

SUMMARY
Brain delivery is one of the major challenges for the neuropharmaceutical industry since an alarming increase in brain disease incidence is going on. Despite major advances in neuroscience, many potential therapeutic agents are denied access to the central nervous system (CNS) because of the existence of a physiological low permeable barrier, the blood–brain barrier (BBB). To obtain an improvement of drug CNS performance, sophisticated approaches such as nanoparticulate systems are rapidly developing. Many recent data demonstrate that drugs could be transported successfully into the brain using colloidal systems after i.v. injection by several mechanisms such as endocytosis or P‐glycoprotein inhibition. This review summarizes the main brain targeted nanoparticulate carriers such as liposomes, lipid nanoparticles, polymeric nanoparticles, and micelles with great potential in drug delivery into the CNS.
Keywords: Alzheimer's disease, Movement disorders/Parkinson's disease, Multiple sclerosis, Neuropsychopharmacology
Introduction
The design of drug delivery systems is an increasingly valuable discipline in pharmaceutical development, allowing rational manipulation of both the pharmacological profiles and concomitant therapeutic indices of drugs [1]. In particular, the development of delivery systems that are able to alter biodistribution, tissue uptake, pharmacokinetics, and pharmacodynamics of therapeutic agents is considered of utmost importance in biomedical research and pharmaceutical industry [2]. The main reason for the extremely rapid growth of research and technology in this field is the realization that substantial improvement of current therapies will necessitate the use of therapeutic modalities allowing for the efficient and site‐specific transport of drugs to the target tissues affected by the disease [3]. This necessity arises primarily due to the enormous barriers that a drug molecule must overcome before it reaches its target site within the body. Generally, the use of colloidal carriers for drug administration may offer several advantages: an increased longevity and stability of the carrier and carrier‐incorporated drug after intravascular administration, an increase of drug bioavailability after oral or parenteral administration, the possibility to obtain a tissue or cellular targeting release, a prolonged activity in target tissue and, above all, the ability to cross physiological barriers. From a pharmacokinetic point of view, the association of drugs with any nanocarrier has pronounced effects: delayed drug absorption, restricted drug biodistribution, decreased volume of drug biodistribution, delayed drug clearance, and retarded drug metabolism. The presence of protective polymer on the carrier surface changes all these parameters still further [4].
Colloidal systems are nanosized structures ranging in size from 1 to 500 nm, which can be obtained starting on polymeric or lipid materials and can encapsulate therapeutic agents, modifying the drug release profile and rate in the human body [1]. Size, surface charge, morphology, and matrix composition play a fundamental role for their in vivo fate and all these features can be modulated according to obtain suitable modified release profile, biological activity, and compatibility. Colloidal systems are administrable for all routes (oral, pulmonary, nasal, and ophthalmic) even for intravenous route being their size below the smallest vessels (5–6 μm in diameter).
Thanks to their potential, a broad range of drug delivery systems is being designed to improve drug delivery for central nervous system (CNS) disorders [5, 6, 7, 8]. The blood–brain barrier (BBB) consists of a monolayer of polarized endothelial cells connected by complex tight junctions. Various cells such as astrocytes, neurons, and pericytes dynamically control the functions of BBB. The brain endothelial cells are separated from these other cells by a basal lamina, whose components such as type IV collagen, laminin, fibronectin, and heparan sulphate may be involved in drug transport. Moreover, the capillaries of the vertebrate brain and spinal cord lack the small pores that allow rapid movement of solutes from circulation into other organs. Microvessels constitute an estimated 95% of the total surface area of the BBB, and they represent the principal route by which chemicals could enter the brain. However, the complex structure of the BBB, the presence of high levels of efflux transport proteins, including P‐glycoprotein (P‐gp) and Multidrug Resistance Protein‐1 (MRP‐1), and the expression of metabolic enzymes pose hurdles for drug–brain entry. The BBB is a unique regulatory system that protects the brain environment by separating it from direct contact with the circulating blood. In doing so, it impedes at the same time the access of a large number of diagnostic and therapeutic agents (belonging to classes such as antibiotics, antiinflammatory and antineoplastic agents, and a variety of CNS—active drugs, especially neuropeptides) into the brain parenchyma in physiological and, often, in pathological conditions. Therefore, restricted permeability of BBB requests more sophisticated approaches with the aim to obtain the improvement of drug performance into the CNS.
One of the possibilities of bypassing this barrier relies on specific properties of nanoparticulate vectors designed to interact with BBB‐forming cells at a molecular level, as a result of which the transport of drugs or other molecules (such as nucleic acids, proteins, or imaging agents) could be achieved without interfering with the normal function of the brain. However, these nanovectors must be optimized to affect their size, shape, and coatings in order to facilitate drug uptake, release and ingress across the BBB.
Size and Surface Properties
The basic property of a drug carrier is longevity in the blood that means an increased stability and half‐life during the course of blood circulation, avoiding glomerular excretion by the kidney and the uptake by the reticulo‐endothelial system (RES) in the liver, spleen, and lung [9]. In fact, because for the body defence system nanocarriers usually represent foreign particles, they become easily opsonized and removed by phagocytosis from the circulation prior to complete their function. This process mainly depends on particle size, charge, and surface properties of the nanocarrier. In general, particles that have a mean diameter of approximatively 100 nm and neutral surface show prolonged blood circulation and a relatively low rate of mononuclear phagocyte system (MPS) uptake [9]. Moreover, chemical modification of nanocarrier surface with certain hydrophilic polymers, such as poly(ethylene glycols) (PEGs), represents the most frequent way to sterically stabilize them by a polymer‐mediated protection and to impart the in vivo longevity to drug carriers [4]. In particular, on the biological level, coating nanocarriers with PEG sterically hinders interactions of blood components with their surface and reduces the adsorption of plasma proteins. PEGs are commonly used as nanoparticulate surface protecting polymers thanks to the fact that they provide a very attractive combination of properties, such as excellent solubility in aqueous solutions, high flexibility of their polymeric chains, low toxicity, immunogenicity, and antigenicity [10, 11]. PEG chains can be physically adsorbed on, or covalently linked to the particle surface, although the use of surface‐adsorbed PEG moieties has the drawback that they could desorb once in the blood stream, leaving holes in the surface coverage where opsonins can bind [12]. PEG covalently bonded to fully formed nanocarriers can be obtained by using PEGylated copolymers or lipids as starting material [6, 13, 14].
