

Abstract
Endocannabinoids and their receptors, mainly the CB1 receptor type, function as a retrograde signaling system in many synapses within the CNS, particularly in GABAergic and glutamatergic synapses. They also play a modulatory function on dopamine (DA) transmission, although CB1 receptors do not appear to be located in dopaminergic terminals, at least in the major brain regions receiving dopaminergic innervation, e.g., the caudate‐putamen and the nucleus accumbens/prefrontal cortex. Therefore, the effects of cannabinoids on DA transmission and DA‐related behaviors are generally indirect and exerted through the modulation of GABA and glutamate inputs received by dopaminergic neurons. Recent evidence suggest, however, that certain eicosanoid‐derived cannabinoids may directly activate TRPV1 receptors, which have been found in some dopaminergic pathways, thus allowing a direct regulation of DA function. Through this direct mechanism or through indirect mechanisms involving GABA or glutamate neurons, cannabinoids may interact with DA transmission in the CNS and this has an important influence in various DA‐related neurobiological processes (e.g., control of movement, motivation/reward) and, particularly, on different pathologies affecting these processes like basal ganglia disorders, schizophrenia, and drug addiction. The present review will address the current literature supporting these cannabinoid‐DA interactions, with emphasis in aspects dealing with the neurochemical, physiological, and pharmacological/therapeutic bases of these interactions.
Keywords: Cannabinoid‐dopamine, Drug addiction, Movement disorders/Parkinson's disease, Schizophrenia
The Control of Neurotransmitter Activity by Endocannabinoids
Endocannabinoids and their receptors play a modulatory function in several physiological processes, mainly in the brain [1, 2, 3] but also in various peripheral processes such as the immune regulation [4], the cardiovascular system [5], the reproductive endocrine processes [6], and the control of energetic metabolism [7]. In the brain, endocannabinoids participate in processes such as the control of movement [8, 9, 10], nociception [11], brain reward [12], learning and memory [13], feeding [14], and emesis [15], as well as they play an important role in various events related to brain development [16, 17]. This large list of brain functions, in which endocannabinoids and their receptors are active, is the result of numerous studies developed during the last 20 years which demonstrated that: (i) changes in these processes are included within the spectrum of pharmacological actions in humans and laboratory animals of those compounds capable to activate or to inhibit the cannabinoid system (reviewed in [18, 19, 20]); (ii) cannabinoid receptors, mainly the CB1 receptor type, as well as their endogenous ligands, mainly anandamide and 2‐arachidonoylglycerol, are abundant in the brain structures involved in the above processes [2, 21]; and/or (iii) mice lacking specific genes of the cannabinoid signaling such as those encoding for the CB1 or CB2 receptor, or for the enzymes N‐arachidonoyl‐phosphatidylethanolamine (NAPE‐PLD) or fatty acid amide hydrolase (FAAH), exhibited behavioral changes compatible with a role of the cannabinoid system in such processes [22, 23, 24]. An important consequence of these brain functions proposed for the cannabinoid signaling system is that it may be considered of therapeutic relevance for different pathologies related to these brain functions [20, 25], which explains the increasing development in this field during the last years.
The involvement of the endocannabinoid system in this large list of brain functions is likely the consequence of its capability to interact with specific neurotransmitters in several brain regions [26]. For many years, most of researchers tried to demonstrate that endocannabinoids and their receptors may function as a novel transmitter system mimicking the process developed in the 1970s with the discovery of the opioid system. However, the action of endocannabinoids in the synaptic function appeared to be more compatible with a modulatory role rather than with a function as a classic transmitter. For example, the frequent, although not exclusive, presynaptic location of CB1 receptors allows endocannabinoids to directly influence presynaptic events, such as synthesis, release or reuptake, for specific transmitters, mainly glutamate, opioid peptides and GABA, since CB1 receptors are frequently located onto neurons containing these neurotransmitters in the brain, but also for acetylcholine and serotonin (for review, see [27]). The combination of numerous pharmacological, electrophysiological and immunohistochemical studies allowed to demonstrate that endocannabinoids function as retrograde signal molecules at the synapse (for review, see [1, 3], and an overview in Figure 1), in particular in glutamatergic and GABAergic synapses, then preventing an excess of excitation or inhibition, respectively [28].
Figure 1.
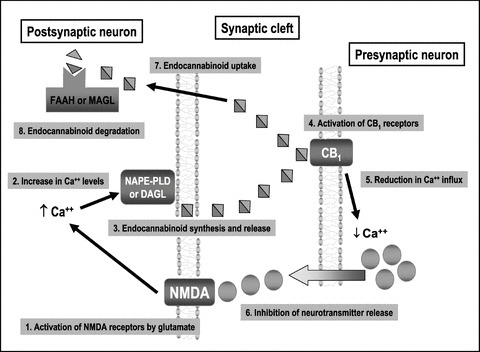
Processes involved in the function of endocannabinoids and CB1 receptors as a retrograde signaling mechanism in glutamatergic neurons.
Dopamine (DA) is one of the neurotransmitters that has been more frequently linked to the action of cannabinoids within the CNS. This can be applied to the case of those dopaminergic neuronal subpopulations, whose cell bodies are located in the reticular formation of the midbrain (e.g., substantia nigra and ventral‐tegmental area), and that project to different forebrain structures, namely, the caudate‐putamen (nigrostriatal pathway; see Figure 2), and the nucleus accumbens/prefrontal cortex complex (mesocorticolimbic pathway). Both neuronal systems would exert a regulatory action on different effector neurons in these structures, then influencing processes such as the control of movement and various cognitive functions, respectively, effects that are among the most relevant pharmacological actions of cannabinoids [8, 9, 10, 12]. It is also important to remark that deficiency or overactivity of these dopaminergic pathways can result in disorders, such as Parkinson's disease (PD) and schizophrenia, respectively, for which various cannabinoid‐related compounds have been proposed as a novel type of therapy [10, 29]. However, despite this close pharmacological interaction, there is little evidence supporting that dopaminergic neurons in the basal ganglia and limbic structures contain CB1 receptors [30, 31]. Most of the researchers agree that cannabinoid effects on DA transmission are frequently indirect and exerted by either postsynaptic or presynaptic mechanisms (reviewed in [32, 33]). For these authors, the abundance of CB1 receptors in GABAergic, glutamatergic or opioidergic projections located in the closest vicinity of dopaminergic neurons [34, 35, 36], facilitates such indirect action. This is also supported by data showing that midbrain dopaminergic neurons, although do not contain CB1 receptors, can, however, produce and release endocannabinoid ligands from their somas and dendrites, then facilitating the retrograde signaling function of these molecules and CB1 receptors in excitatory and inhibitory synapses (reviewed in [37] and see also below).
Figure 2.
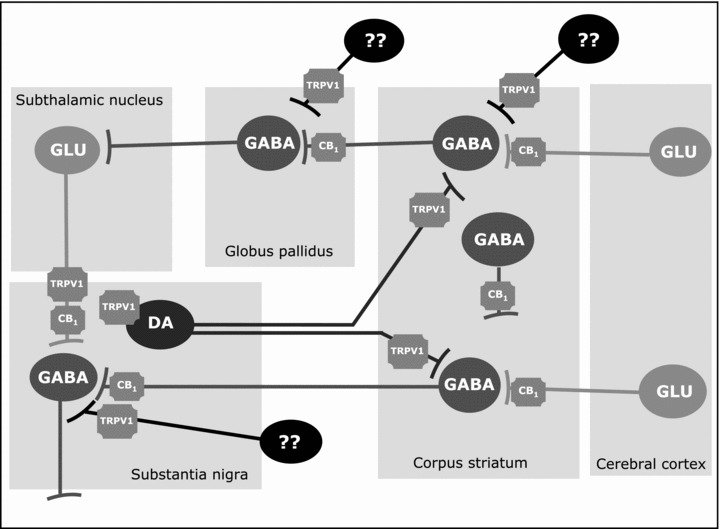
Distribution of CB1 and TRPV1 receptors in different neuronal subpopulations in the basal ganglia circuitry (DA, dopamine; GABA, γ‐aminobutiric acid; GLU, glutamate).
Despite that most of cannabinoid effects on DA transmission seem to be GABA‐ and/or glutamate‐mediated, recent studies have provided an additional mechanism available for those eicosanoid‐derived cannabinoids that have demonstrated some affinity for the TRPV1 receptor (e.g., anandamide, AM404 or N‐arachidonoyl‐dopamine (NADA); reviewed in [38]). These receptors are molecular integrators of nociceptive stimuli, abundant on sensory neurons, but they have been found in dopaminergic neurons within the basal ganglia too [39], thus enabling a direct action of certain cannabinoids on DA function. In fact, there is recent evidence demonstrating that endovanilloid and DA signalling systems are closely linked in the regulation of various neurobiological processes including the control of movement [40, 41]. The case of NADA deserves some comments since it is formed by a molecule of DA linked to arachidonic acid by an amide bond which conferes this molecule properties of endocannabinoid and endovanilloid ligand [38]. NADA seems to be synthesized through the conjugation of an arachidonic acid molecule directly with DA [42], discarding previous hypothesis that suggested that it would be synthesized through the hydroxylation of N‐arachidonoyl‐tyrosine followed by decarboxylation by the same enzymes involved in DA synthesis. Its physiological significance is yet poorly understood, but recent evidence suggests that it can serve as an antioxidant and neuroprotective compound [43].
Finally, a recent study by Oz et al. [44] published during the preparation of this review has provided preliminary evidence that anandamide may inhibit the DA transporter function by a receptor‐independent mechanism, an effect found in heterologous cells and synaptosomal preparations. The anandamide analog methanandamide mimicked this effect, but arachidonic acid was without effect [44]. In addition, inhibition of FAAH or COX‐2 failed to alter the effect of anandamide, thus indicating that this effect is not related to the metabolism of this endocannabinoid [44]. Authors also found that the effect was not attenuated by pertussis toxin, then excluding the involvement of CB1, CB2, or GPR55 receptors, but this does not exclude that TRPV1 receptors may be involved in line with the comments of the above paragraph.
Both aspects, that cannabinoids may alter DA transmission and DA‐related behaviors via an indirect action on GABA and glutamate neurons, and that they can activate TRPV1 receptors located onto dopaminergic neurons, will be key elements in this review. We will concentrate preferentially in reviewing the role(s) played by endocannabinoids on those adult brain functions, where DA is a key regulatory neurotransmitter, namely the control of motor function at the basal ganglia level, and the expression of some cognitive functions, including emotionality, motivation, and brain reward. Additional interactions between cannabinoids and DA have been also claimed for the hypothalamic regulation of pituitary hormone secretion [45], for the expression of key genes during brain development [16, 17], for memory formation [46, 47], sleep regulation [48], and retina function [49] in mammals, as well as some interactions in the brain of invertebrates [50]. However, they will not be addressed in the present review.
Interactions Between Cannabinoids and DA at the Basal Ganglia
Anatomical, physiological and therapeutic evidences indicate that DA is the key regulatory transmitter in the basal ganglia circuitry (reviewed in [51]). So, the activation of DA transmission in this circuitry is generally associated with an increase of movement, whereas the inhibition is followed by hypokinesia. In fact, the basal ganglia disorder with highest prevalence in the human population, PD, is consequence of a progressive degeneration of nigrostriatal dopaminergic neurons resulting in bradykinesia, rigidity, and tremor [52]. Cannabinoids are hypokinetic substances thus producing motor depression and even catalepsy (reviewed in [9]) and it has been largely speculated that this hypokinetic effect of cannabinoids might be produced by reducing dopaminergic activity. This assumption is correct but the role of CB1 receptors, which are not located onto dopaminergic neurons [53], in this effect has not been completely elucidated. Meanwhile, the increasing importance of TRPV1 receptors for the action of certain endocannabinoids [38], as well as the location of these receptors onto dopaminergic neurons [39], have opened interesting novel aspects to discuss about the role and therapeutic potential of the endocannabinoid signaling in the basal ganglia, from both basic and clinical perspectives, aspects that will be addressed below.
