

SUMMARY
Aims: Drugs currently used for the treatment of Alzheimer's disease (AD) partially stabilize patients’ symptoms without modifying disease progression. Brain accumulation of oligomeric species of β‐amyloid (Aβ) peptides, the principal components of senile plaques, is believed to play a crucial role in the development of AD. Based on this hypothesis, huge efforts are being spent to identify drugs able to interfere with proteases regulating Aβ formation from amyloid precursor protein (APP). This article briefly reviews the profile of γ‐secretase inhibitors, compounds that inhibit γ‐secretase, the pivotal enzyme that generates Aβ, and that have reached the clinic. Discussion: Several classes of potent γ‐secretase inhibitors have been designed and synthesized. Preclinical studies have indicated that these compounds are able to lower brain Aβ concentrations and, in some cases, reduce Aβ plaque deposition in transgenic mouse models of AD. The most developmentally advanced of these compounds is semagacestat, presently in Phase III clinical trials. In animals, semagacestat reduced Aβ levels in the plasma, cerebrospinal fluid (CSF), and the brain. However, studies have not reported on its cognitive effects. Studies in both healthy volunteers and patients with AD have demonstrated a dose‐dependent inhibition of plasma Aβ levels, and a recent study in healthy subjects demonstrated a robust, dose‐dependent inhibition of newly generated Aβ in the CSF after single oral doses. Conclusions: Unfortunately, γ‐secretase inhibitors may cause intestinal goblet cell hyperplasia, thymus atrophy, decrease in lymphocytes, and alterations in hair color, effects associated with the inhibition of the cleavage of Notch, a protein involved in cell development and differentiation. Nevertheless, at least other two promising γ‐secretase inhibitors are being tested clinically. This class of drugs represents a major hope to slow the rate of decline of AD.
Keywords: Alzheimer’s disease, β‐Amyloid, Dementia, γ‐Secretase inhibitors
Introduction
Alzheimer's disease (AD), an age‐related progressive neurodegenerative disorder characterized by relatively slow chronic but progressive impairment in cognition, behavior, and functionality, is the most common form of dementia. Accurate AD epidemiological data have been recently released for the United States [1]. These 2009 figures suggested that 5.3 million Americans have AD and >15 million patients with AD worldwide. The prevalence is approximately 13% in people over the age of 65 years, and increases to >40% for those over 85 years of age. It is expected that the number of patients with AD in the United States will increase to 7.4 million by 2020, with 400,000 new cases diagnosed each year caused largely by a significant increase in the aging population. The number of people suffering from AD, that currently affects more than 26 million people worldwide with an expected increase to more than 106 million by 2050 [2], is rising quickly because there are no effective treatments for the disorder available. At present, only cholinesterase inhibitors (ChEIs) and the N‐methyl‐D‐aspartic acid (NMDA)‐receptor antagonist memantine have received Food and Drug Administration (FDA) approval for the symptomatic treatment of AD [3, 4]. Evidence from controlled clinical trials suggests that ChEIs in particular can stabilize patients’ symptoms for periods of time ranging between 1 and 3 years but without modifying progression of the disease [3]. Despite many theoretical considerations suggesting that ChEIs or memantine may have a disease‐modifying effect, only symptomatic effects of these compounds have been proven [3]. Individual ChEIs have additional pharmacological effects besides the inhibition of acetylcholinesterase [5, 6]. However, a clinical benefit of these additional effects has not been convincingly shown [7]. A recent large meta‐analysis based on 59 unique studies showed that both ChEIs and memantine had consistent effects in the domains of cognition and global assessment, but summary estimates showed small effect sizes [8]. Epidemiological studies mainly with cross‐sectional data have suggested that nonsteroidal anti‐inflammatory drugs (NSAIDs), estrogens, HMG‐CoA reductase inhibitors (statins), and tocopherol (vitamin E) may be beneficial to reduce the incidence of AD [9]. However, bias of case selection and several other sources of error are inherent in epidemiological studies and subsequent clinical trials have often been disappointing.
Drugs Targeting β‐Amyloid
In the last decade, advances in understanding the neurobiology of AD have translated into an increase in clinical trials assessing various potential AD treatments [3, 4, 9]. In particular, most pharmaceutical research has been directed against the production and the accumulation of β‐amyloid (Aβ) with the aim of slowing the deterioration rate of patients [10]. In fact, AD involves aberrant protein processing and is characterized by the presence of both intraneuronal protein clusters composed of paired helical filaments of hyperphosphorylated tau protein (neurofibrillary tangles [NFTs]), and extracellular protein aggregates (senile plaques [SPs]). Therefore, Alzheimer's classic pathological description of AD as a “two hallmarks disorder”[11] was confirmed by subsequent observations [12]. The exact relationship between these two neuropathological hallmarks remains unclear and how they may cause neuronal death is still an area of intense research effort [13]. However, these neuropathological hallmarks of AD strongly influenced recent therapeutic approaches [14]. SPs consist of a proteinaceous core composed of 5 to 10 nm amyloid fibrils surrounded by dystrophic neurites, astrocytic processes, and microglial cells. The Aβ peptide consists of 38 to 42 amino acids generated by the cleavage of amyloid precursor protein (APP), a type‐1 transmembrane protein. The SPs are the result of misprocessing of the APP by β‐ and γ‐secretases to form a toxic Aβ peptide of 40 to 42 amino acids that aggregates and initiates a pathogenic self‐perpetuating cascade ultimately leading to neuronal loss and dementia [15]. The main form of Aβ contains 40 amino acids (Aβ40), but the carboxy‐terminal‐extended species, made up of 42 residues (Aβ42), is also produced. This longer form is more prone to aggregate into fibrils and Aβ42 makes up the major component of SPs. Extracellular and perhaps also intracellular Aβ exert neurotoxic effects [16]. Extracellular Aβ peptides cluster in a β‐sheet structure to form SPs. According to the “amyloid cascade hypothesis”[17, 18], the development of SPs is thought to precede and precipitate the formation of NFTs as a result of the cellular changes invoked, and the oligomeric forms of Aβ peptide are the main cause of neuronal death in AD [19]. APP may be metabolically processed according to two pathways. In the so‐called nonamyloidogenic pathway, the α‐secretase enzyme cleaves APP within the Aβ sequence and releases its transmembrane fragment soluble amyloid precursor protein α‐cleaved (sAPPα) that appears to exert neuroprotective activity. In the amyloidogenic pathway, the β‐secretase enzyme releases APP plus a 12‐kDa protein fragment (C99), which in turn is cleaved by the γ‐secretase enzyme giving way to Aβ. Accumulation of toxic, aggregated forms of Aβ seem crucial in the pathogenesis of familial forms of AD [19]. In fact, many studies showed a weak correlation between Aβ deposits and cognitive status [20]. Some other reports showed that cognitively healthy elderly people could have a substantial amyloid burden [21].
The manipulation of the enzymes responsible for the generation of Aβ (α, β, and γ‐secretase) has been intensively pursued and several compounds have reached clinical testing. The last metabolic step in the generation of Aβ involves the enzymatic intramembranous cleavage of APP by a high‐molecular weight complex called γ‐secretase. This article briefly reviews the profile of γ‐secretase inhibitors that have reached the clinic and discusses the clinical issues surrounding this new class of anti‐AD compounds. Therefore, the review did not include active and passive immunization [22], drugs that target the proteases involved in both the nonamyloidogenic and the amyloidogenic pathways of the APP metabolism (α‐secretase activators and β‐secretase inhibitors) [3], Aβ aggregation inhibitors [4], and drugs similar to tarenflurbil (R‐flurbiprofen, FlurizanTM), a compound that recently failed in a large Phase III study, because it is not a γ‐secretase inhibitor but just indirectly modulates its activity (shifting Aβ42 to Aβ38 production) by binding to APP (γ‐secretase modulators) [9]. We reviewed studies from the English literature published before March 2010. We searched through the PubMed database of NCBI (available at http://www.ncbi.nlm.nih.gov) by author and the following keywords: drugs targeting β‐amyloid; γ‐secretase inhibitors; dementia syndromes, and AD.