Then, long circulating nanocarriers can slowly accumulate in pathological sites with leaky vasculature by the well‐known enhanced permeability and retention‐EPR‐effect, and facilitate drug delivery in those areas [10].
To increase the accumulation specifically in the required pathological site, nanocarriers may be actively targeted by incorporating surface grafted recognition moieties that impart an affinity for cellular receptors or components that are present on and/or upregulated by specific cells [15]. The active targeting allows that nanoparticles either are delivered from the blood into the target tissue by endothelial transcytosis or are kept in the microvessels of the target tissue, which usually occurs within a few hours [9]. Specifically, active targeting to the brain could be achieved by attaching on the outer surface of nanocarriers ligands such as peptides, antibodies, antibody fragments, and proteins, with an enhanced binding affinity for brain specific cellular receptors [6, 7, 8]. In Figure 1, the schematic structure of pharmaceutical nanoparticulate carriers is reported, while in Table 1 their main characteristics are summarized.
Figure 1.
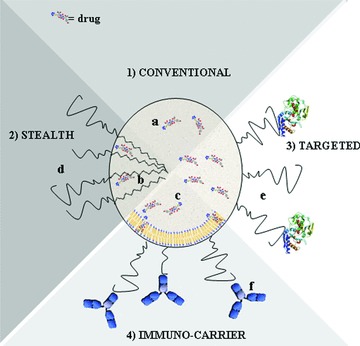
The schematic structure of pharmaceutical nanoparticulate carriers: (a) polymeric or lipid nanoparticles; (b) polymeric micelles, and (c) liposomes). (1) Conventional nanocarrier; (2) long‐circulating nanocarrier. (d) Surface‐attached protecting polymer, usually PEGs, allowing for prolonged circulation of the nanocarrier in the bloodstream; (3) targeted nanocarrier; (e) specific targeting ligand attached to the carrier surface; (4) immunocarrier; (f) specific monoclonal antibody linked to the carrier surface.
Table 1.
Nanocarriers used for brain drug delivery and targeting and their main characteristics
| Nanocarrier | Size | Description | Ref. |
|---|---|---|---|
| Liposomes | 20–100 nm (Multilamellar) 50–1000 nm (Unilamellar) 10–50 nm (Small unilamellar) | Artificial vesicles of spherical shape obtained from nontoxic natural or synthetic phospholipids and/or cholesterol. Drug molecules can be either entrapped in the aqueous space or intercalated into the lipid bilayer. | [1, 17, 18] |
| Lipid nanostructures | 10–500 nm | Spherical solid lipid particles from biocompatible lipids and emulsifiers. SLNs are made from one solid lipid, while NLCs from a mixture of spatially different lipids or from a blend of a solid lipid with a liquid lipid (oil). | [1, 29, 30, 31, 32, 33] |
| Polymeric nanoparticles | 10–1000 nm | Artificial spherical particles from natural or synthetic polymers, where drug is uniformly dispersed (nanospheres) or enclosed by polymer membrane in a vesicular system (nanocapsules). | [1, 42] |
| Polymeric micelles | 10–50 nm | Core‐shell typed colloidal carriers prepared from the self‐assemblies of amphiphilic block or graft copolymers in aqueous solution. | [1, 58, 59, 60] |
Nanoparticulate Carriers For Brain Drug Release
In the last decade, nanotechnologies applied to brain drug delivery assumed great importance due to nanostructure advantages such as subcellular size and the possibility to employ biocompatible and biodegradable materials for their realization [5]. Thus, the pharmaceutical strategies based on technology represent a valid approach for achieve brain drug release.
Various pharmaceutical nanocarriers, such as liposomes, lipid nanoparticles, polymeric nanoparticles and micelles, and some others were developed to achieve brain drug release [6, 7, 8]. For these systems, a long circulating time does not ensure an effective BBB overcome either in physiological or pathological condition and, therefore, additional carrier features are needed in order to deliver drugs in therapeutic concentrations to the CNS. In this respect, exploitation of specific carrier mediated transport systems on the BBB can be considered as a major strategy for delivering drug‐loaded colloidal carriers as well as drugs to the CNS [16].
Liposomes
Liposomes represent the first generation of the novel colloidal carriers, which revolutionized the scenario in parenteral drug delivery [17, 18]. Liposomes consist of one or more phospholipid bilayers enclosing an aqueous space, which are obtained starting from phospholipids such as distearoyl‐sn‐glycero‐phosphoetanolamine (DSPE) and dioleoylphosphatidylcholine‐dipalmitoyl phosphatidylglycerol (DOPC‐DPPG) [18]. Depending upon their size and number of bilayers, liposomes can be classified into three categories: multilamellar (20–100 nm), large unilamellar (LUV, 50–1000 nm), and small unilamellar (10–50 nm) vesicles. Water‐soluble drugs can be included within the aqueous compartments while lipophilic and amphiphilic molecules can be associated with lipid bilayers [16]. The techniques of preparation have often been described in literature [18]. In short, in each method, lipids are dissolved in an organic solvent. A dry lipid film is obtained after evaporation of the solvent and then is dispersed in an aqueous phase. The different preparation procedures differ according to the method used to disperse the lipids (thin lipid film hydration, mechanical methods such as sonication, extrusion, etc.). These structures have long been used as carriers for the delivery of therapeutic agents because of their easy preparation, good biocompatibility, low toxicity, and commercial availability [7]. The successful commercialization of various injectable liposomal products such as Amphotec® (Amphotericin B), Doxil®/Caelyx® (Doxorubicin), and DaunoXome® (Daunorubicin) and a large array of investigational products clearly indicates the potential advantages of liposomes as novel lipid carriers [1].