Cannabinoids, DA and the Basal Ganglia in Healthy Conditions
Behavioral Data
The abundant presence of CB1 receptors and their endogenous ligands in brain regions related to the control of movement, such as the different basal ganglia (e.g., caudate‐putamen, globus pallidus, and substantia nigra) and the cerebellum [2, 21, 34, 35, 36], suggests that the endocannabinoid system plays a prominent role in the control of movement (for review, see [8, 9, 10]). Thus, excluding very low doses which may produce stimulatory effects in some cases, it is generally well accepted that the different cannabinoid agonists originate a dose‐dependent motor inhibition in both humans and laboratory animals, that may produce even catalepsia with the highest doses (reviewed in [8, 9]). Similar results were obtained by administering inhibitors of the endocannabinoid inactivation, the so‐called indirect cannabinoid agonists (reviewed in [9]). These hypokinetic effects were generally reversed by the administration of CB1 receptor antagonists (e.g., rimonabant, despite some differences found depending on species used), while these antagonists produced by themselves a certain degree of hyperlocomotion due to their frequent properties as inverse agonists (reviewed in [8, 9]). In concordance with these last data, mice lacking CB1 receptors exhibited several motor anomalies (see [24] for review), thus supporting the idea that this receptor type is the key cannabinoid receptor involved in motor effects of cannabinoid compounds.
Neurochemical Data
The motor effects of cannabinoid agonists are likely originated because of the capability of these substances to influence the activity of several neurotransmitters, through the activation of CB1 receptors located in specific neuronal subpopulations within the basal ganglia circuitry (see Figure 2). Striatal projection GABAergic neurons and subthalamonigral glutamatergic neurons, both containing CB1 receptors [35, 36, 53, 54] were identified first as the major substrates for the action of cannabinoids. More recent evidence suggest, however, that CB1 receptors are also located in corticostriatal glutamatergic afferences [55, 56] and in some subpopulations of striatal GABA interneurons [54, 55]. It is important to remark that, in the basal ganglia, CB1 receptors are always located in GABA or glutamate‐containing neurons, which means that the first event associated with the activation of these receptors is an alteration in the activity of GABA and glutamate synapses. However, despite dopaminergic neurons within the basal ganglia do not contain CB1 receptors (with the only exception of the developmental period [31, 57]), they can be secondarily altered given that they are preferential targets for GABA and glutamate synapses. This is supported by numerous pharmacological studies that demonstrated how cannabinoid agonists strongly modified the motor effects of some DA‐acting substances. For example, they potentiated reserpine‐induced hypokinesia [58] and dopaminergic antagonist‐induced catalepsy [59], while reduced quinpirole‐induced hyperlocomotion [60] and amphetamine‐induced hyperactivity [61], although these responses may change when low doses of cannabinoid agonists were used (reviewed in [9]). Neurochemical studies also sustain the idea that cannabinoids reduce the activity of nigrostriatal dopaminergic neurons (reviewed in [8, 9, 32]), although, as mentioned above, the lack of CB1 receptors in these neurons would imply that the changes in the activity of dopaminergic neurons were originated by previous changes in GABAergic or glutamatergic influences arising the substantia nigra ([8, 9] for review).
As mentioned above, further investigations have, however, provided new elements to re‐evaluate the idea that the effects of endocannabinoids on DA transmission in the basal ganglia are exclusively indirect. For example, it has been demonstrated that anandamide and some analogs (e.g., AM404 and NADA), but not classic cannabinoids (e.g., Δ9‐THC), may behave as full agonists for the TRPV1 receptors (reviewed in [38]). These receptors have been also identified in the basal ganglia (see Figure 2) in colocalization with tyrosine hydroxylase enzyme, which in these structures, serves as a specific marker for dopaminergic neurons [39]. TRPV1 receptors have been also identified in glutamatergic terminals in the substantia nigra pars compacta [62]. The activation of these receptors with capsaicin or with other potential vanilloid ligands produced hypokinesia in rats [63]. The same effect was seen with anandamide accompanied by a reduction in the activity of dopaminergic terminals in the striatum [64]. This effect of anandamide was reversed by capsazapine, thus indicating that it would be exerted by the activation of TRPV1 receptors [64]. These neurochemical responses were furtherly confirmed in vitro using perfused striatal fragments, but they were not reproduced by classic cannabinoids, such as Δ9‐THC, that do not bind to vanilloid‐like receptors [64], thus indicating that the TRPV1 receptor rather than the CB1 is the key target in these responses. By contrast, other authors [62, 65] found that the activation of TRPV1 receptors in the substantia nigra pars compacta stimulated DA release, although these effects might be mediated by TRPV1 receptors located in glutamatergic neurons rather than by those located in dopaminergic terminals. Other support to the increasing relevance of TRPV1 receptors in the basal ganglia comes from studies conducted in rat models of Huntington's disease, where several cannabinoid‐based compounds, such as AM404, exhibited antihyperkinetic properties, being these effects depending on their capability to activate TRPV1 receptors rather than CB1 receptors [66, 67]. Finally, making the issue even more complex, a recent study by Ferrara and coworkers [68] revealed that N‐acyldopamines, such as NADA, are able to control the activity of dopaminergic terminals in the striatum via novel ion channels other than TRPV1 receptors, an effect that was not observed with anandamide or capsaicin. Importantly, NADA was likely synthesized in the substantia nigra in conditions of hyperactivity [62].
Despite CB1 receptors do not appear to be located in dopaminergic neurons, they colocalize with D1 or D2 receptors in striatal GABAergic projection neurons (striatonigral and striatopallidal pathways, respectively) [69, 70], which facilitate postsynaptic interactions between endocannabinoids and DA at the level of G‐protein/adenylyl cyclase signal transduction [71, 72], even the formation of heteromers between CB1 and D2 receptors, and also adenosine A2a receptors (reviewed in [73]). These CB1, D2, and A2a receptor heteromers were found in the dendritic spines of GABAergic neurons projecting to the globus pallidus, but their functional properties and their role in striatal function are pending of further investigation (reviewed in [73]). This type of postsynaptic mechanisms facilitates the direct interaction between cannabinoids and DA allowing, in this case, a bidirectional regulation, endocannabinoids and DA and viceversa. It is in this context of bidirectional regulation where one may place, at the same time, data showing that: (i) motor effects of CB1 agonists are associated with an activation of DARPP‐32 signaling, which has been linked to intracellular responses elicited by D1 and D2 receptors in the striatal projection neurons, whereas the genetic inactivation of DARPP‐32 attenuated motor effects of cannabinoids [74]; and (ii) dopaminergic D2 receptors control anandamide production in the striatum, thus indicating that the endocannabinoid system serves as an inhibitory feedback mechanism counteracting DA‐induced facilitation of psychomotor activity [71]. In addition, D2 receptors also control Gi/o protein availability for CB1 receptors [75] and facilitate endocannabinoid‐mediated long‐term synaptic depression of GABAergic neurons [76], an effect also seen in the ventral‐tegmental area [77]. A similar interaction of endocannabinoids with D1 receptors has been recently demonstrated [70]. All these observations are concordant with the old idea proposed by Mailleux and Vanderhaeghen [78] that endocannabinoid signaling in the basal ganglia is regulated by DA and viceversa, which might be relevant for a disease like PD, where CB1 receptors and their ligands seem to be up‐regulated in conditions of DA deficiency (see [79, 80, 81] and details later).
Cannabinoids, DA and the Basal Ganglia in Pathological Conditions
Assuming that the endocannabinoid signaling system modulates the activity of DA and other neurotransmitters at the basal ganglia by acting at CB1 and/or TRPV1 receptors, one may postulate that the pharmacological management of this system may serve to normalize DA transmission in conditions of DA deficiency, overactivity or dysregulation, and, subsequently, to alleviate DA‐related motor symptoms in various basal ganglia disorders (for review, see [9, 32, 82]). To date, most studies that have addressed this issue are preclinical and have provided the first experimental evidences using animal models (see [10, 82] for recent reviews). However, in some cases, a few number of clinical trials have been already conducted unfortunately with poorly effective results [83, 84, 85, 86, 87, 88]. As this review is centered in cannabinoid‐DA interactions, we will concentrate here in PD, the major basal ganglia disorder characterized by either dopaminergic degeneration or malfunctioning. However, we will briefly mention that cannabinoids have been also studied in other disorders related to the basal ganglia function, for example in Huntington's disease where direct or indirect (inhibitors of endocannabinoid inactivation) agonists of CB1 receptors, especially if they also behave as TRPV1 receptor agonists, have been proposed as having therapeutic value (reviewed in [89]). Cannabinoid agonists, presumably those activating CB1 receptors, are also effective in Gilles de la Tourette's syndrome, where they reduced tics and improved behavioral problems in patients (reviewed in [90]). Similar studies have been conducted in relation with dystonia [84, 91] and dyskinesia, particularly, the case of levodopa‐induced dyskinesia [92].
As mentioned above, PD is the major basal ganglia disorder characterized by the progressive death of nigral dopaminergic neurons and DA denervation of the striatum. Both CB1 receptor agonists and antagonists have been proposed of therapeutic value in this disorder, alone or as coadjuvants, and addressed to alleviate specific motor symptoms or to delay/arrest the progression of this disease (reviewed in [8, 9, 82, 93]). CB1 receptor agonists have been proposed, for example, for the reduction of tremor associated with the frequent overactivity of the subthalamic nucleus occurring in PD [94], although the few clinical trial conducted to explore this effect in patients did not generate positive results [88]. CB1 receptor agonists were also investigated in relation with the dyskinetic states associated with long‐term therapy of dopaminergic replacement with levodopa, showing positive effects [95]. However, this effect was not observed with the so‐called indirect cannabinoid agonists, e.g., FAAH inhibitors, presumably because they are also able to activate TRPV1 receptors [96]. Only using coadministration with a TRPV1 receptor antagonist, FAAH inhibitors were capable to show antidyskinetic properties, thus indicating that CB1 and TRPV1 receptors have opposite effects in the control of levodopa‐induced dyskinesia [96]. Again the clinical testing of this potential produced controversial results [83, 97].
Positive effects were also found in the case of those plant‐derived cannabinoid agonists having cannabinoid receptor‐independent antioxidant properties, when they were examined for their capability to afford protection against dopaminergic degeneration in experimental models of parkinsonism [98, 99], indicating that they may represent a promising therapy against disease progression in PD. These results provide a neurobiological support for those anecdotal data (e.g., surveys) that indicated that PD patients who self‐medicated with cannabis obtained beneficial effects (see an example in [100]), despite controlled clinical studies did not reproduce these benefits [97]. However, both surveys on cannabis use for PD and clinical studies were always centered in specific symptoms rather than in the disease progression, therefore the issue deserves further clinical testing. Positive effects against disease progression have been recently obtained with CB2 receptor agonists in MPTP‐lesioned animals [101], whereas CB1 receptor‐deficient mice resulted to be more vulnerable to the lesion with 6‐hydroxydopamine than wild‐type animals [102], thus indicating an additional contribution of CB1 and CB2 receptors in neuroprotective effects of cannabinoids in this disorder that would also deserve further clinical investigation.