Therapeutic Potential of γ‐Secretase Inhibitors
γ‐Secretase is an unconventional aspartyl protease that resides and cleaves its substrates within the lipid bilayer. In fact, γ‐secretase complex belongs to a group of proteases called intramembrane cleaving proteases (I‐CLiPs) that are membrane‐embedded enzymes. These enzymes hydrolyze transmembrane substrates and the residues essential to catalysis reside within the boundaries of the lipid bilayer [23]. AD is believed to be caused by a progressive cerebral accumulation of Aβ, and the γ‐secretase activity, which consists of both presenilin‐dependent and presenilin‐independent activities [24, 25, 26], determines the length of Aβ and therefore controls the ratio Aβ42/Aβ40 [27]. In fact, at least four subunit proteins form γ‐secretase: presenilin (PS), nicastrin, anterior pharynx‐defective‐1 (Aph‐1), and presenilin enhancer‐2 (Pen‐2) [28] (Figure 1). Presenilins are of exceptional pathophysiological importance because more than 175 autosomal dominant point mutations are known in these proteins, and most of which cause aggressive early‐onset AD elevating the Aβ42/Aβ40 ratio and interfering with the processing of APP and other γ‐secretase substrates [29, 30]. The catalytic component of γ‐secretase complex is PS, with two aspartate residues forming the active site. The inhibition of the catalytic unit of the γ‐secretase enzymatic complex appears a logical strategy to counteract Aβ accumulation in patients with AD [31, 32]. γ‐Secretase processes a wide range of type‐I integral membrane proteins, some of them with critical cellular functions. However, despite the fact that more than 70 type‐I integral membrane proteins are known to be cleaved by γ‐secretase [33], the physiological function of these proteolytic events is poorly understood. γ‐Secretase displays poor substrate specificity, but a functional γ‐secretase cleavage has been clearly demonstrated for some substrates. Notch proteolysis by γ‐secretase generates an intracellular domain (notch intracellular domain [NICD]) which is essential for many cell differentiation events and neurite outgrowth [34, 35]. Proteolysis of N‐cadherin leads to degradation of the transcriptional factor CBP (cAMP response element‐binding protein), and cleavage of ErB4 inhibits astrocyte differentiation by interacting with repressors of astrocyte gene expression [36, 37]. Cleavage of APP generates an APP intracellular domain (AICD), although its role in signal transduction remains controversial [38]. The long list of substrates processed by γ‐secretase has clear implications for the development of new therapies for AD and, in particular, for the search of γ‐secretase inhibitors. The challenge in AD research has been thus far to find a γ‐secretase inhibitor able to selectively lower Aβ but without interfering with the cleavage of other important substrates.
Figure 1.
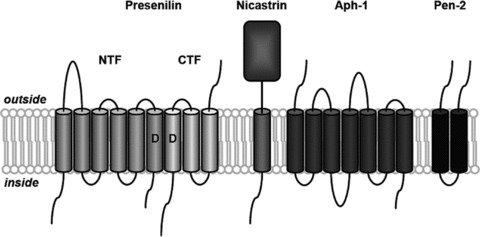
Schematic representation of the γ‐secretase complex. γ‐Secretase is composed of four different integral membrane proteins: presenilin (PS), nicastrin, anterior pharynx‐defective‐1 (Aph‐1), and presenilin enhancer‐2 (Pen‐2). Presenilin undergoes endoproteolysis into an N‐terminal fragment (NTF) and a C‐terminal fragment (CTF) that remain associated. Two conserved aspartates (D) within adjacent transmembrane domains are essential for both presenilin endoproteolysis and γ‐secretase activity.
In 2001, the first in vivo testing of a γ‐secretase inhibitor was reported, the dipeptidic compound DAPT (N‐[N‐(3,5‐difluorophenacetyl)‐L‐alanyl]‐S‐phenylglycine t‐butyl ester) developed by Elan and Eli Lilly [27]. Later data demonstrated that DAPT reversed contextual memory deficit in a transgenic mouse model of AD [39]. Several other nonpeptidic, orally‐available, γ‐secretase inhibitors have been reported to lower brain Aβ concentrations in both transgenic and nontransgenic animals [40]. The first γ‐secretase inhibitor to reach clinical development appears to be BMS‐299897, a sulfonamide derivative synthesized at Bristol‐Myers Squibb and the former SIBIA Neurosciences [40]. Human testing of BMS‐299897 started in 2001 but clinical data have never been fully described. The long‐lasting lack of information on its clinical development may indicate that it has been abandoned [40]. Benzodiazepine analog LY‐411575 and benzolactam semagacestat (LY‐450139), developed at Eli Lilly, are highly potent γ‐secretase inhibitors that have been tested extensively in vivo[41, 42]. At least five other γ‐secretase inhibitors reached the clinic, and most of the information on these other compounds is derived from conference communications: MK‐0752 [43], BMS‐708163 [44], PF‐3084014 (abandoned) [45], and GSI‐953 (begacestat) [46], and E2012 [40] (Table 1). Some γ‐secretase inhibitors appear to spare Notch cleavage in vitro and are relatively well tolerated in man. Three of these compounds affect Aβ levels in the cerebrospinal fluid (CSF) of humans, which is a potential biomarker of the disease. These compounds are Merck & Co Inc's (Whitehouse Station, NJ, USA) MK‐0752, Bristol‐Myers Squibb Co's (New York City, NY, USA) BMS‐708163, and Eli Lilly & Co's (Indianapolis, IN, USA) semagacestat [40]. The best documented and most advanced of these compounds is semagacestat [47].
Table 1.
γ‐Secretase inhibitors in clinical development for the treatment of Alzheimer's disease (AD)
| Compound | Mechanism of action | Side effects | Development status | Company | References |
|---|---|---|---|---|---|
| Semagacestat (LY‐450139) | Decreases newly synthesized Aβ in CSF of AD patients | No significant effects on brain plaque burden in transgenic mice. Lack of data on behavioral effects in animal models of AD. Gastro‐intestinal and skin side effects in AD patients | Phase III | Eli‐Lilly | [42] |
| MK‐0752 | Decreases Aβ40 levels in CSF of healthy volunteers | Inhibits Notch cleavage. Significant gastro‐intestinal toxicity in humans | Abandoneda | Merck | [43] |
| E2012 | Notch sparing | Lenticular opacity in rats | Phase I | Eisai | [40] |
| BMS‐708163 | Notch sparing Decreases Aβ levels in CSF of healthy volunteers | Lack of data on brain plaque deposition in transgenic mice. Lack of data on behavioral effects in animal models of AD | Phase II | Bristol Myers Squibb | [44] |
| PF‐3084014 | Notch sparing. Good brain penetration. Long‐lasting effects on Aβ levels in animals. No rebound effect on plasma Aβ in animals | Lack of data on brain plaque deposition in transgenic mice. Lack of data on behavioral effects in animal models of AD | Abandoneda | Pfizer | [45] |
| GSI‐953 (begacestat) | Improves memory in a transgenic mouse model of AD. | Does not decrease Aβ40 levels in CSF of AD patients | Phase II | Wyeth | [46] |
aIn development as anticancer agent.