In this respect, surface‐pegylated liposomes (PL) have been obtained from modified phospholipids such as methoxypolyethylene glycol‐DSPE (MPEG‐DSPE), and considered for site specific targeting in several brain pathologies through both intracerebral and intravenous administrations, although most of the studies have been focused on tumor therapies, thanks to their low affinity to healthy tissues in vivo, and their capability to extravasate in pathological sites.
Although prednisolone‐loaded PL have been effective against experimental autoimmune encephalomyelitis, a further improvement of PL brain distribution was obtained by conjugating monoclonal antibodies (mAbs) that will be recognized by cell surface receptors in the targeted tissue [19, 20]. These antibody‐directed liposomes, namely immunoliposomes, were prepared through either covalent coupling mAbs directly to the liposome surface, or coupling to the distal tip of PEG chains, thus reducing the PEG chain steric hindrance and facilitating the antibody–target interaction [21]. Many studies regarding immunoliposome utilization in brain drug delivery have also demonstrated their applicability in gene therapy [20, 22, 23]. In this regard, PL coated with transferrin receptor (TfR) mAbs or human insulin receptor (HIR) mAbs have been constructed for the delivery of DNA across the BBB [24]. In particular, it was demonstrated that TR mAbs‐targeted PL with expression plasmid for tyrosine hydrolase (TH) has been shown effective in treating Parkinson's disease (PD) in rat model. This approach has also been used to deliver interfering RNA (shRNA) against epithelial growth factor receptor (EGFR) and resulted in the knock‐down of EGFR expression and increased survival of mice implanted intracranially with brain tumors [24].
Transferrin (Tf) surface‐conjugated liposomes were obtained by covalently coupling of Tf with –NH2 groups present on the surface of stearylamine‐containing liposomes [25]. These structures were able to 17‐fold increase in brain uptake of 5‐fluorouracil due to the presence of Tf receptors on BBB and consequently to a greater access of the Tf‐coupled across the barrier through a receptor‐mediated endocytosis (RME) mechanism.
Taking into account that in many neurological diseases, inflammatory cells can cross an intact BBB, in a recent paper the RGD peptide was utilized as targeting moiety for decorating liposomes due to its capability to combine with integrin receptors that are expressed on the surface of leukocytes [26]. Ferulic acid‐loaded RGD‐liposomes were able to bind to monocytes/neutrophils efficiently and, the FA concentration into brain inflammation‐bearing rats after administration of these systems was 6‐fold higher than that of FA. Moreover, in vitro pharmacodynamics studies demonstrated that FA loaded into RGD‐liposomes exhibited greater antioxidant activity than free drug.
However, complexity associated with the manufacturing of liposomes, difficulties in scale‐up, limited physical stability and enormous cost of the liposomal formulation are the major barriers in the successful commercialization of liposomes [27]. Indeed, some concerns are emerging about the effectiveness of PL upon multiple successive administrations, due to an extremely high clearance rate increase after second administration of stealth liposomes [28].
Lipid Nanoparticles
Lipid‐based nanoparticles, such as solid lipid nanoparticles (SLNs), are an important class of colloidal systems in the area of modified drug delivery technology [29, 30]. More recently modification of SLNs, the so‐called nanostructured lipid carriers (NLCs), have been obtained by mixing spatially different lipids or blending solid lipids with liquid lipids, which show a high drug loading capacity and a long‐term stability during storage. Important advantages deriving from their use include the possibility to control drug release and targeting, and to increase drug stability. Moreover, these do not have toxicity or biodegradability problems, being obtained from physiological lipids (triglycerides, fatty acids, steroids, and waxes, etc.), solid at body temperature, such as trimyristin (Dynasan 114), tripalmitin (Dynasan116), cetyl palmitate, glyceryl behenate, stearic acid, and caprylic/capric triglyceride (Mygliol). The most common stabilizers employed are ionic and nonionic molecules, such as lecithins (Epikuron 200), polysorbates (Tween 80), poloxamers, derivatized fatty acids, and their combinations [6, 29, 30]. SLNs and NLCs have been investigated as possible carriers for modified drug delivery and targeting after intravenous administration or as oral drug carriers to overcome administration problems of drugs that are instable in the gastrointestinal tract or are inadequately absorbed [30, 31, 32]. Furthermore, the possibility to produce SLNs in a large scale at low cost and excellent reproducibility performing high pressure homogenization (HPH) method is not less important [6, 33]. SLNs and NLCs can also be formulated by using the warm oil‐in‐water (o/w) microemulsion and the solvent emulsification/evaporation or diffusion techniques [29, 30].
Generally, when these systems reach the bloodstream, these may avoid the macrophage uptake of MPS due to their small size [29, 34]. In addition, thanks to their lipophilic features, these systems could reach the CNS overcoming the BBB by an endocytosis or a transcytosis mechanism which occurs in the endothelial cells lining the brain blood capillaries, or SLNs could also permeate the tight junctions between the endothelial cells [6, 30, 31, 35]. The absorption of a plasmatic protein, the apoliprotein E, onto nanoparticle surface is supposed to be responsible for the brain SLN uptake by mediating its adherence to the endothelial cells of the BBB [30, 36]. Moreover, retention of SLNs in brain–blood capillaries with absorption to capillary walls could occur and create a higher drug concentration gradient leading to enhanced drug transport across the endothelial cells [6, 30].