By contrast, the blockade of CB1 receptors (e.g., with rimonabant) was recently reported to reduce bradykinesia and other parkinsonian symptoms (e.g., levodopa‐induced dyskinesia; see Figure 3 and below) [103, 104, 105], despite previous studies showing conflicting results [81, 106] and the negative results found in the only clinical trial conducted with CB1 receptor antagonists in parkinsonian patients [87]. It appears that the blockade of CB1 receptors: (i) works more efficiently in certain circumstances, for example in very advanced phases of the disease characterized by extreme nigral damage [103], (ii) would be DA‐independent [104] despite it enhanced the antiparkinsonian efficacy of levodopa [105, 107], and/or (iii) needs the use of low doses of the antagonist [104, 105] (see Figure 3), conditions that were not completely reproduced in the only clinical trial conducted so far with rimonabant which included a group of patients that were all well responders to levodopa [87]. Further studies have demonstrated that this effect of rimonabant would be exerted by enhancing glutamatergic transmission at the striatal level [108]. Therefore, if these preclinical data are finally confirmed with the appropriate clinical testing, it would be possible to have a novel antiparkinsonism agent in a stage of the disease when the classic dopaminergic therapy is generally failed or for the group of patients that have a poor levodopa response. CB1 receptor antagonists have been also proposed for reducing and/or delaying levodopa‐induced dyskinesia (reviewed in [92]), thus indicating the extreme complexity of this circuitry where both CB1 agonists and antagonists may serve for the same type of treatment, a fact presumably related to the presence of these receptors in both excitatory and inhibitory synapses within the basal ganglia circuitry. On the other hand, it is important to remark that the usefulness of CB1 receptor antagonists in this disease agrees with the type of pharmacological strategy expected once several studies have demonstrated an up‐regulation of CB1 receptors and other elements of this signaling system in PD [78, 79, 80, 81]. For some authors, there is an unbalance between DA, which goes down, and endocannabinoids, which go up, at the basal ganglia once nigral damage is already evident (early stages in the development of parkinsonism, when nigral damage does not exist or is minimal, may be, however, associated with down‐regulatory responses [109]), which supports the potential of CB1 receptor antagonists in this disease. This type of response has been observed in rats treated acutely with reserpine [81] or chronically with dopaminergic antagonists [78], or after the damage of nigrostriatal neurons with 6‐hydroxydopamine [78, 79] or MPTP [80] in different laboratory animals. It was also found in patients [80, 110]. In support of the idea of unbalance, the classic dopaminergic replacement therapy with levodopa reversed this endocannabinoid overactivity [80, 111]. On the other hand, it is also important to consider the therapeutic benefits that can offer the antagonists of TRPV1 receptors for the treatment of motor anomalies in PD, given their recently demonstrated role in regulating DA release from nigral neurons [64]. For example, they have been recently found to be necessary for unmasking the antidyskinetic effecs of FAAH inhibitors or other cannabinoid agonists capable to directly or indirectly activate TRPV1 receptors [96]. However, given that they are located in the neuronal subpopulation that degenerates in this disease [97], it is possible that this target may loss interest as soon as the disease progresses, something important in a disease whose first motor symptoms appear when an important loss of dopaminergic neurons has already occurred.
Figure 3.
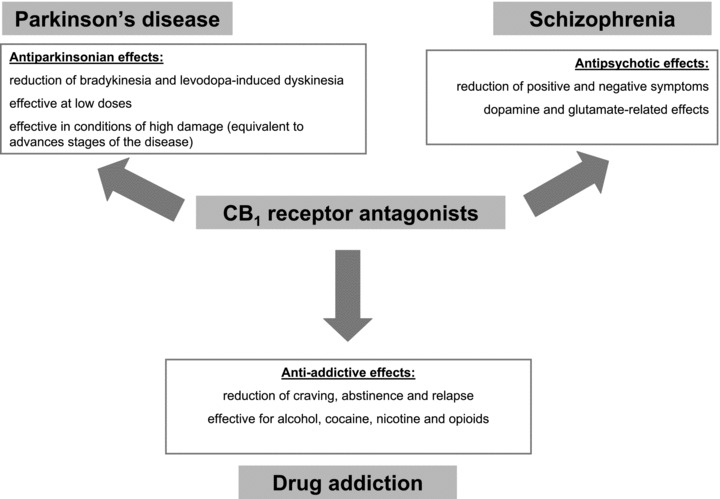
Summary of potential therapeutic applications proposed for CB1 receptor antagonists in pathologies related to dopamine transmission in the CNS.
Interactions Between Cannabinoids and DA at the Corticolimbic Structures
Mesocorticolimbic dopaminergic neurons play a regulatory function in the control of cognitive processes, motivated behavior, the central stress response, and the pleasure produced by natural (e.g., sex, food) or other types (e.g., drugs of abuse) of reinforcers, or by compulsive activities, such as gambling, overeating and sex dependence (for review, see [112, 113]). In fact, the neurotransmitter more studied as a potential target for the pharmacological effects of habit‐forming drugs is DA (for review, see [114]). This also includes the case of cannabis derivatives (for review, see [115, 116]), which acting through cannabinoid receptors, likely the CB1 receptor type, are able to alter mesocorticolimbic DA transmission. Therefore, a first level of interaction between cannabinoids and DA in corticolimbic structures can be found in processes like brain reward and drug abstinence/dysphoric responses [115, 116, 117, 118, 119]. However, the interactions of cannabinoids and DA at this level are probably largest and involve more cortical and subcortical structures and more types of processes. From a pharmacological point of view, cannabinoid agonists (depending on doses and duration of treatment) produce euphoria, stimulate brain reward, are anxiolytic, and decrease motivation and arousal while increasing emotionality, effects that were observed in humans and laboratory animals (reviewed in [120, 121]). These effects were paralleled by alterations in mesocorticolimbic dopaminergic neurons, but it is generally accepted that DA transmission is not the first target for the action of cannabinoid agonists also in these structures, so that the effects would be most likely indirect [120, 121], as in the case of the basal ganglia. Several authors proposed glutamatergic and/or GABAergic inputs to the nucleus accumbens/prefrontal cortex and ventral‐tegmental area as the primary targets for the cannabinoid action in these processes and also as the responsible of DA changes [122, 123, 124]. These glutamatergic and GABAergic subpopulations would contain CB1 receptors regulating presynaptic events and would be ultimately in contact with dopaminergic neurons (for review, see [121]). This includes, for example, the recent demonstration of CB1 receptors in excitatory projections coming from subcortical structures and reaching the bed nucleus of the stria terminalis which, in turn, projects to the ventral‐tegmental area [125]. Finally, there is some evidence of colocalization of CB1 receptors and tyrosine hydroxylase in the ventral‐tegmental area [30], where cell bodies of mesocorticolimbic dopaminergic neurons are located, which opens the possibility of a direct action of cannabinoids on the major substrate for brain reward, motivation/emotionality and other limbic processes.
Cannabinoids, DA, and Corticolimbic Structures in Healthy Conditions
Behavioral Data
As mentioned above, CB1 receptors are located in glutamatergic and/or GABAergic synapses within cortical and subcortical structures and they represent the major substrate for cognitive effects and reinforcing properties of cannabinoids administered to laboratory animals or consumed by human subjects. These pharmacological effects coincide with two key functions described for the cannabinoid signaling system in corticolimbic structures. In relation with the cognitive effects of cannabinoids, it is well known that CB1 receptors are moderately abundant in different cortical structures, in particular they are located in superficial and deep layers, presumably onto GABAergic interneurons [3, 34, 35, 36] and the same happens with their endogenous ligands [21]. These anatomical data suggest a role for the cannabinoid signaling system in the control of sleep‐waking cycle, performance of complex cognitive tasks, working memory, temporal organization of behavior, adaptation of behavioral strategies, sensory perception, and other cognitive functions, whose control resides mainly in the different cortical structures (for review, see [126]). This provides an explanation for major subjective effects and cognitive impairments experienced by cannabis consumers, including: (i) the case of naïve consumers in which cannabinoids reversibly impair cognitive functions, a phenomenon also demonstrated in laboratory animals (reviewed in [127]), and (ii) the particular case of long‐term marijuana abuse where severe irreversible deficits in cognitive function and precipitation of psychiatric disorders, such as psychosis, anxiety or depression, have been postulated, in particular when the abuse of marijuana starts at very early ages during the adolescence (reviewed in [127, 128, 129] and see below). Anyway, the evidence surrounding the cognitive effects of cannabis is always a matter of continuous debate, with studies stating the critical role played by cannabinoids in the development of psychiatric disorders for the general population and others that circumscribe this possibility only in vulnerable individuals (reviewed in [127, 128, 129] and see below).
The cannabinoid signaling system also plays a role in brain reward processes activated by different types of reinforcers, particularly, the addictive drugs, including the case of cannabis consumption itself [130] but also of other different drugs of abuse [131, 132]. This assumption is supported by several evidences. First, several neuroanatomical studies have demonstrated that elements of the cannabinoid signaling system, particularly the CB1 receptors, are abundant in the different brain structures that form the brain reward circuitry [21, 30, 34, 35, 36]. Second, several biochemical studies have demonstrated that these elements, again in particular the CB1 receptor, experience important changes in conditions of acute exposure, chronic consumption, dependence, abstinence, or relapse for most frequently consumed habit‐forming drugs, including the case of opioids [133, 134, 135], cocaine [133, 136], nicotine [136] or alcohol [133, 136, 137, 138, 139]. The importance of a role of the cannabinoid system in addictive processes is that it opens the possibility that the pharmacological management of the cannabinoid signaling may serve to improve behavioral and/or neurochemical anomalies occurring during addictive states (see next section), and for this potential, the interactions between cannabinoids and DA seem to be critical.
Neurochemical Data
Several cannabinoid agonists, mainly Δ9‐THC, the major psychoactive ingredient of cannabis, have been reported to increase mesolimbic dopaminergic activity (reviewed in [115]), as demonstrated in numerous studies conducted with laboratory animals in which the treatment with different cannabinoid agonists elevated D1 receptor density, DA release, and DA metabolism in various limbic structures, as well as it enhanced the firing rate of mesolimbic dopaminergic neurons in the A10 region (reviewed in [129, 140]). The involvement of the CB1 receptor seems critical for these effects and explains why knockout mice for the CB1 receptor exhibited reduced voluntary alcohol consumption [141], morphine self‐administration [142, 143], cocaine‐enhanced locomotion [144], and absence of rewarding effects of nicotine evaluated in the conditioned place preference test [145], although they did not show similar responses for cocaine or nicotine reinforcement in the self‐administration paradigm [143]. Again, DA transmission seems to be critical here because the effects of alcohol, morphine, cocaine, or nicotine on DA release in the nucleus accumbens were completely absent in CB1 receptor knockout mice [141, 142, 144, 146]. In the same direction, an increased endocannabinoid tone has been demonstrated to facilitate the effects of most commonly abused drugs on DA transmission, since DA release was uniformly inhibited by the blockade of CB1 receptors [147]. However, adding complexity to this issue, a recent study by Melis et al. [148] demonstrated that FAAH inhibitors were also effective in the reduction of nicotine‐induced activation of dopaminergic neurons, which contradicts the idea that the endocannabinoid system, and particularly the CB1 receptor, exerts a stimulatory effect on nicotine reinforcing properties. In this case, FAAH inhibitors would act by enhancing the action of certain signaling N‐acylethanolamines (e.g., oleoylethanolamide and palmitoylethanolamide), which are devoid of CB1 activity, but are capable to act at the PPAR‐α nuclear receptors, opening a new avenue in the understanding and perhaps in the treatment of drug addiction.
In parallel to their effects on mesolimbic projections neurons, cannabinoid agonists also augmented the activity of those dopaminergic neurons that coming from the ventral‐tegmental area project specifically to the prefrontal cortex (reviewed in [149]). The prefrontal cortex is involved in many cognitive functions, including working memory, temporal organization of behavior, and adaptation of behavioral strategies, that, as mentioned above, are affected by cannabinoids [149]. This region contains a moderate density of CB1 receptors [34, 35]. The acute administration of cannabinoids increased DA release in rat prefrontal cortex, measured by in vivo microdialysis, but this effect is produced presumably by a previous increase of glutamate release and/or a decrease of GABA, given that CB1 receptors are not located in dopaminergic neurons [150]. By contrast, the repeated administration of cannabinoid agonists decreased DA turnover in the prefrontal cortex but not in the nucleus accumbens and the striatum [151, 152], an effect that persisted even after a drug‐free period of 2 weeks [152].
In general, there are no discrepancies concerning that the activation of CB1 receptors is involved in most of effects produced by cannabinoids on mesocorticolimbic activity. However, as mentioned above, these effects have been considered, so far, as exerted indirectly [132]. Possibly, they would be caused through modifying GABAergic influences to the ventral‐tegmental area and/or the nucleus accumbens, given that CB1 receptors have been found in these GABA neurons rather than in mesocorticolimbic ones [117, 153, 154]. These GABA neurons tonically inhibit DA‐containing neurons and an inhibition of GABA release by cannabinoid agonists, via presynaptic CB1 receptors, would be expected to increase the activity of dopaminergic neurons [123, 124, 154, 155, 156, 157, 158]. Alternatively, CB1 receptors may be located in the excitatory glutamatergic inputs to the GABA‐containing neurons that project from the nucleus accumbens to the ventral‐tegmental area, as reported by Melis and coworkers [159]. In this case, the activation of CB1 receptors would result in a decrease of glutamate release followed by reduction in GABA activity and, again, in an increase in the firing of dopaminergic neurons [124, 160]. An important aspect to remark is that, in both cases—CB1 receptors located in GABA‐ or glutamate‐containing neurons—the activation of these receptors would depend on the release of endocannabinoids by dopaminergic neurons, a fact associated with the increase in the activity of these neurons provoked by abused drugs [121]. According to these authors [121], the release of endocannabinoids and the subsequent activation of presynaptic CB1 receptors by these signaling lipids may represent a common phenomenon associated with the action of a wide variety of habit‐forming drugs. Finally, it is also important to consider the study published by Wenger and coworkers [30] who, as mentioned above, demonstrated for the first time, using double immunohystochemistry, that CB1 receptors colocalize with tyrosine hydroxylase in the nucleus accumbens. This opens the possibility of a direct action of cannabinoids on the major neurochemical substrate of brain reward, dysphoria‐mediated drug craving and drug relapse, although not all researchers agree with this possibility (reviewed in [121, 140]).