Aβ, β‐amyloid; CSF, cerebrospinal fluid.
Semagacestat
Semagacestat is 3‐fold more potent in inhibiting APP cleavage than Notch cleavage [42]. In experimental animals, the effects of semagacestat on Aβ levels in brain, CSF, and plasma were well characterized in transgenic mice [48], nontransgenic mice [49], guinea pigs [50], and dogs [51]. However, the drug failed to show a statistically significant effect on brain plaque deposition in chronic studies in transgenic mice expressing mutated human APPV717F (PDAPP mice) [52]. More importantly, no data are available on the cognitive or behavioral effects of the drug in animal models of AD.
In a Phase I study, semagacestat was administered to healthy men and women, aged 45 years and above, for up to 14 days at doses of 5, 20, 40, and 50 mg once daily [53]. Two subjects in the 50‐mg‐dose group developed possibly drug‐related adverse events and discontinued treatment (significant increases in serum amylase and lipase with moderate abdominal pain and diarrhea positive for occult blood). The 50‐mg dose caused a maximal 40% reduction in total plasma Aβ that returned to baseline within 8 h. However, after returning to baseline, plasma Aβ levels increased to about 300% of baseline values at 15 h before slowly declining again. At lower doses, smaller and shorter decreases in plasma Aβ were observed, although the subsequent plasma Aβ increases were similar. No significant changes in CSF Aβ levels were detected in this study [53]. A second Phase I study evaluated the safety and tolerability and biomarker responses to single oral doses of 60, 100, or 140 mg in 31 healthy male and female volunteers (40 years and older) [54]. No clinically significant adverse events or laboratory changes were observed in this study. A dose‐dependent decrease in plasma Aβ40 levels was demonstrated with maximum inhibition (–73%) at 6 h after the administration of the 140‐mg dose. Again, a rebound effect on plasma Aβ40 levels was observed at 8–12 h after administration and lasted for at least 24 h. CSF concentrations of Aβ were unchanged 4 h after drug administration [54].
Semagacestat has been evaluated also in AD patients in Phase II studies. In a randomized, placebo‐controlled trial, 70 patients received the drug for 6 weeks (30 mg once a day for 1 week followed by 40 mg once a day for 5 weeks) [55]. Six patients taking the drug reported diarrhea and a 76‐year‐old man on semagacestat had gastrointestinal bleeding associated to a Barrett esophagus. Approximately 4 months after discontinuing treatment, this older patient developed endocarditis and approximately 1 month thereafter, died. In the semagacestat‐treated group, circulating CD69, T lymphocytes, eosinophils, and serum concentrations of potassium and inorganic phosphorus showed statistically significant changes. Plasma Aβ40 concentrations of patients taking the compound decreased significantly by 38% compared to baseline. Aβ40 concentrations in CSF did not decrease significantly in this study. In another Phase II study, 51 individuals with mild‐to‐moderate AD were randomized to receive placebo (n = 15) or semagacestat [100 mg (n = 22) or 140 mg (n = 14)] for 14 weeks, with 43 patients completing the treatment phase [56]. Patients on semagacestat received 60 mg/day for 2 weeks, then 100 mg/day for 6 weeks, and then either 100 or 140 mg/day for 6 additional weeks. There were 7 cases with skin rashes and 3 reports of hair color change in the drug treatment groups. There were 3 adverse event‐related discontinuations, including 1 transient bowel obstruction. Compared to placebo, Aβ40 plasma concentrations were reduced by 58% in the 100‐mg group and 65% in the 140‐mg group. No significant reduction was seen in CSF Aβ levels in this study. No differences were seen in cognitive or functional measures between placebo‐ and semagacestat‐treated patients [56]. No data were reported on the effects of semagacestat on Aβ42 levels. More recently, the results of a study on the effects of semagacestat on Aβ synthesis and clearance in CSF of AD patients were reported [57]. CSF was collected hourly for 36 h with a lumbar catheter. Semagacestat significantly decreased newly generated Aβ in a dose‐dependent fashion, with inhibition of Aβ generation of 47%, 52%, and 84% over the first 12‐h period with doses of 100, 140, and 280 mg, respectively. However, ELISA determinations revealed that there was a late rebound of CSF Aβ42 levels compared to placebo in the 20–36‐h period especially after the highest dose of 280 mg [57].
At present, two ongoing Phase III clinical trials were listed on the NIH's clinical trials registry. In March 2008, the first trial, called Interrupting Alzheimer's Dementia by EvaluatiNg Treatment of AmyloId PaThologY (IDENTITY) trial, was a Phase III, randomized, double‐blind, placebo‐controlled, parallel‐assignment, multicenter clinical trial (clinicaltrials.gov identifier: NCT00594568; H6L‐MC‐LFAN) in patients with mild‐to‐moderate AD (expected n = 1500). Patients would be treated with semagacestat (100 or 140 mg p.o., q.d.) for 21 months, with the option of enrolling in an open‐label extension trial for further treatment. Patients taking symptomatic treatments for AD were permitted to continue treatment. The trial incorporates a “randomized delayed start” design, which means that patients initially assigned to the placebo arm will be administered semagacestat sometime before the end of the 21‐month period to assess the effects on disease progression. The primary outcome measures of efficacy are the Alzheimer's Disease Assessment Scale‐Cognition (ADAS‐cog) for cognition and the Alzheimer's Disease Cooperative Study‐Activities of Daily Living scale (ADCS‐ADL) for functionality. Secondary endpoints include Aβ levels in plasma and CSF and other brain biomarkers determined by neuroimaging [58]. Eli Lilly estimated the trial would be completed by May 2011.
The second trial, called IDENTITY‐2, was also a Phase III, randomized, double‐blind, placebo‐controlled, parallel‐assignment, multicenter clinical trial (clinicaltrials.gov identifier: NCT00762411; H6L‐MC‐LFBC) in patients with mild‐to‐moderate AD (expected n = 1100). Patients would be treated with semagacestat (60 mg p.o., q.d., titrated to 140 mg p.o., q.d.) for 21 months, with the option of enrolling in an open‐label extension trial for further treatment. Patients taking symptomatic treatments for AD were permitted to continue treatment. The trial also incorporates a randomized delayed start design, similar to IDENTITY. The primary outcome measures of efficacy is the ADAS‐Cog scale for cognition and the ADCS‐ADL scale for functionality. Secondary endpoints include other dementia rating scales, Aβ levels in plasma and CSF, and other brain biomarkers determined by neuroimaging. The dose titration based on patient tolerability was designed to provide a more “real‐world simulation” of semagacestat [59]. Eli Lilly estimated the trial would be completed by March 2012. Finally, in December 2009, Eli Lilly launched an open‐label extension called Identity XT (ClinicalTrials.gov Identifier: NCT01035138) for AD patients who completed one of two semagacestat Phase III double‐blind studies, IDENTITY, or IDENTITY 2 (H6L‐MC‐LFAN or H6L‐MC‐LFBC), with an estimated enrollment of 1700 patients and an estimated study completion by January 2014 [60].
MK‐0752
Merck is developing a γ‐secretase inhibitor (MK‐0752) that does not distinguish between APP and Notch. A Phase I study evaluated the safety, tolerability, pharmacokinetics, and pharmacodynamics of single oral doses (from 110 to 1000 mg) of MK‐0752 in 27 healthy young men [43]. The drug was generally well tolerated. Drug plasma levels increased proportionally to the dose peaked at 3–4 h and then declined with a half‐life of about 20 h. Doses of 500 mg significantly inhibited for 12 h Aβ40 concentrations in CSF with a peak effect of –35%. After 1000 mg, CSF Aβ40 inhibition was sustained over 24 h. Plasma Aβ40 concentrations also showed a dose‐dependent decrease but were followed by a later rebound over baseline levels.