Several drugs were encapsulated in such particles to either modify the biodistribution or for brain targeting [6, 37]. Clozapine, a lipophilic antipsychotic drug, was successfully carried into the brain after incorporation in sterylamine‐based SLNs on intravenous and intraduodenal administration [38]. Nitrendipine‐SLNs were also found to be taken up to a greater extent by the brain as compared with nitrendipine suspension, after i.v. administration [39].
Recently, riluzole‐loaded SLNs with great potential as drug delivery systems for amyotrophic lateral sclerosis (ALS) were produced [35]. These particles showed a greater efficacy than free riluzole on rats that were immunized using the experimental allergic encephalomyelitis (EAE), and a smaller accumulation of trapped riluzole than free one in not target organs.
Brain‐targeted SLNs were developed by coating the surface with thiamine that increased the unidirectional uptake transfer constant to the BBB at brain perfusion time intervals between 45 s and 120 s, demonstrating that the thiamine ligand on the SLNs may facilitate binding and/or association with BBB thiamine transporters [40]. Recently, Tf‐conjugated SLNs with a great potential in the treatment of brain diseases such as cerebral malaria, were fabricated [41].
Polymeric Nanoparticles
Polymeric nanoparticles are colloidal systems, which can be realized starting from biocompatible and biodegradable copolymers with low water solubility [1, 42]. Example of synthetic polymers already used to prepare these particles are poly(alkylcyanoacrylate) (PACA), polyesters such as poly(lactide) (PLA), poly(D,L‐lactide‐co‐glycolide) (PLGA), and some others such as natural proteins or polysaccharides. PLA and PLGA nanoparticles have been mostly prepared by emulsification–diffusion, solvent emulsion–evaporation, interfacial deposition, and nanoprecipitation method, while the PACA nanoparticles are prepared mostly by emulsion polymerization, interfacial polymerization, and nanoprecipitation [1, 42]. All these techniques have been recently described in literature [42]. Surface decoration of these nanoparticles with physically adsorbed or covalently linked hydrophilic polymers, such as PEGs and polysaccharides, increases their residence time in the systemic circulation while the inclusion of tissue‐recognition ligands enables targeted delivery to the brain [43, 44, 45]. It has been widely demonstrated that polymeric nanoparticles are able to cross the BBB probably by endocytosis followed by transcytosis through the endothelial cells lining the blood capillaries of the brain, although other processes such as tight junction modulation or P‐glycoprotein inhibition can also occur and contribute to the suitable brain drug release [46].
Several studies unequivocally demonstrated that Polysorbate 80 (PS80)‐coated PACA nanoparticles containing dalargin were able to penetrate into the brain and to produce its antinociceptive effect, after oral administration [47, 48]. The capability of these nanoparticles to carry entrapped drugs across the BBB was demonstrated by using several other drugs, such as doxorubicin [49, 50]. A possible mechanism is that PS80‐coated PACA nanoparticles adsorb ApoE and B from the bloodstream after i.v. injection, and therefore use the low‐density lipoprotein receptor for transcytosis across the BBB [51].
PEG‐PLA nanoparticles with an increased permeability across the BBB by absorptive mediated transcytosis were obtained by using cationic bovine serum albumin (CBSA) as a brain specific targeting moiety, thanks to its greater degree of selectivity to brain tissue as compared to other organs (liver, heart, and lung) [52].
Brain‐targeted nanoparticles were obtained by using modified PLGA copolymer derivatized with the peptide H2N‐Gly‐l‐Phe‐d‐Thr‐Gly‐l‐Phe‐l‐Leu‐l‐Ser(O‐β‐d‐Glucose)‐CONH2, which bear some resemblance to the synthetic opioid peptide MMP‐2200 [53, 54]. In particular, these particles were able to deliver, after intravenous administration, the model drug loperamide into the CNS and to ensure a strong and long‐lasting pharmacological effect, greater than that previously observed with other nanoparticulate carriers. Moreover, their biodistribution showed a localization into the CNS in a quantity about 100 times the amount of the other known nanoparticles that target the brain.
PEG‐chitosan (CS) nanoparticles conjugated with OX26 using the avidin‐biotin approach were also developed, and evaluated their in vivo effectiveness [55].
Recently, brain‐targeted nanoparticles based on human serum albumin (HSA) were obtained by covalently coupling to Tf or TfR mAbs and the obtained particles were able to increase BBB transport of loperamide [56]. However, some open questions still remain on the efficiency of mAb against specific receptors to transport compounds across the BBB, because some studies demonstrated that although the total amount of drug delivery after i.v. injection of fusion molecules is high, most of it stays associated with brain capillary endothelial cells [57].
Polymeric Micelles
Polymeric micelles have recently attracted great attention as novel typology of drug delivery nanostructures [58, 59, 60]. These systems are obtained in aqueous environment from the autoaggregation of amphiphilic copolymers, whose hydrophobic portions are obtained by using molecules such as polypropylene glycols (PPG), PLA, polycaprolactone (PCL), fatty acids (C12–C18), or phospholipids, while the hydrophilic portions are obtained mainly from PEGs. However, to obtain amphiphilic graft copolymers with multigrafted branches, in which the hydrophilic/hydrophobic balance can be readily controlled by adjusting the relative grafting density and derivatization with targeting moieties, poly(N‐vinyl‐2‐pyrrolidone) (PVP), chitosan or α,β‐poly(N‐2‐hydroxyethyl)‐dl‐aspartamide (PHEA) can be used [60, 61]. Polymeric micelles show a core‐shell architecture with dimensions ranging from 10 to 100 nm, and can solubilize high amounts of hydrophobic drugs (more than 20–30% w/w) preventing early drug release and degradation. The external hydrophilic shell is responsible for micelle stabilization in aqueous media, prolongs their circulation time in bloodstream avoiding interactions with RES system and provides accumulation in particular body region with leaky vasculature [11]. Several studies have shown that surface‐PEGylated micelles are able to penetrate and accumulate into both healthy rat brain and brain glioma [60].