Cannabinoids, DA and Corticolimbic Structures in Pathological Conditions
As in the case of the basal ganglia, the capability of the cannabinoid system to influence DA transmission in corticolimbic structures supports that the pharmacological management of this system might have therapeutic value in those diseases involving anomalies of mesocorticolimbic DA transmission, among them, addictive states, and schizophrenia and other psychosis. The working hypothesis is that normalizing DA transmission, with either cannabinoid agonists or antagonists depending on the type of dysfunction, would result in reducing addictive processes or producing antypsychotic effects.
As mentioned earlier, the endocannabinoid transmission has been related to many signs of drug addiction. For example, it can be related to cases of individual vulnerability for drug abuse, a fact supported by recent data showing a higher occurrence of this disorder in individuals bearing specific genetic variants of the CB1 receptor or the FAAH enzyme, particularly for the case of alcohol and opioids [161, 162, 163]. However, most of the evidence supporting a relation of the cannabinoid system with drug addiction involve processes of craving, degree of dependence and intensity of abstinence, or risk to relapse for different types of drugs [119, 164, 165, 166, 167, 168]. The importance of this fact is that it opens the possibility of using cannabinoid‐related substances in the treatment of different aspects of drug addiction (for review, see [131, 132, 137]). These effects would be exerted, among others, by normalizing DA transmission, in particular in those addictive responses more directly related to changes in DA activity such as reinforcement/relapse [118] or withdrawal/dysphoria‐mediated drug craving [117]. Most of pharmacological studies have concentrated in the potential of CB1 receptor antagonists and the most‐studied compound has been rimonabant [169] (see Figure 3). For example, the blockade of CB1 receptors with this antagonist impaired the perception of reinforcing potential of different habit‐forming drugs [170], indicating that positive incentive and/or motivational processes (and relapsing properties too) could be under a permissive control of CB1 receptor‐related mechanisms. This has been described for many drugs including the case of alcohol [141, 167, 171, 172], morphine [164, 165, 173, 174] and nicotine [146, 175, 176], and specifically for relapsing properties in the case of cocaine [166, 177]. This concept has been recently extended to physical dependence/abstinence for most of these drugs (reviewed in [169]). This pharmacological potential shown by rimonabant (and other CB1 receptor antagonists) in preclinical studies led to an exhaustive clinical testing, particularly for tobacco addiction (STRATUS clinical trial, see [169, 178]) and, more recently, for alcohol dependence (ACTOL clinical trial, see [179]), although the results were not positive enough. Major problems with rimonabant and other similar antagonists are that they behave also as inverse agonists, and, given the ubiquitous distribution of CB1 receptors within the CNS, the use of this type of compounds in the clinic is frequently associated with the occurrence of psychiatric side effects. Therefore, the matter is pending of the development of neutral CB1 receptor antagonists [169].
Cannabinoids–DA interactions are also important in another disorder related to corticolimbic structures such as schizophrenia. There are multiple lines of research relating cannabinoids to DA transmission in schizophrenia. For example, the brain structures involved in the pathogenesis of schizophrenia (limbic areas, prefrontal cortex), structures that are densely innervated by dopaminergic terminals, contain also a moderate but significant density of CB1 receptors [34, 35, 36]. They also contain relevant amounts of endocannabinoids [21]. This may explain why cannabis consumption has been associated with the induction or enhancement of psychosis in various cohort studies, and, then, proposed as a potential risk factor for the development of this disorder, in particular in the case of heavy abusers (e.g., frequent consumers or consumers of high Δ9‐THC cannabis), early consumers (e.g., adolescence is a particularly vulnerable period) and/or predisposed individuals (e.g., subjects with family history or prodromal symptoms, or bearing the Val allele of COMT) (reviewed in [127, 128, 129]). Cannabis consumption has been also associated with more frequent and earlier relapses (reviewed in [180]). However, as mentioned above, the association between cannabis and psychosis is not completely understood (see [127, 128, 129, 181, 182] for review). It is also important to remark, given the objective of this review, that, for some authors, cannabinoid agonists, independently of their capability to induce or aggravate psychotic episodes, seem to be capable to interfere with classic antipsychotics, thus reducing their capability to block D2 receptors and/or exacerbating their extrapyramidal symptoms [183, 184]. However, for some authors, this can be a sort of self‐medication to diminish neuroleptic‐side effects [129, 181]. That cannabis/cannabinoids may cause or exacerbate psychoses may be related to the facilitatory effects of cannabinoids on DA transmission at the nucleus accumbens [181], given that overactivity of DA transmission in this structure is a key pathological event in schizophrenia. The inhibitory effect of cannabinoids on GABA and glutamate transmission may also collaborate in these responses [129].
Another relevant observation concerning the possible relation of cannabis/cannabinoids with the pathogenesis of schizophrenia was obtained in genetic studies, which revealed the association of a polymorphism of the CB1 receptor gene with increased susceptibility to develop schizophrenia [181, 185, 186, 187], although there are studies showing no association (reviewed in [181]). For some authors, this is an important support to the idea that the cannabinoid signaling system plays a role in the pathogenesis of schizophrenia, at least in a subgroup of schizophrenic individuals [181]. This is the so‐called cannabinoid hypothesis of schizophrenia, which would consist of an impairment in the activity of this signaling system (higher CB1 receptor density or endocannabinoid levels, responses that have been found in schizophrenic patients; see below) in cortical and subcortical (limbic) structures. These changes would be associated with hyperactivity of dopaminergic neurons (positive symptoms) and hypoactivity of glutamate neurons (negative symptoms) (for review, see [32]).
On the other hand, despite the evidence indicating that cannabis consumption or cannabinoid signaling dysregulation may be adverse elements collaborating in the development of schizophrenia or reducing the efficacy of classic antipsychotic therapies, recent evidences have also indicated that certain cannabinoid‐related compounds may serve as novel therapeutic agents in schizophrenia. This includes first the possibility that psychotic patients use cannabis to overcome the unpleasant feelings produced by classic antipsychotic therapy (reviewed in [181]). For example, Voruganti et al. [188], using in vivo SPECT analysis, demonstrated that cannabis smoking in schizophrenic patients produced an immediate calming effect, although followed by a worsening of psychotic symptoms [188]. The calming effect correlated with a reduction in striatal D2 receptor binding that the authors interpreted as suggestive of increased DA activity [188]. Similar results were furtherly obtained by Bossong et al. [189], but not by Stokes et al. [190]. More recent evidences have indicated that the management of the cannabinoid signaling may even provide antipsychotic effects, with two compounds, cannabidiol and rimonabant, being the most studied. The antipsychotic potential of cannabidiol has been recently demonstrated even in the clinical area [191], although it is possible that its effects are not mediated by the activation of CB1 or CB2 receptors for which this phytocannabinoid has poor affinity, but through its capability to modulate TRPV1 receptors or to interfere with endocannabinoid inactivation. As regards to rimonabant or other similar CB1 receptor antagonists (see Figure 3), their antipsychotic potential was recently examined in preclinical models [192, 193] at the light of the so‐called cannabinoid hypothesis of schizophrenia mentioned above, according to which the cannabinoid signaling system would be elevated in schizophrenic patients. In this respect, schizophrenic patients have elevated anandamide levels in CSF [194, 195] and blood [196]. Using analyses in postmortem human cortical and subcortical structures or neuroimaging procedures, several authors demonstrated that the density of CB1 receptors is also elevated [197, 198, 199, 200], although this was not found in other study [201] and, even, a last study described a decrease in CB1 receptor binding [202]. The preclinical studies revealed that the antipsychotic potential of rimonabant was related to alterations in DA [203] and glutamate [193] transmissions in cortical structures. However, rimonabant did not differ from placebo in reducing positive and negative symptoms in a placebo‐controlled clinical trial conducted with patients having schizophrenia and schizoaffective disorder [204].
Concluding Remarks
Along this review, we have reviewed the classic knowledge and the recent advances on cannabinoid‐DA interactions paying emphasis in two processes where DA has been proposed as a key neurotransmitter, such as the basal ganglia function and the corticolimbic processes. We have explored the mechanisms underlying these interactions, which represent the way for plant‐derived cannabinoids to interfere with these processes. In most of the cases, we have concluded that dopaminergic neurons do not contain CB1 receptors, with some exceptions, but these receptors are located on neurons present in regions innervated by dopaminergic neurons, which allows relevant bidirectional interactions. Finally, we have reviewed those diseases characterized by either deficiency, dysregulation or overactivity of DA transmission and where cannabinoids might be of therapeutic potential possibly through actions that facilitate, among others, a normalization of DA transmission. These diseases included basal ganglia disorders (e.g., Parkinson's disease), drug addiction, and schizophrenia and related‐psychoses.
Funding
This work has been supported by grants from CIBERNED (CB06/05/0089), MICINN (SAF2009/11847), and CAM (S‐SAL‐0261/2006). These agencies had no further role in study design, the collection, analysis, and interpretation of data, in the writing of the report, or in the decision to submit the paper for publication.
Authors Contributions
The three authors shared the decision on the different contents to be included in this review article, the management of literature searches and the design of graphical materials. Javier Fernández‐Ruiz wrote the different versions of the manuscript, which were revised by the other authors. All authors have approved the final version for submission.
Conflict of Interest
All authors declare that they have no conflicts of interest.
Acknowledgments
Authors are indebted to all coworkers who participated in the studies of our group mentioned in this review, and to Borja Gallardo and Yolanda García‐Movellán for technical and administrative assistance.