Although MK‐0752 was initially developed as a treatment for AD, there is increasing interest in the applicability of this drug to the treatment of cancer. Indeed, MK‐0752 has been shown to inhibit γ‐secretase‐mediated cleavage of Notch with an IC50 of 55 nM. Increasing evidence implicates the Notch pathway in normal T‐cell lymphopoiesis and the pathogenesis of several human malignancies. Prolonged activation of the Notch signal transduction pathway occurs in more than 50% of patients with T‐cell acute lymphoblastic leukemias and is important in the pathogenesis of the disease. Preclinical studies indicate that pharmacologic inhibition of γ‐secretase activity suppresses T‐cell acute lymphoblastic leukemias cell growth and induces apoptosis by preventing cleavage of Notch, thus preventing prolonged activation in downstream pathways. Studies with MK‐0752 in pediatric and adult patients with T‐cell acute lymphoblastic leukemias and acute myeloid leukemia reported that drug was associated with gastrointestinal toxicity and fatigue without substantive clinical activity [60, 61]. An intermittent dosing schedule appears to reduce toxicity while demonstrating adequate target inhibition, warranting further evaluation [61, 62].
BMS‐708163
BMS‐708163 is a potent, Notch‐sparing, γ‐secretase inhibitor in development at Bristol‐Myers Squibb. In vitro, the drug shows a 193‐fold selectivity versus Notch cleavage [44]. Studies in rats and dogs have shown that BMS‐708163 is able to decrease brain and CSF Aβ40 levels without causing Notch‐related gastrointestinal and lymphoid toxicity [44]. Studies in healthy young subjects have indicated that BMS‐708163 is well tolerated up to 400 mg after single administration and up to 150 mg/day after multiple doses for 28 days [44]. After oral administration, BMS‐708163 appears to be quickly absorbed (Tmax= 1–2 h), to produce systemic exposure proportional to the dose (up to 200 mg) and to be slowly eliminated (terminal half‐life ≈ 40 h). Single oral doses of BMS‐708163 decrease dose dependently Aβ40 levels in CSF with a peak inhibition of about 55% after 400 mg [44]. A multicenter, randomized, double‐blind, placebo‐controlled, Phase II trial of the safety, tolerability, and pharmacodynamic and pharmacokinetic effects of oral BMS‐708163 (25 to 125 mg q.d. for 12 weeks) in 200 patients with mild‐to‐moderate AD began in February 2009. At that time, the trial was estimated to complete in September 2010 (clinicaltrials.gov identifier: NCT00810147).
PF‐3084014
PF‐3084014 is a potent, Notch‐sparing, γ‐secretase inhibitor in development at Pfizer. In a cell‐free assay, PF‐3084014 appears to be a potent, noncompetitive but reversible inhibitor of human γ‐secretase activity with an IC50 of 6.2 nM [45]. In a whole‐cell assay, PF‐3084014 displays an IC50 of 1.3 nM. In fetal thymus organ culture assay, PF‐3084014 appears to be a weak inhibitor of Notch signaling with an IC50 of 1915 nM. The APP to Notch selectivity ratio is 1.473. In guinea pigs, dose‐response inhibition of total Aβ levels was observed in plasma, CSF, and brain after subcutaneous administration (0.03–10 mg/kg). At the highest dose (10 mg/kg), Aβ levels were reduced by 70% in brain and plasma, and by 50% in CSF, which was maintained at 30 h postdose. No late rebound effects on plasma Aβ were observed. Drug levels in the brain were similar to that measured in plasma. Studies in young (plaque‐free) Tg2576 transgenic mice showed that brain, CSF, and plasma levels of Aβ were inhibited dose dependently following doses of 1 to 18 mg/kg. At the highest dose (18 mg/kg), Aβ levels were reduced by 78% in brain, 72% in CSF, and 92% in plasma. Aβ40 was most potently inhibited in all compartments. Aβ42 showed approximately 20% less reduction than Aβ40 in all compartments. In a Phase I study in healthy volunteers, single doses of 1–120 mg were safe and well tolerated, and the maximum tolerated dose was not identified. The plasma half‐life of the drug was approximately 19 h [63]. The analysis of the pharmacokinetic and pharmacodynamic data derived from another Phase I study employing multiple doses has recently led to the decision to end development of the compound for AD [64]. In this study, plasma drug concentrations and plasma Aβ levels were collected from 18 healthy volunteers that received 40 or 90 mg once daily for 14 days. Pharmacokinetic/pharmacodynamic modeling of these data yielded the finding that exposure levels needed to reach the prespecified inhibition of plasma Aβ40 levels were 2‐ to 3‐fold higher than exposure limits based on animal toxicology data. As a result, much higher doses than those previously used would be needed to attain a high probability of pharmacological activity in man. PF‐3084014 has recently entered clinical studies as an anticancer agent. A Phase I study in patients with advanced solid tumors and leukemia, with an estimated enrollment of 60, is currently recruiting patients. The open‐label, dose‐escalation study is designed to evaluate safety (ClinicalTrials.gov Identifier: NCT00878189).
GSI‐953 (Begacestat)
GSI‐953 is a potent γ‐secretase inhibitor in development at Wyeth (Madison, NJ, USA). This compound inhibits Aβ production with low nM potency in both cellular (Aβ1–42 IC50= 15 nM) and cell‐free (IC50= 8 nM) assays [65]. Cellular assays of Notch cleavage reveal that this compound is 15‐fold selective for the inhibition of APP cleavage [65]. In Tg2576 mice, high doses of GSI‐953 significantly reduced Aβ1–40 levels in brain, CSF, and plasma [65]. At lower doses, GSI‐953 significantly reduced Aβ1–40 only in brain and plasma but not in CSF. Importantly, this compound has been reported to reverse contextual memory deficits in Tg2576 transgenic mice [65].
The pharmacokinetic, pharmacodynamic, and tolerability profile of GSI‐953 after single oral administration has been recently described in young subjects and in AD patients [46]. GSI‐953 was well tolerated and no dose‐limiting adverse events were observed. The drug was rapidly absorbed in both young subjects and AD patients (Tmax= 1–2 h). Drug plasma levels increased proportionally to the dose. Plasma elimination half‐life was also similar in the two populations (7–8 h). Drug concentrations in CSF were 10‐fold lower than plasma in both young and AD subjects. For both young healthy subjects and AD patients, a biphasic pattern of plasma Aβ40 concentrations was observed, with an initial reduction below baseline for approximately 4 h, followed by a second phase of increased concentrations above baseline lasting up to 48 h before returning to baseline. Maximum inhibition in plasma Aβ40 concentrations was observed at 2 h with a 28% reduction in AD subjects and 33% reduction in young subjects. Notably, initial reductions in plasma concentrations were less pronounced for Aβ42 than Aβ40. Maximum reductions in plasma Aβ42 were only 7% in AD subjects and 17% in young subjects. Subsequent increases in Aβ42 above baseline were similar to those of Aβ40 in magnitude and duration. No significant effects of the drug on CSF Aβ40 levels were observed in either AD patients or young volunteers.