Of particular interest is the class of Pluronic® block copolymers (also known as Poloxamers), that are known to modulate the P‐Glycoprotein (Pgp) efflux transport system of the BBB [32, 62]. It was demonstrated that incorporation of low molecular mass drugs into Pluronic micelles can facilitate brain delivery by increasing the drug solubility and stability in plasma, and can improve drug pharmacokinetics and biodistribution.
Biologically active polymeric micelles, the surface of which is anchored with Tat molecules, have been successfully obtained by self‐assembling of the transcriptional activator peptide (TAT)‐PEG‐block‐cholesterol (TAT‐PEG‐β‐Chol) and proposed for antibiotic delivery across the BBB for treatment of brain infections due to their capability to enter the brain [63, 64].
Looking for the success strategy of PS80 surface coating for achieving brain uptake, potential brain‐targeted polymeric micelles were obtained starting from an autoassembling amphiphilic graft copolymer with a hydrophilic PHEA backbone, a hydrophobic PLA side chains and covalently linked PS80[61].
Conclusions
A central problem in the current treatment of brain disorders is to reach a suitable drug amount to the brain, due to the presence of the BBB. Advances in novel colloidal systems for drug delivery have progressed rapidly in recent years, and therapeutically efficient formulations have been already developed for drugs that were unable to cross the BBB. Some examples of nanoparticulate systems such as liposomes, lipid nanoparticles, polymeric nanoparticles and micelles with controlled size, surface charge, and specific ligands, are summarized in Table 2. These systems are showing a great potential as drug carriers to the brain thanks to many advantages associated with their use, and are becoming an alternative to the present surgical and conventional methods.
Table 2.
Summary of nanoparticulate systems used as brain drug delivery and targeting
| Nanocarrier | Drug | Targeting ligand | Relevant results | Ref. |
|---|---|---|---|---|
| Pegylated‐liposomes | Prednisolone | – | Brain accumulation of the injected pegylated liposomes in rats with experimental autoimmune encephalomyelitis (EAE), reaching values of up to 4.5‐fold higher than in healthy control animals. No apoptosis in resident cells such as astrocytes, oligodendrocytes or microglia in spinal cord, which rules out some important unwanted side effects. | [19] |
| Pegylated‐liposomes | RNAi | mAbs | Weekly i.v. RNAi gene therapy directed against the human EGFR by the pegylated liposomes gene transfer technology causes an 88% increase in survival time in adult mice with intracranial human brain cancer. | [20] |
| Immuno‐liposomes | DNA | TfR‐mAbs, HIR‐mAbs | Tyrosine hydrolase (TH) gene therapy with liposomes resulted in a complete normalization of striatal TH enzyme activity ipsilateral to the lesioned nigral–striatal dopaminergic track of rat brain. | [24] |
| Immuno‐liposomes | 5‐fluorouracil | Tf | An average of 10‐fold increase in the brain drug uptake was observed after the not‐targeted liposomal administration, while the Tf‐coupled liposomes caused a 17‐fold increase. | [25] |
| Immuno‐liposomes | Ferulic acid | RGD peptide | RGD‐coated liposomes exhibit in vivo brain targeting ability with 6‐fold concentration FA in brain compared with FA solution and 3‐fold in comparison of plain liposomes. | [26] |
| SLNs | Clozapine | – | Positively charged clozapine SLNs enhanced the bioavailability of clozapine from 3.1‐ to 4.5‐fold on intraduodenal administration. The AUC of clozapine SLNs showed higher uptake in RES organs and brain after intravenous administration than clozapine suspension. | [38] |
| SLNs | Nitrendipine | – | Effective bioavailability of nitredipine‐SLNs were 2.81–5.35 folds greater after i.d. administration in comparison with drug‐suspension. In tested organs, the AUC of drug‐SLNs were higher than those of drug‐suspension especially in brain, heart and RES organs. | [39] |
| SLNs | Riluzole | – | Riluzole entrapped into SLNs that reaches the brain was 3‐fold greater than free riluzole, 16h postinjection, and showed a smaller accumulation in the RES organs. In addition, rats treated with riluzole‐loaded SLNs showed clinical signs of EAE later than those treated with free riluzole. | [35] |
| SLNs | – | Thiamine | Association of thiamine‐targeted nanoparticles with the BBB thiamine transporter, and accumulation at the BBB, increasing brain uptake during perfusion time frames. | [40] |
| SLNs | Quinine dihydrochloride | Tf | Enhancement of brain uptake of quinine dihydrochloride into Tf‐targeted SLNs, as demonstrated by the recovery of a higher percentage of the dose from the brain after in vivo administration of Tf‐coupled SLNs compared with unconjugated SLNs or drug solution. | [41] |