References
- 1. Heifets BD, Castillo PE. Endocannabinoid signaling and long‐term synaptic plasticity. Annu Rev Physiol 2009;71:283–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Breivogel CS, Sim‐Selley LJ. Basic neuroanatomy and neuropharmacology of cannabinoids. Int Rev Psychiatry 2009;21:113–121. [DOI] [PubMed] [Google Scholar]
- 3. Kano M, Ohno‐Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid‐mediated control of synaptic transmission. Physiol Rev 2009;89:309–380. [DOI] [PubMed] [Google Scholar]
- 4. Pandey R, Mousawy K, Nagarkatti M, Nagarkatti P. Endocannabinoids and immune regulation. Pharmacol Res 2009;60:85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Randall MD. Endocannabinoids and the haematological system. Br J Pharmacol 2007;152:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maccarrone M. Endocannabinoids and reproductive endocrinology. Curr Opin Investig Drugs 2009;10:305–310. [PubMed] [Google Scholar]
- 7. Matias I, Di Marzo V. Endocannabinoids and the control of energy balance. Trends Endocrinol Metab 2007;18:27–37. [DOI] [PubMed] [Google Scholar]
- 8. Gerdeman GL, Fernández‐Ruiz J. The endocannabinoid system in the physiology and pathophysiology of the basal ganglia In: Kofalvi A, editor. Cannabinoids and the brain. New York , NY : Springer‐Verlag, 2008;423–483. [Google Scholar]
- 9. Fernández‐Ruiz J, González S. Cannabinoid control of motor function at the basal ganglia In: Pertwee RG. editor. Handbook of experimental pharmacology – 168 – cannabinoids. Heidelberg (Germany) : Springer‐Verlag, 2005;479–507. [DOI] [PubMed] [Google Scholar]
- 10. Fernández‐Ruiz J. The endocannabinoid system as a target for the treatment of motor dysfunction. Br J Pharmacol 2009;156:1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guindon J, Hohmann AG. The endocannabinoid system and pain. CNS Neurol Disord Drug Targets 2009;8:403–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Solinas M, Goldberg SR, Piomelli D. The endocannabinoid system in brain reward processes. Br J Pharmacol 2008;154:369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Riedel G, Davies SN. Cannabinoid function in learning, memory and plasticity In: Pertwee RG, editor. Handbook of experimental pharmacology – 168 – cannabinoids. Heidelberg (Germany) : Springer‐Verlag, 2005;445–477. [DOI] [PubMed] [Google Scholar]
- 14. Kirkham TC. Cannabinoids and appetite: Food craving and food pleasure. Int Rev Psychiatry 2009;21:163–171. [DOI] [PubMed] [Google Scholar]
- 15. Storr MA, Sharkey KA. The endocannabinoid system and gut‐brain signalling. Curr Opin Pharmacol 2007;7:575–582. [DOI] [PubMed] [Google Scholar]
- 16. Fernández‐Ruiz J, Berrendero F, Hernández ML, Ramos JA. The endogenous cannabinoid system and brain development. Trends Neurosci 2000;23:14–20. [DOI] [PubMed] [Google Scholar]
- 17. Ramos JA, Gómez M, De Miguel R. Effects on development In: Pertwee RG, editor. Handbook of experimental pharmacology – 168 cannabinoids. Heidelberg (Germany) : Springer‐Verlag, 2005;643–656. [DOI] [PubMed] [Google Scholar]
- 18. Pertwee RG. Ligands that target cannabinoid receptors in the brain: From THC to anandamide and beyond. Addict Biol 2008;13:147–159. [DOI] [PubMed] [Google Scholar]
- 19. Pertwee RG. CB1 and CB2 receptor pharmacology In: Kofalvi A, editor. Cannabinoids and the brain. Springer‐Verlag, 2008;91–100. [Google Scholar]
- 20. Kogan NM, Mechoulam R. Cannabinoids in health and disease. Dialogues Clin Neurosci 2007;9:413–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bisogno T, Berrendero F, Ambrosino G, Cebeira M, Ramos JA, Fernández‐Ruiz JJ, Di Marzo V. Brain regional distribution of endocannabinoids: Implications for their biosynthesis and biological function. Biochem Biophys Res Comm 1999;256:377–380. [DOI] [PubMed] [Google Scholar]
- 22. Kunos G, Bátkai S. Novel physiologic functions of endocannabinoids as revealed through the use of mutant mice. Neurochem Res 2001;26:1015–1021. [DOI] [PubMed] [Google Scholar]
- 23. Schlosburg JE, Kinsey SG, Lichtman AH. Targeting fatty acid amide hydrolase (FAAH) to treat pain and inflammation. AAPS J 2009;11:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Valverde O, Karsak M, Zimmer A. Analysis of the endocannabinoid system by using CB1 cannabinoid receptor knockout mice In: Pertwee RG, editor. Handbook of experimental pharmacology – 168 cannabinoids. Heidelberg (Germany) : Springer‐Verlag, 2005;117–145. [DOI] [PubMed] [Google Scholar]
- 25. Di Marzo V. Targeting the endocannabinoid system: To enhance or reduce? Nat Rev Drug Discov 2008;7:438–455. [DOI] [PubMed] [Google Scholar]
- 26. Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 2003;83:1017–1066. [DOI] [PubMed] [Google Scholar]
- 27. Schlicker E, Kathmann M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci 2001;22:565–572. [DOI] [PubMed] [Google Scholar]
- 28. Lovinger DM. Presynaptic modulation by endocannabinoids. Handb Exp Pharmacol 2008;184:435–477. [DOI] [PubMed] [Google Scholar]
- 29. Vinod KY, Hungund BL. Cannabinoid‐1 receptor: A novel target for the treatment of neuropsychiatric disorders. Expert Opin Ther Targets 2006;10:203–210. [DOI] [PubMed] [Google Scholar]
- 30. Wenger T, Moldrich G, Furst S. Neuromorphological background of cannabis addiction. Brain Res Bull 2003;61:125–128. [DOI] [PubMed] [Google Scholar]
- 31. Hernández ML, Berrendero F, Suarez I, et al Cannabinoid CB1 receptors colocalize with tyrosine hydroxylase in cultured fetal mesencephalic neurons and their activation increases the levels of this enzyme. Brain Res 2000;857:56–65. [DOI] [PubMed] [Google Scholar]
- 32. Van Der Stelt M, Di Marzo V. The endocannabinoid system in the basal ganglia and in the mesolimbic reward system: Implications for neurological and psychiatric disorders. Eur J Pharmacol 2003;480:133–150. [DOI] [PubMed] [Google Scholar]
- 33. Laviolette SR, Grace AA. The roles of cannabinoid and dopamine receptor systems in neural emotional learning circuits: Implications for schizophrenia and addiction. Cell Mol Life Sci 2006;63:1597–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Herkenham M, Lynn AB, Little MD, Melvin LS, Johnson MR, De Costa DR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J Neurosci 1991;11:563–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mailleux P, Vanderhaeghen JJ. Distribution of neuronal cannabinoid receptor in the adult rat brain: A comparative receptor binding radioautography and in situ hybridization histochemistry. Neuroscience 1992;48:655–668. [DOI] [PubMed] [Google Scholar]
- 36. Tsou K, Brown S, Sañudo‐Peña MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 1998;83:393–411. [DOI] [PubMed] [Google Scholar]
- 37. Seutin V. Dopaminergic neurones: Much more than dopamine? Br J Pharmacol 2005;146:167–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Starowicz K, Nigam S, Di Marzo V. Biochemistry and pharmacology of endovanilloids. Pharmacol Ther 2007;114:13–33. [DOI] [PubMed] [Google Scholar]
- 39. Mezey E, Toth ZE, Cortright DN, et al Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1‐like immunoreactivity, in the central nervous system of the rat and human. Proc Natl Acad Sci USA 2000;97:3655–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tzavara ET, Li DL, Moutsimilli L, et al Endocannabinoids activate transient receptor potential vanilloid 1 receptors to reduce hyperdopaminergia‐related hyperactivity: Therapeutic implications. Biol Psychiatry 2006;59:508–515. [DOI] [PubMed] [Google Scholar]
- 41. Micale V, Cristino L, Tamburella A, Petrosino S, Leggio GM, Drago F, Di Marzo V. Altered responses of dopamine D3 receptor null mice to excitotoxic or anxiogenic stimuli: Possible involvement of the endocannabinoid and endovanilloid systems. Neurobiol Dis 2009;36:70–80. [DOI] [PubMed] [Google Scholar]
- 42. Hu SS, Bradshaw HB, Benton VM, et al The biosynthesis of N‐arachidonoyl dopamine (NADA), a putative endocannabinoid and endovanilloid, via conjugation of arachidonic acid with dopamine. Prostaglandins Leukot Essent Fatty Acids 2009;81:291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bobrov MY, Lizhin AA, Andrianova EL, Gretskaya NM, Frumkina LE, Khaspekov LG, Bezuglov VV. Antioxidant and neuroprotective properties of N‐arachidonoyldopamine. Neurosci Lett 2008;431:6–11. [DOI] [PubMed] [Google Scholar]
- 44. Oz M, Jaligam V, Galadari S, Petroianu G, Shuba YM, Shippenberg TS. The endogenous cannabinoid, anandamide, inhibits dopamine transporter function by a receptor‐independent mechanism. J Neurochem 2010;112:1454–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wenger T, Moldrich G. The role of endocannabinoids in the hypothalamic regulation of visceral function. Prostaglandins Leukot Essent Fatty Acids 2002;66:301–307. [DOI] [PubMed] [Google Scholar]
- 46. Costanzi M, Battaglia M, Rossi‐Arnaud C, Cestari V, Castellano C. Effects of anandamide and morphine combinations on memory consolidation in cd1 mice: Involvement of dopaminergic mechanisms. Neurobiol Learn Mem 2004;81:144–149. [DOI] [PubMed] [Google Scholar]
- 47. Nasehi M, Sahebgharani M, Haeri‐Rohani A, Zarrindast MR. Effects of cannabinoids infused into the dorsal hippocampus upon memory formation in 3‐days apomorphine‐treated rats. Neurobiol Learn Mem 2009;92:391–399. [DOI] [PubMed] [Google Scholar]
- 48. Murillo‐Rodríguez E, Vázquez E, Millán‐Aldaco D, Palomero‐Rivero M, Drucker‐Colin R. Effects of the fatty acid amide hydrolase inhibitor URB597 on the sleep‐wake cycle, c‐Fos expression and dopamine levels of the rat. Eur J Pharmacol 2007;562:82–91. [DOI] [PubMed] [Google Scholar]
- 49. Yazulla S. Endocannabinoids in the retina: From marijuana to neuroprotection. Prog Retin Eye Res 2008;27:501–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stefano GB, Salzet B, Rialas CM, et al Morphine‐ and anandamide‐stimulated nitric oxide production inhibits presynaptic dopamine release. Brain Res 1997;763:63–68. [DOI] [PubMed] [Google Scholar]
- 51. Smith Y, Villalba R. Striatal and extrastriatal dopamine in the basal ganglia: An overview of its anatomical organization in normal and Parkinsonian brains. Mov Disord 2008;23:S534–S547. [DOI] [PubMed] [Google Scholar]
- 52. Obeso JA, Marin C, Rodriguez‐Oroz C, et al The basal ganglia in Parkinson's disease: Current concepts and unexplained observations. Ann Neurol 2008;64:S30–S46. [DOI] [PubMed] [Google Scholar]
- 53. Herkenham M, Lynn AB, De Costa BR, Richfield EK. Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res 1991;547;267–264. [DOI] [PubMed] [Google Scholar]
- 54. Fusco FR, Martorana A, Giampa C, et al Immunolocalization of CB1 receptor in rat striatal neurons: A confocal microscopy study. Synapse 2004;53:159–167. [DOI] [PubMed] [Google Scholar]
- 55. Uchigashima M, Narushima M, Fukaya M, Katona I, Kano M, Watanabe M. Subcellular arrangement of molecules for 2‐arachidonoyl‐glycerol‐mediated retrograde signaling and its physiological contribution to synaptic modulation in the striatum. J Neurosci 2007;27:3663–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Köfalvi A, Rodrigues RJ, Ledent C, Mackie K, Vizi ES, Cunha RA, Sperlagh B. Involvement of cannabinoid receptors in the regulation of neurotransmitter release in the rodent striatum: A combined immunochemical and pharmacological analysis. J Neurosci 2005;25:2874–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lau T, Schloss P. The cannabinoid CB1 receptor is expressed on serotonergic and dopaminergic neurons. Eur J Pharmacol 2008;578:137–141. [DOI] [PubMed] [Google Scholar]