E2012
E2012 is a novel γ‐secretase inhibitor claimed to lower Aβ by modulating γ‐secretase without inferring with Notch processing [40]. It was developed by Eisai in a partnership with Torrey‐Pines Therapeutics (La Jolla, CA, USA). In mid‐2006, the compound entered Phase I clinical development. In February 2007, the Phase I study was put on hold because lenticular opacity was observed in a high‐dose group of a 13‐week safety preclinical study in rats. At the time when the study was suspended, no medical concerns were reported. Lenticular opacity was not observed in a later 13‐week safety study in monkeys [40]. An additional 13‐week multiple dosing study in rats did not reveal ocular toxicity and the clinical hold on E2012 was lifted in April 2008. The drug is now headed back into a Phase 1 clinical trial [40].
Efficacy of γ−Secretase Inhibitors
The hypothesis that Aβ is the key pathologic factor affecting the disease process has been strongly questioned. The most recent challenge derives from a recently published article: although immunization with preaggregated Aβ42 (AN‐1792; formerly in development by Elan Corp plc and Wyeth) resulted in almost complete clearance of SPs from the brain of patients with AD, plaque removal did not prevent progressive neurodegeneration [66]. Aβ may have a physiological role in modulating synaptic plasticity [67] and hippocampal neurogenesis [68]. Aβ deposition could simply represent a host response to an upstream pathophysiologic process [20] or serving a protective function [69] likely as an antioxidant/metal chelator [70, 71]. Nevertheless, pharmaceutical companies have devoted huge efforts and finances to the development of agents that block γ‐secretase with several compounds being actively studied in humans, including AD patients [40]. Semagacestat is the most advanced γ‐secretase inhibitor in clinical development with two Phase III trials ongoing [47].
The Aβ reduction in plasma or CSF required to modify the rate of AD progression remains largely unknown [40]. A study in transgenic mice suggested that single doses of DAPT causing 25 to 30% reductions in soluble brain Aβ levels were associated with attenuation of cognitive deficit [39]. As outlined above, semagacestat produced significant and long‐lasting inhibition of brain Aβ levels at relatively low doses in transgenic mice. However, in nontransgenic animals (mice, guinea pigs, and dogs), the effect of the drug appears to be much more complex with inhibitory or stimulatory effects depending on the dose and the time. In healthy human volunteers, semagacestat dose‐dependently inhibited Aβ in CSF with approximately 80% inhibition of newly formed Aβ and by approximately 50% inhibition of total Aβ levels at high doses. However, some rebound effects have been observed, particularly on Aβ42 levels in CSF. The potential clinical consequences of the rebound in this neurotoxic Aβ species are not known.
The effects of chronic administration of γ‐secretase on brain Aβ deposition have been documented in a few studies [32]. The first published study was with respect to MRK‐560, a potent γ‐secretase inhibitor (IC50 = 4.3 nM) investigated by Merck. In this study, MRK‐560 (3 mg/kg/day p.o. for 3 months) decreased the number of Aβ deposits by 49% in Tg2576 mice. The mean size of the remaining SPs remained unchanged suggesting that MRK‐560 did not alter deposit growth once initiated, which may suggest that an earlier intervention with γ‐secretase inhibitors would be more effective on brain Aβ pathology [72]. Another recent study reported that LY‐411575 (3 mg/kg/day p.o. for 3 months) reduced neocortical plaque area by 80% in transgenic mice (bearing the Swedish mutation). This group also reported on LY‐411575 (3 mg/kg/day p.o. for 2 months) in different transgenic mice (bearing both Swedish and “London”[Val717Ile] mutations), which demonstrate more diffuse and AD‐like plaque pathology. Aβ deposition was reduced by only 31% in the neocortex but by 79% in the caudate putamen, which has a lower Aβ load than the neocortex [73]. These observations suggest that γ‐secretase inhibitors effectively reduce further Aβ formation in brains already containing SPs and that the efficacy is inversely correlated with the initial Aβ load. At present, only one study describing the effects of chronic semagacestat treatment had been reported (3, 10, or 30 mg/kg/day p.o. for 5 months in PDAPP mice) [74]. Quantitative analysis of Aβ immunohistochemistry data did not demonstrate significant changes in total plaque burden between treatment groups. However, the median plaque burden was reduced by 43 and 48% in the cortex and hippocampus, respectively, of treated animals compared with controls. Unfortunately, the study was not fully published but these data suggested that the inhibitory effects of semagacestat on plaque burden were not dramatic, although median inhibition provides more robust data [74].
Studies on the cognitive and behavioral effects of γ‐secretase inhibitors in animal models of AD are very limited [41]. While single oral administration of DAPT or begacestat or the acute intrahippocampal administration of L‐685458 (Merck's peptidic γ‐secretase inhibitor) may improve contextual or spatial memory in rats [75] or in Tg2576 transgenic mice [39, 65], data on the cognitive effects of prolonged administration of oral γ‐secretase inhibitors is lacking. No data on the behavioral effects of semagacestat are available from either acute or chronic dosing studies. It is possible that any beneficial effects on learning and memory caused by the antagonism of brain Aβ production and deposition may be more than offset by other biochemical changes resulting from γ‐secretase inhibition, such as the chronic accumulation of the neurotoxic C‐terminal fragments of APP in neurons and the cerebral cortex [76]. This hypothesis is supported by the observation that conditional inactivation of PS‐1 is accompanied by beneficial effects on cognition in 3‐month‐old transgenic mice but not in 6‐month‐old animals [77]. The lack of chronic behavioral data represents the major weakness of the preclinical documentation of semagacestat and may undermine the confidence on the cognitive efficacy potential of the drug in AD patients [47].
Safety of γ‐Secretase Inhibitors
Although γ‐secretase has in many ways been an attractive target for AD therapeutics, γ‐secretase inhibition may have safety drawbacks. The PS knockout in mice resulted in an early embryonal lethality with the phenotype presenting skeletal and CNS defects similar to those of a Notch knockout [78]. Indeed, γ‐secretase inhibitors block proteolysis of Notch‐1, a particular γ‐secretase substrate, the Notch receptor by inhibiting cleavage at site 3 (“S3”) [79]. Physiological cleavage of Notch leads to release of the NICD that is translocated to the nucleus where it regulates transcription of target genes involved in cell development and in differentiation of adult self‐renewing cells. The inhibition of Notch cleavage has been associated with goblet cell hyperplasia in intestinal epithelium and changes in the immune system with a decrease of lymphocytes in the spleen and thymus [80, 81]. Unfortunately, semagacestat does not spare Notch cleavage in vitro and this may have pharmacological and clinical consequences [47]. In the first Phase II clinical trial, T‐lymphocyte and eosinophil counts were found to be slightly but statistically increased in semagacestat‐treated patients compared to placebo‐treated subjects. A decrease in an early marker (CD69) of activated mature T cells and thymocytes was also found. However, this change was no longer present 90 days after treatment discontinuation [55]. Indeed, clinical trials in AD patients have identified a relatively high rate of gastrointestinal side effects (nausea, vomiting, and diarrhea) that could be Notch mediated. Much more data have to be collected from human studies to establish the separation between well‐tolerated and toxic doses of γ‐secretase inhibitors in chronic treatment of AD patients. Recently, hair color changes in animals have been linked to the inhibition of Notch processing [82]. Interestingly, alterations in hair color have also been observed in a clinical study with semagacestat [56]. The use of a Notch‐related toxicity biomarker such as adipsin may be useful for the early detection of potential toxicity [83]. Compounds have been described and are being developed that specifically modulate the proteolytic activity of γ‐secretase. These compounds, called γ‐secretase modulators, shift the production of Aβ42 to Aβ38 and do not affect Notch cleavage, meaning they may be more attractive from a safety point of view than the traditional γ‐secretase inhibitors [32].
