. Olanow CW. Tolcapone and hepatotoxic effects. Tasmar Advisory Panel. Arch Neurol 2000;57:263–267. [DOI] [PubMed] [Google Scholar]
- 27. Borges N. Tolcapone in Parkinson's disease: Liver toxicity and clinical efficacy. Expert Opin Drug Saf 2005;4:69–73. [DOI] [PubMed] [Google Scholar]
- 28. Nunes T, Machado R, Rocha JF, et al Pharmacokinetic‐pharmacodynamic interaction between nebicapone, a novel COMT inhibitor, and controlled release levodopa/benserazide: A single‐centre, phase I, double‐blind, randomized, placebo‐controlled, four‐way crossover study in healthy subjects. Clin Ther 2009;31:2258–2271. [DOI] [PubMed] [Google Scholar]