- 58. Moss DE, McMaster SB, Rogers J. Tetrahydrocannabinol potentiates reserpine‐induced hypokinesia. Pharmacol Biochem Behav 1981;15:779–783. [DOI] [PubMed] [Google Scholar]
- 59. Anderson JJ, Kask AM, Chase TN. Effects of cannabinoid receptor stimulation and blockade on catalepsy produced by dopamine receptor antagonists. Eur J Pharmacol 1996;295:163–168. [DOI] [PubMed] [Google Scholar]
- 60. Marcellino D, Carriba P, Filip M, et al Antagonistic cannabinoid CB1/dopamine D2 receptor interactions in striatal CB1/D2 heteromers. A combined neurochemical and behavioral analysis. Neuropharmacology 2008;54:815–823. [DOI] [PubMed] [Google Scholar]
- 61. Gorriti MA, Rodriguez de Fonseca F, Navarro M, Palomo T. Chronic (−)‐Δ9‐tetrahydrocannabinol treatment induces sensitization to the psychomotor effects of amphetamine in rats. Eur J Pharmacol 1999;365:133–142. [DOI] [PubMed] [Google Scholar]
- 62. Marinelli S, Di Marzo V, Florenzano F, et al N‐arachidonoyl‐dopamine tunes synaptic transmission onto dopaminergic neurons by activating both cannabinoid and vanilloid receptors. Neuropsychopharmacology 2007;32:298–308. [DOI] [PubMed] [Google Scholar]
- 63. Di Marzo V, Lastres‐Becker I, Bisogno T, De Petrocellis L, Milone A, Davis JB, Fernandez‐Ruiz JJ. Hypolocomotor effects in rats of capsaicin and two long chain capsaicin homologues. Eur J Pharmacol 2001;420:123–131. [DOI] [PubMed] [Google Scholar]
- 64. De Lago E, De Miguel , Lastres‐Becker I, Ramos JA, Fernández‐Ruiz JJ. Involvement of vanilloid‐like receptors in the effects of anandamide on motor behavior and nigrostriatal dopaminergic activity: In vivo and in vitro evidence. Brain Res 2004;1007:152–159. [DOI] [PubMed] [Google Scholar]
- 65. Marinelli S, Di Marzo V, Berretta N, Matias I, Maccarrone M, Bernardi G, Mercuri NB. Presynaptic facilitation of glutamatergic synapses to dopaminergic neurons of the rat substantia nigra by endogenous stimulation of vanilloid receptors. J Neurosci 2003;23:3136–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lastres‐Becker I, Hansen HH, Berrendero F, et al Alleviation of motor hyperactivity and neurochemical deficits by endocannabinoid uptake inhibition in a rat model of Huntington's disease. Synapse 2002;44:23–35. [DOI] [PubMed] [Google Scholar]
- 67. Lastres‐Becker I, De Miguel R, De Petrocellis L, Makriyannis A, Di Marzo V, Fernández‐Ruiz JJ. Compounds acting at the endocannabinoid and/or endovanilloid systems reduce hyperkinesia in a rat model of Huntington's disease. J Neurochem 2003;84:1097–1109. [DOI] [PubMed] [Google Scholar]
- 68. Ferreira SG, Lomaglio T, Avelino A, Cruz F, Oliveira CR, Cunha RA, Köfalvi A. N‐acyldopamines control striatal input terminals via novel ligand‐gated cation channels. Neuropharmacology 2009;56:676–683. [DOI] [PubMed] [Google Scholar]
- 69. Hermann H, Marsicano G, Lutz B. Coexpression of the cannabinoid receptor type 1 with dopamine and serotonin receptors in distinct neuronal subpopulations of the adult mouse forebrain. Neuroscience 2002;109:451–460. [DOI] [PubMed] [Google Scholar]
- 70. Martín AB, Fernandez‐Espejo E, Ferrer B, et al Expression and function of CB1 receptor in the rat striatum: Localization and effects on D1 and D2 dopamine receptor‐mediated motor behaviors. Neuropsychopharmacology 2008;33:1667–1679. [DOI] [PubMed] [Google Scholar]
- 71. Giuffrida A, Parsons LH, Kerr TM, Rodriguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci 1999;2:358–363. [DOI] [PubMed] [Google Scholar]
- 72. Meschler JP, Howlett AC. Signal transduction interactions between CB1 cannabinoid and dopamine receptors in the rat and monkey striatum. Neuropharmacology 2001;40:918–926. [DOI] [PubMed] [Google Scholar]
- 73. Ferré S, Goldberg SR, Lluis C, Franco R. Looking for the role of cannabinoid receptor heteromers in striatal function. Neuropharmacology 2009;56:226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Andersson M, Usiello A, Borgkvist A, et al Cannabinoid action depends on phosphorylation of dopamine‐ and cAMP‐regulated phosphoprotein of 32 kDa at the protein kinase A site in striatal projection neurons. J Neurosci 2005;25:8432–8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gonzalez B, Paz F, Florán L, Aceves J, Erlij D, Florán B. Cannabinoid agonists stimulate [3H]GABA release in the globus pallidus of the rat when Gi protein‐receptor coupling is restricted: Role of dopamine D2 receptors. J Pharmacol Exp Ther 2009;328:822–828. [DOI] [PubMed] [Google Scholar]
- 76. Kreitzer AC, Malenka RC. Endocannabinoid‐mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature 2007;445:643–647. [DOI] [PubMed] [Google Scholar]
- 77. Pan B, Hillard CJ, Liu QS. D2 dopamine receptor activation facilitates endocannabinoid‐mediated long‐term synaptic depression of GABAergic synaptic transmission in midbrain dopamine neurons via cAMP‐protein kinase A signaling. J Neurosci 2008;28:14018–14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mailleux P, Vanderhaeghen JJ. Dopaminergic regulation of cannabinoid receptor mRNA levels in the rat caudate‐putamen: An in situ hybridization study. J Neurochem 1993;61:1705–1712. [DOI] [PubMed] [Google Scholar]
- 79. Gubellini P, Picconi B, Bari M, et al Experimental parkinsonism alters endocannabinoid degradation: Implications for striatal glutamatergic transmission. J Neurosci 2002;22:6900–6907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lastres‐Becker I, Cebeira M, De Ceballos M, Zeng B‐Y, Jenner P, Ramos JA, Fernández‐Ruiz J. Increased cannabinoid CB1 receptor binding and activation of GTP‐binding proteins in the basal ganglia of patients with Parkinson's syndrome and of MPTP‐treated marmosets. Eur J Neurosci 2001;14:1827–1832. [DOI] [PubMed] [Google Scholar]
- 81. Di Marzo V, Hill MP, Bisogno T, Crossman AR, Brotchie JM. Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of Parkinson's disease. FASEB J 2000;14:1432–1438. [DOI] [PubMed] [Google Scholar]
- 82. García‐Arencibia M, García C, Fernández‐Ruiz J. Cannabinoids and Parkinson's disease. CNS Neurol Disord Drug Targets 2009;8:432–439. [DOI] [PubMed] [Google Scholar]
- 83. Sieradzan KA, Fox SH, Hill M, Dick JP, Crossman AR, Brotchie JM. Cannabinoids reduce levodopa‐induced dyskinesia in Parkinson's disease: A pilot study. Neurology 2001;57:2108–2111. [DOI] [PubMed] [Google Scholar]
- 84. Fox SH, Kellett M, Moore AP, Crossman AR, Brotchie JM. Randomised, double‐blind, placebo‐controlled trial to assess the potential of cannabinoid receptor stimulation in the treatment of dystonia. Mov Disord 2002;17:145–149. [DOI] [PubMed] [Google Scholar]
- 85. Müller‐Vahl KR, Schneider U, Koblenz A, Jobges M, Kolbe H, Daldrup T, Emrich HM. Treatment of Tourette's syndrome with Δ9‐tetrahydrocannabinol (THC): A randomized crossover trial. Pharmacopsychiatry 2002;35:57–61. [DOI] [PubMed] [Google Scholar]
- 86. Müller‐Vahl KR, Schneider U, Prevedel H, Theloe K, Kolbe H, Daldrup T, Emrich HM. Δ9‐Tetrahydrocannabinol (THC) is effective in the treatment of tics in Tourette syndrome: A 6‐week randomized trial. J Clin Psychiatry 2003;64:459–465. [DOI] [PubMed] [Google Scholar]
- 87. Mesnage V, Houeto JL, Bonnet AM, et al Neurokinin B, neurotensin, and cannabinoid receptor antagonists and Parkinson's disease. Clin Neuropharmacol 2004;27:108–110. [DOI] [PubMed] [Google Scholar]
- 88. Frankel JP, Hughes A, Lees AJ, Stern GM. Marijuana for parkinsonian tremor. J Neurol Neurosurg Psychiatry 1990;53:436–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Pazos MR, Sagredo O, Fernández‐Ruiz J. The endocannabinoid system in Huntington's disease. Curr Pharm Des 2008;14:2317–2325. [DOI] [PubMed] [Google Scholar]
- 90. Müller‐Vahl KR. Cannabinoids reduce symptoms of Tourette's syndrome. Expert Opin Pharmacother 2003;4:1717–1725. [DOI] [PubMed] [Google Scholar]
- 91. Jabusch HC, Schneider U, Altenmüller E. Δ9‐tetrahydrocannabinol improves motor control in a patient with musician's dystonia. Mov Disord 2004;19:990–991. [DOI] [PubMed] [Google Scholar]
- 92. Fabbrini G, Brotchie JM, Grandas F, Nomoto M, Goetz CG. Levodopa‐induced dyskinesias. Mov Disord 2007;22:1379–1389. [DOI] [PubMed] [Google Scholar]
- 93. Brotchie JM. CB1 cannabinoid receptor signalling in Parkinson's disease. Curr Opin Pharmacol 2003;3:54–61. [DOI] [PubMed] [Google Scholar]
- 94. Sañudo‐Peña MC, Tsou K, Walker JM. Motor actions of cannabinoids in the basal ganglia output nuclei. Life Sci 1999;65:703–713. [DOI] [PubMed] [Google Scholar]
- 95. Ferrer B, Asbrock N, Kathuria S, Piomelli D, Giuffrida A. Effects of levodopa on endocannabinoid levels in rat basal ganglia: Implications for the treatment of levodopa‐induced dyskinesias. Eur J Neurosci 2003;18:1607–1614. [DOI] [PubMed] [Google Scholar]
- 96. Morgese MG, Cassano T, Cuomo V, Giuffrida A. Anti‐dyskinetic effects of cannabinoids in a rat model of Parkinson's disease: Role of CB1 and TRPV1 receptors. Exp Neurol 2007;208:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Carroll CB, Bain PG, Teare L, et al Cannabis for dyskinesia in Parkinson disease: A randomized double‐blind crossover study. Neurology 2004;63:1245–1250. [DOI] [PubMed] [Google Scholar]
- 98. Lastres‐Becker I, Molina‐Holgado F, Ramos JA, Mechoulam R, Fernández‐Ruiz J. Cannabinoids provide neuroprotection against 6‐hydroxydopamine toxicity in vivo and in vitro: Relevance to Parkinson's disease. Neurobiol Dis 2005;19:96–107. [DOI] [PubMed] [Google Scholar]
- 99. García‐Arencibia M, González S, De Lago E, Ramos JA, Mechoulam R, Fernández‐Ruiz J. Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson's disease: Importance of antioxidant and cannabinoid receptor‐independent properties. Brain Res 2007;1134:162–170. [DOI] [PubMed] [Google Scholar]
- 100. Venderová K, Růzicka E, Vorísek V, Visnovský P. Survey on cannabis use in Parkinson's disease: Subjective improvement of motor symptoms. Mov Disord 2004;19:1102–1106. [DOI] [PubMed] [Google Scholar]
- 101. Price DA, Martinez AA, Seillier A, et al WIN55,212–2, a cannabinoid receptor agonist, protects against nigrostriatal cell loss in the 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine mouse model of Parkinson's disease. Eur J Neurosci 2009;29:2177–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Pérez‐Rial S, García‐Gutiérrez MS, Molina JA, et al Increased vulnerability to 6‐hydroxydopamine lesion and reduced development of dyskinesias in mice lacking CB1 cannabinoid receptors. Neurobiol Aging 2009; in press. [DOI] [PubMed] [Google Scholar]
- 103. Fernández‐Espejo E, Caraballo I, De Fonseca FR, El Banoua F, Ferrer B, Flores JA, Galan‐Rodriguez B. Cannabinoid CB1 antagonists possess antiparkinsonian efficacy only in rats with very severe nigral lesion in experimental parkinsonism. Neurobiol Dis 2005;18:591–601. [DOI] [PubMed] [Google Scholar]
- 104. González S, Scorticati C, Garcia‐Arencibia M, De Miguel R, Ramos JA, Fernández‐Ruiz J. Effects of rimonabant, a selective cannabinoid CB1 receptor antagonist, in a rat model of Parkinson's disease. Brain Res 2006;1073–1074:209–219. [DOI] [PubMed] [Google Scholar]