Conclusions
γ‐Secretase is a rationale target for the treatment of AD because it regulates the final step of the Aβ formation. One of the components of the γ‐secretase, PS, is of exceptional biological importance since point mutations of this protein cause the most frequent and aggressive forms of familial AD. Although γ‐secretase is a complicated and unusual proteolytic complex, several classes of potent inhibitors have been discovered. At least six compounds have entered clinical development. In order to rationally guide their clinical development, efforts are being pursued to characterize their pharmacodynamic profile, not only by measuring Aβ concentrations in plasma and CSF over time, but also by estimating the effects of these drugs on newly synthesized Aβ and its clearance with recently available isotopic techniques. Studies in both transgenic and nontransgenic animals have indicated that γ‐secretase inhibitors, administered by the oral route, are able to lower brain Aβ concentrations. However, scanty data are available on the effects of these compounds on soluble Aβ oligomers, the neurotoxic molecular species believed to be responsible for neuronal death in AD. Studies on the effects of γ‐secretase inhibitors on brain Aβ deposition after prolonged administration are also few. Behavioral and cognitive studies after chronic administration in animal models of AD have not been published. The major contribution to side effects of γ‐secretase inhibitors is thought to originate from the interference with intestinal and lymphatic cell differentiation associated with the inhibition of Notch cleavage. Fortunately, some γ‐secretase inhibitors appear to spare Notch cleavage in vitro and are well tolerated in vivo. One γ‐secretase inhibitor (semagacestat) has been properly characterized from the pharmacodynamic point of view in initial Phase I and II studies and now is in Phase III clinical testing. We will know if semagacestat does work in the next couple of years.
It was forecasted that by 2014 there would be at least two new anti‐AD drugs on the market. One could be Wyeth/Elan Corp's bapineuzumab (humanized mAb) and the other could be semagacestat [84], although the recent discontinuation of the highest dose in a Phase III trial of bapineuzumab [85] and the mixed clinical data we have reported here for semagacestat, may cast doubt on these predictions. Nevertheless, the approval of either one of these agents could have a considerable impact on the way AD is treated in the future as they target the underlying cause of the disease and may therefore be used in combination with other agents to stop or slow the disease progression. It is expected that the commercial market will at least double in size upon entry of any new agents and it is believed that a small molecule compound (like semagacestat) with disease‐modifying properties could easily reach US$7 billion in sales in the United States alone [86]. However, semagacestat could be a relatively high development risk based on its poor selectivity on Notch cleavage, lack of cognitive, and behavioral data in preclinical animal models of AD and complex time‐ and dose‐dependent pharmacodynamics with preclinical and clinical indications of late Aβ42 rebounds in the CSF. Nevertheless, it and other γ‐secretase inhibitors represent a major hope to slow the rate of decline of AD and to modify the natural history of this devastating disease.
Conflicts of Interest
No Disclosures to Report.
Acknowledgments
This work was supported by the Ministero della Salute, IRCCS Research Program 2009–2011, Line 2: “Malattie complesse.” It was also supported by the Italian Longitudinal Study on Aging (ILSA) (Italian National Research Council – CNR‐Targeted Project on Ageing – Grants 9400419PF40 and 95973PF40). The funding agencies had no role in design or conduct of the study.
References
- 1. Alzheimer's Association . Alzheimer's disease facts and figures. Alzheimer Dementia 2009;53:234–270. [DOI] [PubMed] [Google Scholar]
- 2. Brookmeyer R, Johnson E, Ziegler‐Graham K, Arrighi HM. Forecasting the global burden of Alzheimer's disease. Alzheimer Dementia 2008;4:316–323. [DOI] [PubMed] [Google Scholar]
- 3. Panza F, Solfrizzi V, Frisardi V, et al Disease‐modifying approach to the treatment of Alzheimer's disease: From alpha‐secretase activators to gamma‐secretase inhibitors and modulators. Drugs Aging 2009;26:537–555. [DOI] [PubMed] [Google Scholar]
- 4. Frisardi V, Solfrizzi V, Imbimbo BP, et al Towards disease‐modifying treatment of Alzheimer's disease: Drugs targeting beta‐amyloid. Curr Alzheimer Res 2010;7:40–55. [DOI] [PubMed] [Google Scholar]
- 5. Maelicke A, Samochocki M, Jostock R, Fehrenbacher A, Ludwig J, Albuquerque EX, Zerlin M. Allosteric sensitization of nicotinic receptors by galantamine, a new treatment strategy for Alzheimer's disease. Biol Psychiatry 2001;49:279–288. [DOI] [PubMed] [Google Scholar]
- 6. Greig NH, Utsuki T, Yu Q, et al A new therapeutic target in Alzheimer's disease treatment: Attention to butyrylcholinesterase. Curr Med Res Opin 2001;17:159–165. [DOI] [PubMed] [Google Scholar]
- 7. Wilkinson DG, Francis PT, Schwam E, Payne‐Parrish J. Cholinesterase inhibitors used in the treatment of Alzheimer's disease: The relationship between pharmacological effects and clinical efficacy. Drugs Aging 2004;21:453–478. [DOI] [PubMed] [Google Scholar]
- 8. Raina P, Santaguida P, Ismaila A, et al Effectiveness of cholinesterase inhibitors and memantine for treating dementia: Evidence review for a clinical practice guideline. Ann Intern Med 2008;148:379–397. [DOI] [PubMed] [Google Scholar]
- 9. Panza F, Solfrizzi V, Frisardi V, et al Beyond the neurotransmitter focused approach in treating Alzheimer's Disease: Drugs targeting β‐amyloid and tau protein. Aging Clin Exp Res 2009;21:386–406. [DOI] [PubMed] [Google Scholar]
- 10. Opar A. Mixed results for disease‐modification strategies for Alzheimer's disease. Nat Rev Drug Discov 2008;7:717–718. [DOI] [PubMed] [Google Scholar]
- 11. Alzheimer A. Uber eine eigenartige erkrankung der hirnrinde. Allg Zeitschrift Psych 1907;64:146–148. [Google Scholar]
- 12. Terry RD. Alzheimer's Disease at mid century (1927–1977) In: Jucker M, Beyreuther K, Haas C, Nitsch R, editors. Alzheimer: 100 years and beyond. Berlin Heidelberg : Springer‐Verlag, 2006;58–61. [Google Scholar]