| PACA nanoparticles | Dalargin | PS80 | Measurement of in vivo central antinociceptive effect of dalargin along with a dose response curve was observed after 60 min of oral administration of PS80‐targeted nanoparticles to mice. | [47, 48] |
| PACA nanoparticles | Doxorubicin | PS80 | High brain drug concentrations (>6 μg/g) were achieved with the PS80‐coated nanoparticles, while with both uncoated nanoparticles and drug saline solution were always below the detection limit ((<0.1 μg/g). However, both types of nanoparticles prevented the drug accumulation in the heart. | [49] |
| PACA nanoparticles | MRZ 2/576 | PS80 | Intravenous administration of the drug bound to PS80‐targeted PACA nanoparticles prolongs the duration of the anticonvulsive activity in mice up to 210 min and after probenecid pre‐treatment up to 270 min compared to 150 min with probenecid and MRZ 2/576 alone | [50] |
| PEG‐PLA nanoparticles | – | CBSA | CBSA‐nanoparticles did not impact the integrity of BBB endothelial tight junctions and also showed little toxicity against brain capillary endothelial cells (BCECs). The permeability of CBSA‐nanoparticles was about 7.76 times higher than that of BSA‐nanoparticles. | [52] |
| PLGA nanoparticles | Loperamide | H2N‐Gly‐l‐Phe‐d‐Thr‐Gly‐l‐Phe‐l‐Leu‐l‐Ser(O‐β‐d‐Glucose)‐CONH2 peptide | 13% of the injected dose of loperamide‐loaded into peptide‐modified nanoparticles was found in the brain 4 h postinjection, while fluorescent peptide‐modified nanoparticles ∼about 9% of injected dose per gram of tissue at 0.25 hours after administration. | [53, 54] |
| PEG‐CS nanoparticles | Z‐DEVD‐FMK peptide | OX26 | Translocation of PEG‐CS/OX26 nanoparticles into the brain tissue after iv administration to mice. | [55] |
| HSA nanoparticles | Loperamide | Tf or TfR‐mAbs | Significant antinociceptive effects induced by loperamide‐loaded targeted HSA nanoparticles in mice after iv injection, and only marginal effects with control loperamide‐loaded HSA nanoparticles. | [56] |
| PHEA‐PLA micelles | – | PS80 | Escape from the macrophage uptake and internalization into neuroblastoma cells of PS80‐targeted PHEA‐PLA micelles. | [61] |
| PEG‐β‐Chol micelles | – | TAT | Brain penetration of fluorescent probe‐loaded TAT‐PEG‐β‐Chol micelles in hippocampus sections of rats 2 h after i.v. injection, while free probe did no cross the BBB. | [63, 64] |
Conflict of Interest
The authors have no conflict of interest.
References
- 1. Prokop A, Davidson JM. Nanovehicular intracellular delivery systems. J Pharm Sci 2008;97:3518–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moses MA, Brem H, Langer R. Advancing the field of drug delivery: Taking aim at cancer. Cancer Cell 2003;4:337–341. [DOI] [PubMed] [Google Scholar]
- 3. Kabanov AV, Batrakova EV. New technologies for drug delivery across the blood brain barrier. Current Pharm Design 2004;10:1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Juillerat‐Jeanneret L. The targeted delivery of cancer drugs across the blood–brain barrier: Chemical modifications of drugs or drug‐nanoparticles Drug Discov Today 2008;13:1099–1106. [DOI] [PubMed] [Google Scholar]
- 5. Kaur IP, Bhandari R, Bhandari S, Kakkar V. Potential of solid lipid nanoparticles in brain targeting. J Control Release 2008;127:97–109. [DOI] [PubMed] [Google Scholar]
- 6. Denora N, Trapani A, Laquintana V, Lopedota A, Trapani G. Recent advances in medicinal chemistry and pharmaceutical technology‐strategies for drug delivery to the brain. Curr Topics Med Chem 2009;9:182–196. [DOI] [PubMed] [Google Scholar]
- 7. Gabathuler R. Approaches to transport therapeutic drugs across the blood–brain barrier to treat brain diseases. Neurobiol Dis 2010;37:48–57 [DOI] [PubMed] [Google Scholar]
- 8. Li S‐D, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm 2008;5:496–504. [DOI] [PubMed] [Google Scholar]
- 9. Torchilin VP. Multifunctional nanocarriers. Adv Drug Del Rev 2006;58:1532–1555. [DOI] [PubMed] [Google Scholar]
- 10. Van Butsele K, Jerome R, Jerome C. Functional amphiphilic and biodegradable copolymers for intravenous vectorisation. Polymer 2007;48:7431–7443. [Google Scholar]
- 11. Park JH, Lee S, Kim JH, Park K, Kim K, Kwon IC. Polymeric nanomedicine for cancer therapy. Prog Polymer Sci 2008;33:113–137. [Google Scholar]
- 12. Owens DE 3rd, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm 2006;307:93–102. [DOI] [PubMed] [Google Scholar]
- 13. Ben‐Shabat S, Kumar N, Domb AJ. PEG‐PLA block copolymer as potential drug carrier: Preparation and characterization. Macromol Biosci 2006;6:1019–1025. [DOI] [PubMed] [Google Scholar]