- 105. Kelsey JE, Harris O, Cassin J. The CB1 antagonist rimonabant is adjunctively therapeutic as well as monotherapeutic in an animal model of Parkinson's disease. Behav Brain Res 2009;203:304–307. [DOI] [PubMed] [Google Scholar]
- 106. Meschler JP, Howlett AC, Madras BK. Cannabinoid receptor agonist and antagonist effects on motor function in normal and 1‐methyl‐4‐phenyl‐1,2,5,6‐tetrahydropyridine (MPTP)‐treated non‐human primates. Psychopharmacology 2001;156:79–85. [DOI] [PubMed] [Google Scholar]
- 107. Cao X, Liang L, Hadcock JR, et al Blockade of cannabinoid type 1 receptors augments the antiparkinsonian action of levodopa without affecting dyskinesias in 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine‐treated rhesus monkeys. J Pharmacol Exp Ther 2007;323:318–326. [DOI] [PubMed] [Google Scholar]
- 108. García‐Arencibia M, Ferraro L, Tanganelli S, Fernández‐Ruiz J. Enhanced striatal glutamate release after the administration of rimonabant to 6‐hydroxydopamine‐lesioned rats. Neurosci Lett 2008;438:10–13. [DOI] [PubMed] [Google Scholar]
- 109. García‐Arencibia M, García C, Kurz A, et al Cannabinoid CB1 receptors are early down‐regulated followed by a further up‐regulation in the basal ganglia of mice with deletion of specific park genes. J Neural Transm Suppl 2009;73:269–275. [DOI] [PubMed] [Google Scholar]
- 110. Pisani A, Fezza F, Galati S, et al High endogenous cannabinoid levels in the cerebrospinal fluid of untreated Parkinson's disease patients. Ann Neurol 2005;57:777–7779. [DOI] [PubMed] [Google Scholar]
- 111. Maccarrone M, Gubellini P, Bari M, et al Levodopa treatment reverses endocannabinoid system abnormalities in experimental parkinsonism. J Neurochem 2003;85:1018–1025. [DOI] [PubMed] [Google Scholar]
- 112. Nieoullon A. Dopamine and the regulation of cognition and attention. Prog Neurobiol 2002;67:53–83. [DOI] [PubMed] [Google Scholar]
- 113. Wise RA. Dopamine and reward: The anhedonia hypothesis 30 years on. Neurotox Res 2008;14:169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Pierce RC, Kumaresan V. The mesolimbic dopamine system: The final common pathway for the reinforcing effect of drugs of abuse? Neurosci Biobehav Rev 2006;30:215–238. [DOI] [PubMed] [Google Scholar]
- 115. Gardner EL. Endocannabinoid signaling system and brain reward: Emphasis on dopamine. Pharmacol Biochem Behav 2005;81:263–284. [DOI] [PubMed] [Google Scholar]
- 116. Fattore L, Fadda P, Spano MS, Pistis M, Fratta W. Neurobiological mechanisms of cannabinoid addiction. Mol Cell Endocrinol 2008;286:S97–S107. [DOI] [PubMed] [Google Scholar]
- 117. Diana M, Melis M, Gessa GL. Increase in meso‐prefrontal dopaminergic activity after stimulation of CB1 receptors by cannabinoids. Eur J Neurosci 1998;10:2825–2830. [DOI] [PubMed] [Google Scholar]
- 118. Tanda G, Pontieri FE, Di Chiara G. Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science 1997;276:2048–2050. [DOI] [PubMed] [Google Scholar]
- 119. Diana M, Melis M, Muntoni AL, Gessa GL. Mesolimbic dopaminergic decline after cannabinoid withdrawal. Proc Nat Acad Sci USA 1998;95:10269–10273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Lupica CR, Riegel AC, Hoffman AF. Marijuana and cannabinoid regulation of brain reward circuits. Br J Pharmacol 2004;143:227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Lupica CR, Riegel AC. Endocannabinoid release from midbrain dopamine neurons: A potential substrate for cannabinoid receptor antagonist treatment of addiction. Neuropharmacology 2005;48:1105–1116. [DOI] [PubMed] [Google Scholar]
- 122. Cheer JF, Kendall DA, Mason R, Marsden CA. Differential cannabinoid‐induced electrophysiological effects in rat ventral tegmentum. Neuropharmacology 2003;44:633–641. [DOI] [PubMed] [Google Scholar]
- 123. Pistis M, Porcu G, Melis M, Diana M, Gessa GL. Effects of cannabinoids on prefrontal neuronal responses to ventral tegmental area stimulation. Eur J Neurosci 2001;14:96–102. [DOI] [PubMed] [Google Scholar]
- 124. Pistis M, Muntoni AL, Pillolla G, Gessa GL. Cannabinoids inhibit excitatory inputs to neurons in the shell of the nucleus accumbens: An in vivo electrophysiological study. Eur J Neurosci 2002;15:1795–1802. [DOI] [PubMed] [Google Scholar]
- 125. Massi L, Elezgarai I, Puente N, Reguero L, Grandes P, Manzoni OJ, Georges F. Cannabinoid receptors in the bed nucleus of the stria terminalis control cortical excitation of midbrain dopamine cells in vivo. J Neurosci 2008;28:10496–10508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Iversen L. Cannabis and the brain. Brain 2003;126:1252–1270. [DOI] [PubMed] [Google Scholar]
- 127. Henquet C, Di Forti M, Morrison P, Kuepper R, Murray RM. Gene‐environment interplay between cannabis and psychosis. Schizophr Bull 2008;34:1111–11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Hall W, Degenhardt L. Cannabis use and the risk of developing a psychotic disorder. World Psychiatry 2008;7:68–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. D'Souza DC. Cannabinoids and psychosis. Int Rev Neurobiol 2007;78:289–326. [DOI] [PubMed] [Google Scholar]
- 130. González S, Cebeira M, Fernández‐Ruiz J. Cannabinoid tolerance and dependence: A review of studies in laboratory animals. Pharmacol Biochem Behav 2005;81:300–318. [DOI] [PubMed] [Google Scholar]
- 131. Parolaro D, Rubino T. The role of the endogenous cannabinoid system in drug addiction. Drug News Perspect 2008;21:149–157. [PubMed] [Google Scholar]
- 132. Maldonado R, Valverde O, Berrendero F. Involvement of the endocannabinoid system in drug addiction. Trends Neurosci 2006;29:225–232. [DOI] [PubMed] [Google Scholar]
- 133. González S, Fernández‐Ruiz JJ, Sparpaglione V, Parolaro D, Ramos JA. Chronic exposure to morphine, cocaine or ethanol in rats produced different effects in brain cannabinoid CB1 receptor binding and mRNA levels. Drug Alcoh Depend 2002;66:77–84. [DOI] [PubMed] [Google Scholar]
- 134. González S, Schmid PC, Fernández‐Ruiz JJ, Krebsbach R, Schmid HHO, Ramos JA. Region‐dependent changes in endocannabinoid transmission in the brain of morphine‐dependent rats. Addict Biol 2003;8:159–166. [DOI] [PubMed] [Google Scholar]
- 135. Vigano D, Cascio MG, Rubino T, Fezza F, Vaccani A, Di Marzo V, Parolaro D. Chronic morphine modulates the contents of the endocannabinoid, 2‐arachidonoyl glycerol, in rat brain. Neuropsychopharmacol 2003;28:1160–1167. [DOI] [PubMed] [Google Scholar]
- 136. González S, Cascio MG, Fernandez‐Ruiz J, Fezza F, Di Marzo V, Ramos JA. Changes in endocannabinoid contents in the brain of rats chronically exposed to nicotine, ethanol or cocaine. Brain Res 2002;954:73–81. [DOI] [PubMed] [Google Scholar]
- 137. Basavarajappa BS, Hungund BL. Neuromodulatory role of the endocannabinoid signaling system in alcoholism: An overview. Prostag Leukot Essent Fatty Acids 2002;66:287–299. [DOI] [PubMed] [Google Scholar]
- 138. Rubio M, Fernández‐Ruiz J, De Miguel R, Maestro B, Michael Walker J, Ramos JA. CB1 receptor blockade reduces the anxiogenic‐like response and ameliorates the neurochemical imbalances associated with alcohol withdrawal in rats. Neuropharmacology 2008;54:976–988. [DOI] [PubMed] [Google Scholar]
- 139. Rubio M, McHugh D, Fernández‐Ruiz J, Bradshaw H, Walker JM. Short‐term exposure to alcohol in rats affects brain levels of anandamide, other N‐acylethanolamines and 2‐arachidonoyl‐glycerol. Neurosci Lett 2007;421:270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Fernández‐Ruiz J, De Miguel R, Hernández ML, Cebeira M, Ramos JA. Endocannabinoids and dopamine‐related functions in the CNS In: Onaivi ES, Sugiura T, Di Marzo V, editors. Endocannabinoids: The brain and body's marijuana and beyond. Boca Raton , FL : CRC Press, 2006;261–290. [Google Scholar]
- 141. Hungund BL, Szakall I, Adam A, Basavarajappa BS, Vadasz C. Cannabinoid CB1 receptor knockout mice exhibit markedly reduced voluntary alcohol consumption and lack alcohol‐induced dopamine release in the nucleus accumbens. J Neurochem 2003;84:698–704. [DOI] [PubMed] [Google Scholar]
- 142. Mascia MS, Obinu MC, Ledent C, Parmentier M, Bohme GA, Imperato A, Fratta W. Lack of morphine‐induced dopamine release in the nucleus accumbens of cannabinoid CB1 receptor knockout mice. Eur J Pharmacol 1999;383:R1‐R2. [DOI] [PubMed] [Google Scholar]
- 143. Cossu G, Ledent C, Fattore L, Imperato A, Bohme GA, Parmentier M, Fratta W. Cannabinoid CB1 receptor knockout mice fail to self‐administer morphine but not other drugs of abuse. Behav Brain Res 2001;118:61–65. [DOI] [PubMed] [Google Scholar]
- 144. Li X, Hoffman AF, Peng XQ, Lupica CR, Gardner EL, Xi ZX. Attenuation of basal and cocaine‐enhanced locomotion and nucleus accumbens dopamine in cannabinoid CB1‐receptor‐knockout mice. Psychopharmacology (Berl) 2009;204:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Castañé A, Valjent E, Ledent C, Parmentier M, Maldonado R, Valverde O. Lack of CB1 cannabinoid receptors modifies nicotine behavioural responses, but not nicotine abstinence. Neuropharmacology 2002;43:857–867. [DOI] [PubMed] [Google Scholar]
- 146. Cohen C, Perrault G, Voltz C, Steinberg R, Soubrie P. SR141716, a central cannabinoid (CB1) receptor antagonist, blocks the motivational and dopamine‐releasing effects of nicotine in rats. Behav Pharmacol 2002;13:451–463. [DOI] [PubMed] [Google Scholar]
- 147. Cheer JF, Wassum KM, Sombers LA, et al Phasic dopamine release evoked by abused substances requires cannabinoid receptor activation. J Neurosci 2007;27:791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Melis M, Pillolla G, Luchicchi A, Muntoni AL, Yasar S, Goldberg SR, Pistis M. Endogenous fatty acid ethanolamides suppress nicotine‐induced activation of mesolimbic dopamine neurons through nuclear receptors. J Neurosci 2008;28:13985–13994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Egerton A, Allison C, Brett RR, Pratt JA. Cannabinoids and prefrontal cortical function: Insights from preclinical studies. Neurosci Biobehav Rev 2006;30:680–695. [DOI] [PubMed] [Google Scholar]
- 150. Pistis M, Ferraro L, Pira L, Flore G, Tanganelli S, Gessa GL, Devoto P. Δ9‐tetrahydrocannabinol decreases extracellular GABA and increases extracellular glutamate and dopamine levels in the rat prefrontal cortex: An in vivo microdialysis study. Brain Res 2002;948:155–158. [DOI] [PubMed] [Google Scholar]
- 151. Jentsch JD, Verrico CD, Le D, Roth RH. Repeated exposure to Δ9‐tetrahydrocannabinol reduces prefrontal cortical dopamine metabolism in the rat. Neurosci Lett 1998;246:169–172. [DOI] [PubMed] [Google Scholar]
- 152. Verrico CD, Jentsch JD, Roth RH. Persistent and anatomically selective reduction in prefrontal cortical dopamine metabolism after repeated, intermittent cannabinoid administration to rats. Synapse 2003;49:61–66. [DOI] [PubMed] [Google Scholar]
- 153. Szabo B, Muller T, Koch H. Effects of cannabinoids on dopamine release in the corpus striatum and the nucleus accumbens in vitro. J Neurochem 1999;73:1084–1089. [DOI] [PubMed] [Google Scholar]
- 154. Szabo B, Siemes S, Wallmichrath I. Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. Eur J Neurosci 2002;15:2057–2061. [DOI] [PubMed] [Google Scholar]