- 13. Small SA, Duff K. Linking Aβ and tau in late‐onset Alzheimer's disease: A dual pathway hypothesis. Neuron 2008;60:534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Giacobini E, Becker RE. One hundred years after the discovery of Alzheimer's disease. A turning point for therapy? J Alzheimers Dis 2007;12:37–52. [DOI] [PubMed] [Google Scholar]
- 15. Walter J, Kaether C, Steiner H, Haass C. The cell biology of Alzheimer's disease: Uncovering the secrets of secretases. Curr Opin Neurobiol 2001;11:585–590. [DOI] [PubMed] [Google Scholar]
- 16. Ohyagi Y, Asahara H, Chui DH, et al Intracellular Abeta42 activates p53 promoter: A pathway to neurodegeneration in Alzheimer's disease. FASEB J 2005;19:255–257. [DOI] [PubMed] [Google Scholar]
- 17. Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci 1991;12:383–388. [DOI] [PubMed] [Google Scholar]
- 18. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science 2002;297:353–356. [DOI] [PubMed] [Google Scholar]
- 19. Shankar GM, Li S, Mehta TH, et al Amyloid‐β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 2008;14:837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Castellani RJ, Lee HG, Zhu X, Perry G, Smith MA. Alzheimer disease pathology as a host response. J Neuropathol Exp Neurol 2008;67:523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Knopman DS, Parisi JE, Salviati A, et al Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol 2003;62:1087–1095. [DOI] [PubMed] [Google Scholar]
- 22. Wisniewski T, Konietzko U. Amyloid‐beta immunisation for Alzheimer's disease. Lancet Neurol 2008;7:805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wolfe MS, Kopan R. Intramembrane proteolysis: Theme and variations. Science 2004;305:1119–1123. [DOI] [PubMed] [Google Scholar]
- 24. Armogida M, Petit A, Vincent B, Scarzello S, Da Costa CA, Checler F. Endogenous β‐amyloid production in presenilin deficient embryonic mouse fibroblasts. Nature Cell Biol 2001;3:1030–1033. [DOI] [PubMed] [Google Scholar]
- 25. Wilson CA, Doms RW, Zheng H, Lee VM. Presenilins are not required for Ab42 production in the early secretory pathway. Nature Neurosci 2002;5:849–855. [DOI] [PubMed] [Google Scholar]
- 26. Lai MT, Crouthamel MC, DiMuzio J, et al A presenilin‐independent aspartyl protease prefers the gamma‐42 site cleavage. J Neurochem 2006;96:118–125. [DOI] [PubMed] [Google Scholar]
- 27. Dovey HF, John V, Anderson JP, et al Functional γ‐secretase inhibitors reduce β‐amyloid peptide levels in brain. J Neurochem 2001;76:173–181. [DOI] [PubMed] [Google Scholar]
- 28. Wolfe MS. Inhibition and modulation of gamma‐secretase for Alzheimer's disease. Neurotherapeutics 2008;5:391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lleo A, Berezovska O, Growdon JH, Hyman BT. Clinical, pathological, and biochemical spectrum of Alzheimer disease associated with PS‐1 mutations. Am J Geriatr Psychiat 2004;12:146–156. [DOI] [PubMed] [Google Scholar]
- 30. Bentahir M, Nyabi O, Verhamme J, et al Presenilin clinical mutations can affect γ‐secretase activity by different mechanisms. J Neurochem 2006;96:732–742. [DOI] [PubMed] [Google Scholar]
- 31. Olson RE, Albright CF. Recent progress in the medicinal chemistry of γ‐secretase inhibitors. Curr Top Med Chem 2008;8:17–33. [DOI] [PubMed] [Google Scholar]
- 32. Imbimbo BP. Therapeutic potential of gamma‐secretase inhibitors and modulators. Curr Top Med Chem 2008;8:54–61. [DOI] [PubMed] [Google Scholar]
- 33. Guardia‐Laguarta C, Pera M, Lleó A. gamma‐Secretase as a therapeutic target in Alzheimer's disease. Curr Drug Targets 2010;11:506–517. [DOI] [PubMed] [Google Scholar]
- 34. De Strooper B, Annaert W, Cupers P, et al A presenilin‐1‐dependent γ‐secretase‐like protease mediates release of Notch intracellular domain. Nature 1999;398:518–522. [DOI] [PubMed] [Google Scholar]
- 35. Sestan N, Artavanis‐Tsakonas S, Rakic P. Contact‐dependent inhibition of cortical neurite growth mediated by notch signaling. Science 1999;286:741–746. [DOI] [PubMed] [Google Scholar]
- 36. Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK. A CBP binding transcriptional repressor produced by the PS1/γ‐cleavage of Ncadherin is inhibited by PS1 FAD mutations. Cell 2003;114:635–645. [DOI] [PubMed] [Google Scholar]
- 37. Ni CY, Murphy MP, Golde TE, Carpenter G. γ‐Secretase cleavage and nuclear localization of ErbB‐4 receptor tyrosine kinase. Science 2001;294:2179–2181. [DOI] [PubMed] [Google Scholar]
- 38. Muller T, Meyer HE, Egensperger R, Marcus K. The amyloid precursor protein intracellular domain (AICD) as modulator of gene expression, apoptosis, and cytoskeletal dynamics‐relevance for Alzheimer's disease. Prog Neurobiol 2008;85:393–406. [DOI] [PubMed] [Google Scholar]
- 39. Comery TA, Martone RL, Aschmies S, et al Acute γ‐secretase inhibition improves contextual fear conditioning in the Tg2576 mouse model of Alzheimer's disease. J Neurosci 2005;25:8898–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Imbimbo BP. Alzheimer's disease: γ‐Secretase inhibitors. Drug Disc Today-Ther Strategies 2008;5:169–175. [Google Scholar]
- 41. Best JD, Jay MT, Otu F, et al Quantitative measurement of changes in amyloid‐β(40) in the rat brain and cerebrospinal fluid following treatment with the γ‐secretase inhibitor LY‐411575 [N2‐[(2S)‐2‐(3,5‐difluorophenyl)‐2‐hydroxyethanoyl]‐N1‐[(7S)‐5‐methyl‐6‐oxo‐6,7‐dihydro‐5H‐dibenzo[b,d]azepin‐7‐yl]‐L‐alaninamide]. J Pharmacol Exp Ther 2005;313:902–908. [DOI] [PubMed] [Google Scholar]
- 42. Gitter BD, Czilli DL, Li W, et al Stereoselective inhibition of amyloid β peptide secretion by LY450139 dihydrate, a novel functional gamma secretase inhibitor [Abstract P4.339]. Neurobiol Aging 2004;25(Suppl 2):S571. [Google Scholar]
- 43. Rosen LB, Stone JA, Plump A, et al The β secretase inhibitor MK‐0752 acutely and significantly reduces CSF Aβ40 concentrations in humans [Abstract O4–03‐02]. Alzheimer Dement 2006;2(Suppl. 1):S79. [Google Scholar]
- 44. Albright CF, Dockens R, Olson RE, et al BMS‐708163, a potent and selective γ‐secretase inhibitor, decreases CSF Aβ at safe and tolerable doses in animals and humans [Abstract HT‐01‐05]. International Conference on Alzheimer's Disease 2008, 26–31 July, Chicago, IL, USA.