- 14. Hu Y, Xie J, Wah Tong Y, Wang C‐H. Effect of PEG conformation and particle size on the cellular uptake efficiency of nanoparticles with the HepG2 cells. J Contl Release 2007;118:7–17. [DOI] [PubMed] [Google Scholar]
- 15. Byrne JD, Betancourt T, Brannon‐Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Del Rev 2008;60:1615–1626. [DOI] [PubMed] [Google Scholar]
- 16. Ricci M, Blasi P, Giovagnoli S, Rossi C. Delivering drugs to the central nervous system: A medicinal chemistry or a pharmaceutical technology issue Curr Med Chem 2006;13:1707–1725. [DOI] [PubMed] [Google Scholar]
- 17. Bawarski WE, Chidlowsky E, Bharali DJ, Mousa SA. Emerging nanopharmaceuticals. Nanomedicine 2008;4:273–282. [DOI] [PubMed] [Google Scholar]
- 18. Shapira I, Budman DR, Bradley T, Gralla R. Evolving lipid‐based delivery systems in the management of neoplastic disease. Oncol Rev 2009;3:113–124. [Google Scholar]
- 19. Schmidt J, Metselaar JM, Wauben MH, Toyka KV, Storm G, Gold R. Drug targeting by long‐circulating liposomal glucocorticosteroids increases therapeutic efficacy in a model of multiple sclerosis. Brain 2003;126:1895–1904. [DOI] [PubMed] [Google Scholar]
- 20. Zhang Y, Zhang Y‐F, Bryant J, Charles A, Boado RJ, Pardridge WM. Intravenous RNA interference gene therapy targeting the human epidermal growth factor receptor prolongs survival in intracranial brain cancer. Clin Cancer Res 2004;10:3667–3677. [DOI] [PubMed] [Google Scholar]
- 21. Schnyder A, Huwyler J. Drug transport to brain with targeted liposomes. J NeuroRx 2005;2:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu C, Zhang Y, Zhang YF, Yi Li J, Boado RJ, Pardridge WM. Organ‐specific expression of the lacZ gene controlled by the opsin promoter after intravenous gene administration in adult mice. J Gene Med 2004;6:906–912. [DOI] [PubMed] [Google Scholar]
- 23. Schlachetzki F, Zhang YF, Boado RJ, Pardridge WM. Gene therapy of the brain. Neurology 2004;62:1275–1281. [DOI] [PubMed] [Google Scholar]
- 24. Pardridge WM. shRNA and siRNA delivery to the brain. Adv Drug Deliv Rev 2007;59:141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Soni V, Kohli DV, Jain SK. Transferrin‐conjugated liposomal system for improved delivery of 5‐fluorouracil to brain. J Drug Target 2008;16:73–78. [DOI] [PubMed] [Google Scholar]
- 26. Qin J, Chen D, Hu H, Cui Q, Qiao M, Chen B. Surface modification of RGD‐liposomes for selective drug delivery to monocytes/neutrophils in brain. Chem Pharm Bull 2007;55:1192–1197. [DOI] [PubMed] [Google Scholar]
- 27. Bunjes H, Siekmann B. Manufacture, characterization and application of solid lipid nanoparticles as drug delivery system In: Deleers M, Pathak Y, Thassu YD, editors. Nanoparticulate drug delivery systems. New York: Informa Healthcare, 2007;213–268. [Google Scholar]
- 28. Ishida T, Harada M, Wang X‐Y, Ichihara M, Irimura K, Kiwada H. Accelerated blood clearance of PEGylated liposomes following preceding liposome injection: Effects of lipid dose and PEG surface‐density and chain length of the first‐dose liposomes. J Contl Release 2005;105:305–317. [DOI] [PubMed] [Google Scholar]
- 29. Wissing SA, Kayser O, Muller RH. Solid lipid nanoparticles for parenteral drug delivery. Adv Drug Deliv Rev 2004;56:1257–1272. [DOI] [PubMed] [Google Scholar]
- 30. Joshi MD, Müller RH. Lipid nanoparticles for parenteral delivery of actives. Eur J Pharm Biopharm 2009;71:161–172. [DOI] [PubMed] [Google Scholar]
- 31. Reddy JS, Venkateswarlu V. Novel delivery systems for drug targeting to the brain. Drugs Future 2004;29:63–83. [Google Scholar]
- 32. Yang S, Zhu J, Lu Y, Liang B, Yang C. Body distribution of camptothecin solid lipid nanoparticles after oral administration. Pharm Res 1999;16:751–757. [DOI] [PubMed] [Google Scholar]
- 33. Bondì ML, Craparo EF. Solid lipid nanoparticles for applications in gene therapy: A review of the state of the art. Expert Opin Drug Del 2010;7:7–18. [DOI] [PubMed] [Google Scholar]
- 34. Kreuter J. Nanoparticulate systems for brain delivery of drugs. Adv Drug Deliv Rev 2001;47:65–81. [DOI] [PubMed] [Google Scholar]
- 35. Bondì ML, Craparo EF, Giammona G, Drago F. Brain‐targeted solid lipid nanoparticles (SLNs) containing riluzole: Preparation, characterisation and biodistribution. Nanomedicine 2010;5:25–32. [DOI] [PubMed] [Google Scholar]
- 36. Blasi P, Giovagnoli S, Schoubben A, Ricci M, Rossi C. Solid lipid nanoparticles for targeted brain drug delivery. Adv Drug Del Rev 2007;59:454–477. [DOI] [PubMed] [Google Scholar]
- 37. Panyam J, Chavanpatil M. Lipid‐derived nanoparticles for brain‐targeted drug delivery, PCT WO2008024753, 2008.