- 155. Cheer JF, Marsden CA, Kendall DA, Mason R. Lack of response suppression follows repeated ventral tegmental cannabinoid administration: An in vitro electrophysiological study. Neuroscience 2000;99:661–667. [DOI] [PubMed] [Google Scholar]
- 156. Hoffman AF, Lupica CR. Direct actions of cannabinoids on synaptic transmission in the nucleus accumbens: A comparison with opioids. J Neurophysiol 2001;85:72–83. [DOI] [PubMed] [Google Scholar]
- 157. Manzoni OJ, Bockaert J. Cannabinoids inhibit GABAergic synaptic transmission in mice nucleus accumbens. Eur J Pharmacol 2001;412:R3–R5. [DOI] [PubMed] [Google Scholar]
- 158. Sperlágh B, Windisch K, Andó RD, Sylvester Vizi E. Neurochemical evidence that stimulation of CB1 cannabinoid receptors on GABAergic nerve terminals activates the dopaminergic reward system by increasing dopamine release in the rat nucleus accumbens. Neurochem Int 2009;54:452–457. [DOI] [PubMed] [Google Scholar]
- 159. Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci 2004;24:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ. Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci 2001;21:109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Ponce G, Hoenicka J, Rubio G, et al Association between cannabinoid receptor gene (CNR1) and childhood attention deficit/hyperactivity disorder in Spanish male alcoholic patients. Mol Psychiatry 2003;8:466–467. [DOI] [PubMed] [Google Scholar]
- 162. Zhang PW, Ishiguro H, Ohtsuki T, et al Human cannabinoid receptor 1: 5′ exons, candidate regulatory regions, polymorphisms, haplotypes and association with polysubstance abuse. Mol Psychiatry 2004;9:916–931. [DOI] [PubMed] [Google Scholar]
- 163. Sipe JC, Chiang K, Gerber AL, Beutler E, Cravatt BF. A missense mutation in human fatty acid amide hydrolase associated with problem drug use. Proc Natl Acad Sci USA 2002;99:8394–8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. De Vries TJ, Homberg JR, Binnekade R, Raaso H, Schoffelmeer AN. Cannabinoid modulation of the reinforcing and motivational properties of heroin and heroin‐associated cues in rats. Psychopharmacology 2003;168:164–169. [DOI] [PubMed] [Google Scholar]
- 165. Fattore L, Spano MS, Cossu G, Deiana S, Fratta W. Cannabinoid mechanism in reinstatement of heroin‐seeking after a long period of abstinence in rats. Eur J Neurosci 2003;17:1723–1726. [DOI] [PubMed] [Google Scholar]
- 166. De Vries TJ, Shaham Y, Homberg JR, et al A cannabinoid mechanism in relapse to cocaine seeking. Nature Med 2001;7:1151–1154. [DOI] [PubMed] [Google Scholar]
- 167. Colombo G, Orrù A, Lai P, et al The cannabinoid CB1 receptor antagonist, rimonabant, as a promising pharmacotherapy for alcohol dependence: Preclinical evidence. Mol Neurobiol 2007;36:102–112. [DOI] [PubMed] [Google Scholar]
- 168. Tanda G, Loddo P, Di Chiara G. Dependence of mesolimbic dopamine transmission on Δ9‐tetrahydrocannabinol. Eur J Pharmacol 1999;376:23–26. [DOI] [PubMed] [Google Scholar]
- 169. Beardsley PM, Thomas BF, McMahon LR. Cannabinoid CB1 receptor antagonists as potential pharmacotherapies for drug abuse disorders. Int Rev Psychiatry 2009;21:134–142. [DOI] [PubMed] [Google Scholar]
- 170. Le Foll B, Goldberg SR. Cannabinoid CB1 receptor antagonists as promising new medications for drug dependence. J Pharmacol Exp Ther 2005;312:875–883. [DOI] [PubMed] [Google Scholar]
- 171. Freedland CS, Sharpe AL, Samson HH, Porrino LJ. Effects of SR141716A on ethanol and sucrose self‐administration. Alcohol Clin Exp Res 2001;25:277–282. [PubMed] [Google Scholar]
- 172. Serra S, Brunetti G, Pani M, Vacca G, Carai MA, Gessa GL, Colombo G. Blockade by the cannabinoid CB1 receptor antagonist, SR 141716, of alcohol deprivation effect in alcohol‐preferring rats. Eur J Pharmacol 2002;443:95–97. [DOI] [PubMed] [Google Scholar]
- 173. Mas‐Nieto M, Pommier B, Tzavara ET, et al Reduction of opioid dependence by the CB1 antagonist SR141716A in mice: Evaluation of the interest in pharmacotherapy of opioid addiction. Br J Pharmacol 2001;132:1809–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Solinas M, Panlilio LV, Antoniou K, Pappas LA, Goldberg SR. The cannabinoid CB1 antagonist N‐piperidinyl‐5‐(4‐chlorophenyl)‐1‐(2,4‐dichlorophenyl)‐4‐methylpyrazole‐3‐carboxamide (SR‐141716A) differentially alters the reinforcing effects of heroin under continuous reinforcement, fixed ratio, and progressive ratio schedules of drug self‐administration in rats. J Pharmacol Exp Ther 2003;306:93–102. [DOI] [PubMed] [Google Scholar]
- 175. Shoaib M. The cannabinoid antagonist AM251 attenuates nicotine self‐administration and nicotine‐seeking behaviour in rats. Neuropharmacology 2008;54:438–444. [DOI] [PubMed] [Google Scholar]
- 176. Cohen C, Kodas E, Griebel G. CB1 receptor antagonists for the treatment of nicotine addiction. Pharmacol Biochem Behav 2005;81:387–395. [DOI] [PubMed] [Google Scholar]
- 177. Xi ZX, Gilbert JG, Peng XQ, Pak AC, Li X, Gardner EL. Cannabinoid CB1 receptor antagonist AM251 inhibits cocaine‐primed relapse in rats: Role of glutamate in the nucleus accumbens. J Neurosci 2006;26:8531–8536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Harris DS, Anthenelli RM. Expanding treatment of tobacco dependence. Curr Psychiatry Rep 2005;7:344–351. [DOI] [PubMed] [Google Scholar]
- 179. Soyka M, Koller G, Schmidt P, et al.; ACTOL Study Investigators . Cannabinoid receptor 1 blocker rimonabant (SR 141716) for treatment of alcohol dependence: Results from a placebo‐controlled, double‐blind trial. J Clin Psychopharmacol 2008;28:317–324. [DOI] [PubMed] [Google Scholar]
- 180. Luzi S, Morrison PD, Powell J, Di Forti M, Murray RM. What is the mechanism whereby cannabis use increases risk of psychosis? Neurotox Res 2008;14:105–112. [DOI] [PubMed] [Google Scholar]
- 181. Müller‐Vahl KR, Emrich HM. Cannabis and schizophrenia: Towards a cannabinoid hypothesis of schizophrenia. Expert Rev Neurother 2008;8:1037–1048. [DOI] [PubMed] [Google Scholar]
- 182. Di Forti M, Morrison PD, Butt A, Murray RM. Cannabis use and psychiatric and cognitive disorders: The chicken or the egg? Curr Opin Psychiatry 2007;20:228–234. [DOI] [PubMed] [Google Scholar]
- 183. Negrete JC, Gill K. Cannabis and schizophrenia In: Nahas GG, Sutin KM, Harvey DJ, Agurell S, editors. Marihuana and medicine. New Jersey : Humana Press, 1999;671–683. [Google Scholar]
- 184. Knudsen P, Vilmar T. Cannabis and neuroleptic agents in schizophrenia. Acta Psychiatr Scand 1984;69:162–174. [DOI] [PubMed] [Google Scholar]
- 185. Leroy S, Griffon N, Bourdel MC, Olie JP, Poirier MF, Krebs MO. Schizophrenia and the cannabinoid receptor type 1 (CB1): Association study using a single‐base polymorphism in coding exon 1. Am J Med Genet 2001;105:749–752. [DOI] [PubMed] [Google Scholar]
- 186. Ujike H, Takaki M, Nakata K, et al CNR1, central cannabinoid receptor gene, associated with susceptibility to hebephrenic schizophrenia. Mol Psychiatry 2002;7:515–518. [DOI] [PubMed] [Google Scholar]
- 187. Martínez‐Gras I, Hoenicka J, Ponce G, et al (AAT)n repeat in the cannabinoid receptor gene, CNR1: Association with schizophrenia in a Spanish population. Eur Arch Psychiatry Clin Neurosci 2006;256:437–441. [DOI] [PubMed] [Google Scholar]
- 188. Voruganti LN, Slomka P, Zabel P, Mattar A, Awad AG. Cannabis induced dopamine release: An in‐vivo SPECT study. Psychiatry Res 2001;107:173–177. [DOI] [PubMed] [Google Scholar]
- 189. Bossong MG, Van Berckel BN, Boellaard R, et al Δ9‐tetrahydrocannabinol induces dopamine release in the human striatum. Neuropsychopharmacology 2009;34:759–766. [DOI] [PubMed] [Google Scholar]
- 190. Stokes PR, Mehta MA, Curran HV, Breen G, Grasby PM. Can recreational doses of THC produce significant dopamine release in the human striatum? Neuroimage 2009;48:186–190. [DOI] [PubMed] [Google Scholar]
- 191. Zuardi AW, Crippa JA, Hallak JE, Moreira FA, Guimarães FS. Cannabidiol, a Cannabis sativa constituent, as an antipsychotic drug. Braz J Med Biol Res 2006;39:421–429. [DOI] [PubMed] [Google Scholar]
- 192. Ballmaier M, Bortolato M, Rizzetti C, Zoli M, Gessa G, Heinz A, Spano P. Cannabinoid receptor antagonists counteract sensorimotor gating deficits in the phencyclidine model of psychosis. Neuropsychopharmacology 2007;32:2098–2107. [DOI] [PubMed] [Google Scholar]
- 193. Tzavara ET, Degroot A, Wade MR, Davis RJ, Nomikos GG. CB1 receptor knockout mice are hyporesponsive to the behavior‐stimulating actions of d‐amphetamine: Role of mGlu5 receptors. Eur Neuropsychopharmacol 2009;19:196–204. [DOI] [PubMed] [Google Scholar]
- 194. Leweke FM, Giuffrida A, Wurster U, Emrich HM, Piomelli D. Elevated endogenous cannabinoids in schizophrenia. Neuroreport 1999;10:1665–1669. [DOI] [PubMed] [Google Scholar]
- 195. Giuffrida A, Leweke FM, Gerth CW, et al Cerebrospinal anandamide levels are elevated in acute schizophrenia and are inversely correlated with psychotic symptoms. Neuropsychopharmacology 2004;29:2108–2114. [DOI] [PubMed] [Google Scholar]
- 196. De Marchi N, De Petrocellis L, Orlando P, Daniele F, Fezza F, Di Marzo V. Endocannabinoid signalling in the blood of patients with schizophrenia. Lipids Health Dis 2003;2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Dean B, Sundram S, Bradbury R, Scarr E, Copolov D. Studies on [3H]CP‐55940 binding in the human central nervous system: Regional specific changes in density of cannabinoid‐1 receptors associated with schizophrenia and cannabis use. Neuroscience 2001;103:9–15. [DOI] [PubMed] [Google Scholar]
- 198. Zavitsanou K, Garrick T, Huang XF. Selective antagonist [3H]SR141716A binding to cannabinoid CB1 receptors is increased in the anterior cingulate cortex in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2004;28:355–360. [DOI] [PubMed] [Google Scholar]
- 199. Newell KA, Deng C, Huang XF. Increased cannabinoid receptor density in the posterior cingulate cortex in schizophrenia. Exp Brain Res 2006;172:556–560. [DOI] [PubMed] [Google Scholar]
- 200. Berding G, Schneider U, Gielow P, et al Feasibility of central cannabinoid CB1 receptor imaging with [124I]AM281 PET demonstrated in a schizophrenic patient. Psychiatry Res 2006;147:249–256. [DOI] [PubMed] [Google Scholar]
- 201. Koethe D, Llenos IC, Dulay JR, Hoyer C, Torrey EF, Leweke FM, Weis S. Expression of CB1 cannabinoid receptor in the anterior cingulate cortex in schizophrenia, bipolar disorder, and major depression. J Neural Transm 2007;114:1055–1063. [DOI] [PubMed] [Google Scholar]
- 202. Eggan SM, Hashimoto T, Lewis DA. Reduced cortical cannabinoid 1 receptor messenger RNA and protein expression in schizophrenia. Arch Gen Psychiatry 2008;65:772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Tzavara ET, Davis RJ, Perry KW, et al The CB1 receptor antagonist SR141716A selectively increases monoaminergic neurotransmission in the medial prefrontal cortex: Implications for therapeutic actions. Br J Pharmacol 2003;138:544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Meltzer HY, Arvanitis L, Bauer D, Rein W; Meta‐Trial Study Group . Placebo‐controlled evaluation of four novel compounds for the treatment of schizophrenia and schizoaffective disorder. Am J Psychiatry 2004;161:975–984. [DOI] [PubMed] [Google Scholar]