- 38. Manjunath K, Venkateswarlu V. Pharmacokinetics, tissue distribution and bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduodenal administration. J Contl Release 2005;107:215–228. [DOI] [PubMed] [Google Scholar]
- 39. Manjunath K, Venkateswarlu V. Pharmacokinetics, tissue distribution and bioavailability of nitrendipine solid lipid nanoparticles after intravenous and intraduodenal administration. J Drug Target 2006;14:632–645. [DOI] [PubMed] [Google Scholar]
- 40. Lockman PR, Oyewumi MO, Koziara JM, Roder KE, Mumper RJ, Allen DD. Brain uptake of thiamine‐coated nanoparticles. J Contl Release 2003;93:271–282. [DOI] [PubMed] [Google Scholar]
- 41. Gupta Y, Jain A, Jain SK. Transferrin‐conjugated solid lipid nanoparticles for enhanced delivery of quinine dihydrochloride to the brain. J Pharm Pharmacol 2007;59:935–940. [DOI] [PubMed] [Google Scholar]
- 42. Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloid Surf B 2010;75:1–18. [DOI] [PubMed] [Google Scholar]
- 43. Wilson B, Samanta MK, Santhi K, Kumar KPS, Paramakrishnan N, Suresh B. Poly(n‐butylcyanoacrylate) nanoparticles coated with polysorbate 80 for the targeted delivery of rivastigmine into the brain to treat Alzheimer's disease. Brain Res 2008;1200:159–168. [DOI] [PubMed] [Google Scholar]
- 44. Mohamed F, Van Der Walle CF. Engineering biodegradable polyester particles with specific drug targeting and drug release properties. J Pharm Sci 2008;97:71–87. [DOI] [PubMed] [Google Scholar]
- 45. Hu K, Li J, Shen Y, Lu W, Gao X, Zhang Q, Jiang X. Lactoferrin‐conjugated PEG–PLA nanoparticles with improved brain delivery: In vitro and in vivo evaluations. J Contr Release 2009;134:55–61. [DOI] [PubMed] [Google Scholar]
- 46. Silva GA. Nanotechnology approaches for drug and small molecule delivery across the blood brain barrier. Surg Neurol 2007;67:113–116. [DOI] [PubMed] [Google Scholar]
- 47. Das D, Lin S. Double‐coated poly (butylcynanoacrylate) nanoparticulate delivery systems for brain targeting of dalargin via oral administration. J Pharm Sci 2005;94:1343–1353. [DOI] [PubMed] [Google Scholar]
- 48. Smith MW, Gumbleton M. Endocytosis at the blood‐brain barrier: From basic understanding to drug delivery strategies. J Drug Targ 2006;14:191–214. [DOI] [PubMed] [Google Scholar]
- 49. Gulyaev AE, Gelperina SE, Skida AS, Antropov GY, Kreuter J. Significant transport of doxorubicin into the brain with polysorbate 80‐coated nanoparticles. Pharm Res 1999;16:1564–1569. [DOI] [PubMed] [Google Scholar]
- 50. Friese A, Seiller E, Quack G, Lorenz B, Kreuter J. Increase of the duration of the anticonvulsive activity of a novel NMDA receptor antagonist using poly(butylcyanoacrylate) nanoparticles as a parenteral controlled release system. Eur J Pharm Biopharm 2000;49:103–109. [DOI] [PubMed] [Google Scholar]
- 51. Kreuter J, Shamenkov D, Petrov V, Ramge P, Cychutek K, Koch‐Brandt C, Alyautdin R. Apolipoprotein‐mediated transport of nanoparticle‐bound drugs across the blood‐brain barrier. J Drug Target 2002;10:317–325. [DOI] [PubMed] [Google Scholar]
- 52. Lu W, Tan Y‐Z, Hu K‐L, Jiang X‐G. Cationic albumin conjugated pegylated nanoparticle with its transcytosis ability and little toxicity against blood‐brain barrier. Int J Pharm 2005;295:247–260. [DOI] [PubMed] [Google Scholar]
- 53. Tosi G, Costantino L, Rivasi F, et al Targeting the central nervous system: In vivo experiments with peptide‐derivatized nanoparticles loaded with Loperamide and Rhodamine‐123. J Control Release 2007;122:1–9. [DOI] [PubMed] [Google Scholar]
- 54. Vergoni AV, Tosi G, Tacchi R, Vandelli MA, Bertolini A, Costantino L. Nanoparticles as drug delivery agents specific for CNS: In vivo biodistribution. Nanomed: Nanotechnol, Biol Med 2009;5:369–377. [DOI] [PubMed] [Google Scholar]
- 55. Aktas Y, Yemisci M, Andrieux K, et al Development and brain delivery of chitosan‐PEG nanoparticles functionalized with the monoclonal antibody OX26. Bioconjug Chem 2005;16:1503–1511. [DOI] [PubMed] [Google Scholar]
- 56. Ulbrich K, Hekmatara T, Herbert E, Kreuter J. Transferrin‐ and transferrin‐receptor monoclonal antibody‐modified nanoparticles enable drug delivery across the blood–brain barrier (BBB). Eur J Pharm Biopharm 2009;71:251–256. [DOI] [PubMed] [Google Scholar]
- 57. Gosk S, Vermehren C, Storm G, Moos T. Targeting anti‐transferrin receptor antibody (OX26) and OX26‐conjugated liposomes to brain capillary endothelial cells using in situ perfusion. J Cereb Blood Flow Metab 2004;24:1193–1204. [DOI] [PubMed] [Google Scholar]
- 58. Allen TM, Cullis PR. Drug delivery systems: Entering the mainstream Science 2004;303:1818–1822. [DOI] [PubMed] [Google Scholar]
- 59. Aliabadi HM, Lavasanifar A. Polymeric micelles for drug delivery. Expert Opin Drug Deliv 2006;3:139–162. [DOI] [PubMed] [Google Scholar]
- 60. Torchilin VP. Micellar nanocarriers: Pharmaceutical perspectives. Pharm Res 2007;24:1–16. [DOI] [PubMed] [Google Scholar]
- 61. Craparo EF, Ognibene MC, Casaletto MP, Pitarresi G, Teresi G, Giammona G. Biocompatible polymeric micelles with polysorbate 80 for brain targeting. Nanotechnology 2008;19:485603 (12pp). [DOI] [PubMed] [Google Scholar]
- 62. Batrakova EV, Kabanov AV. Pluronic block copolymers: Evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J Contl Release 2008;130:98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu L, Guo K, Lu J, et al Biologically active core/shell nanoparticles self‐assembled from cholesterol‐terminated PEG‐TAT for drug delivery across the blood–brain barrier. Biomaterials 2008;29:1509–1517. [DOI] [PubMed] [Google Scholar]
- 64. Liu L, Venkatraman SS, Yang YY, et al Polymeric micelles anchored with TAT for delivery of antibiotics across the blood‐brain barrier. Biopolym – Peptide Sci Sect 2008;90:617–623. [DOI] [PubMed] [Google Scholar]