- 45. Wood KM, Lanz TA, Coffman KJ, et al Efficacy of the novel γ‐secretase inhibitor, Pf‐3084014, in reducing aβ in brain, CSF, and plasma in guinea pigs and Tg2576 mice [Abstract P2–375]. Alzheimer Dementia 2008;4(Suppl 2):T482. [Google Scholar]
- 46. Frick G, Raje S, Wan H, et al GSI‐953, a potent and selective γ‐secretase inhibitor ‐Modulation of β amyloid peptides and plasma and cerebrospinal fluid pharmacokinetic/pharmacodynamic relationships in humans [Abstract P4–366]. Alzheimer Dementia 2008;4(Suppl 2):T781. [Google Scholar]
- 47. Imbimbo BP, Peretto I. Semagacestat, a gamma‐secretase inhibitor for the potential treatment of Alzheimer's disease. Curr Opin Investig Drugs 2009;10:721–730. [PubMed] [Google Scholar]
- 48. Boggs LN, Fuson KS, Gitter BD, et al In vivo characterization of LY450139, a novel, stereoselective, functional γ‐secretase inhibitor [Abstract P1–419]. Neurobiol Aging 2004;25(Suppl 2):218. [Google Scholar]
- 49. May PC, Yang Z, Li WY, Hyslop PA, Siemers E, Boggs LN. Multi‐compartmental pharmaco‐dynamic assessment of the functional γ‐secretase inhibitor LY450139 in PDAPP transgenic mice and nontransgenic mice [Abstract 03–06‐08]. Neurobiol Aging 2004;25(Suppl 2):S65. [Google Scholar]
- 50. Lanz TA, Karmilowicz MJ, Wood KM, et al Concentration‐dependent modulation of Aβ in vivo and in vitro using the γ‐secretase inhibitor, LY‐ 450139. J Pharmacol Exp Ther 2006;319:924–933. [DOI] [PubMed] [Google Scholar]
- 51. Cocke PJ, Boggs LN, Ness DK, May PC. Comparison of cisternal and lumbar cerebrospinal fluid amyloid beta concentrations following oral administration of LY450139 dihydrate to Beagle dogs [Abstract P1‐017]. Alzheimers Dement 2008;4(Suppl 2):T210. [Google Scholar]
- 52. Ness DK, Boggs LN, Hepburn DL, et al Reduced β‐amyloid burden, increased C‐99 concentrations and evaluation of neuropathology in the brains of PDAPP mice given LY450139 dihydrate daily by gavage for 5 months [Abstract P2–053]. Neurobiol Aging 2004;25(Suppl 2):S238. [Google Scholar]
- 53. Siemers E, Skinner M, Dean RA, et al Safety, tolerability, and changes in amyloid beta concentrations after administration of a γ‐secretase inhibitor in volunteers. Clin Neuropharmacol 2005;28:126–132. [DOI] [PubMed] [Google Scholar]
- 54. Siemers ER, Dean RA, Friedrich S, Ferguson‐Sells L, Gonzales C, Farlow MR, May PC. Safety, tolerability, and effects on plasma and cerebrospinal fluid amyloid‐beta after inhibition of gamma‐secretase. Clin Neuropharmacol 2007;30:317–325. [DOI] [PubMed] [Google Scholar]
- 55. Siemers ER, Quinn JF, Kaye J, et al Effects of a γ‐secretase inhibitor in a randomized study of patients with Alzheimer disease. Neurology 2006;66:602–604. [DOI] [PubMed] [Google Scholar]
- 56. Fleisher AS, Raman R, Siemers ER, et al Phase 2 safety trial targeting amyloid beta production with a gamma‐secretase inhibitor in Alzheimer disease. Arch Neurol 2008;65:1031–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bateman RJ, Siemers ER, Mawuenyega KG, et al A gamma‐secretase inhibitor decreases amyloid‐beta production in the central nervous system. Ann Neurol 2009;66:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. ClinicalTrial.gov . Effect of LY450139 on the long term progression of Alzheimer's disease. Available from: http://clinicaltrials.gov/ct2/show/NCT00594568?term=NCT00594568&rank=1. [Assessed 14 March 2010.
- 59. ClinicalTrial.gov . Effects of LY450139, on the Progression of Alzheimer's Disease as Compared With Placebo (IDENTITY‐2). Available from: http://clinicaltrials.gov/ct2/show/NCT00762411?term=IDENTITY&rank=9. [Assessed 14 March 2010.
- 60. ClinicalTrial.gov . A Study in Semagacestat for Alzheimer's Patients (Identity XT). Available from: http://clinicaltrials.gov/ct2/show/NCT01035138?term=IDENTITY&rank=8. [Assessed 14 March 2010.
- 61. Deangelo DJ, Stone RM, Silverman LB, et al A phase I clinical trial of the notch inhibitor MK‐0752 in patients with T‐cell acute lymphoblastic leukemia/lymphoma (T‐ALL) and other leukemias [Abstract 6585]. J Clin Oncol 2006;24(Suppl 18):S357. [Google Scholar]
- 62. Krop IE, Kosh M, Fearen I, et al Phase I pharmacokinetic, and pharmacodynamic trial of the novel oral Notch inhibitor MK‐0752 in patients with advanced breast cancer and other solid tumors [Abstract 6094]. Breast Cancer Res Treat 2006;100(Suppl. 1):S287. [Google Scholar]
- 63. Brodney M. Discovery of PF‐3084014: a novel γ‐secretase inhibitor for the treatment of Alzheimer's disease [Abstract MEDI 233]. 238th American Chemistry Society National Meeting. Washington, DC, August 16–20, 2009.
- 64. Qiu R, Willavize S, Fullerton T, Gastonguay MR. Modeling and simulation of plasma Aβ in humans after multiple oral doses of PF‐3084014, a potent gamma secretase inhibitor [Abstract P1‐261]. 12th International Conference of Alzheimer's Disease and Related Disorders (ICAD). Vienna, July 11–16, 2009. Alzheimer's & Dementia 2009;59(Suppl 1):P253.
- 65. Martone RL, Zhou H, Atchison K, et al Begacestat (GSI‐953): A novel, selective thiophene sulfonamide inhibitor of amyloid precursor protein gamma‐secretase for the treatment of Alzheimer's disease. J Pharmacol Exp Ther 2009;331:598–608. [DOI] [PubMed] [Google Scholar]
- 66. Holmes C, Boche D, Wilkinson D, et al Long‐term effects of Aβ42 immunisation in Alzheimer's disease: Follow‐up of a randomised, placebo‐controlled phase I trial. Lancet 2008;372:216–223. [DOI] [PubMed] [Google Scholar]
- 67. Puzzo D, Privitera L, Leznik E, Fà M, Staniszewski A, Palmeri A, Arancio O. Picomolar amyloid‐β positively modulates synaptic plasticity and memory in hippocampus. J Neurosci 2008;28:14,537–14,545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen Y, Dong C. Aβ40 promotes neuronal cell fate in neural progenitor cells. Cell Death Differ 2009;16:386–394. [DOI] [PubMed] [Google Scholar]
- 69. Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Amyloid beta: The alternate hypothesis. Curr Alzheimer Res 2006;3:75–80. [DOI] [PubMed] [Google Scholar]
- 70. Hayashi T, Shishido N, Nakayama K, Nunomura A, Smith MA, Perry G, Nakamura M. Lipid peroxidation and 4‐hydroxy‐2‐nonenal formation by copper ion bound to amyloid‐beta peptide. Free Radic Biol Med 2007;43:1552–1559. [DOI] [PubMed] [Google Scholar]
- 71. Nakamura M, Shishido N, Nunomura A, et al Three histidine residues of amyloid‐beta peptide control the redox activity of copper and iron. Biochemistry 2007;46:12,737–12,743. [DOI] [PubMed] [Google Scholar]
- 72. Best JD, Smith DW, Reilly MA, et al The novel γ secretase inhibitor N‐[cis‐4‐[(4 chlorophenyl)sulfonyl]‐4‐(2,5‐difluorophenyl)cyclohexyl]‐1,1,1‐trifluoromethanesulfonamide (MRK‐560) reduces amyloid plaque deposition without evidence of notch‐related pathology in the TG2576 mouse. J Pharmacol Exp Ther 2007;320:552–558. [DOI] [PubMed] [Google Scholar]
- 73. Abramowski D, Wiederhold KH, Furrer U, et al Dynamics of Aβ turnover and deposition in different β‐amyloid precursor protein transgenic mouse models following γ‐secretase inhibition. J Pharmacol Exp Ther 2008;327:411–424. [DOI] [PubMed] [Google Scholar]
- 74. Ness DK, Boggs LN, Hepburn DL, et al Reduced β‐amyloid burden, increased C‐99 concentrations and evaluation of neuropathology in the brains of PDAPP mice given LY450139 dihydrate daily by gavage for 5 months. Neurobiol Aging 2004;25(Suppl 2):P2–053 [abstract]. [Google Scholar]
- 75. Dash PK, Moore AN, Orsi SA. Blockade of γ‐secretase activity within the hippocampus enhances long‐term memory. Biochem Biophys Res Commun 2005;338:777–782. [DOI] [PubMed] [Google Scholar]
- 76. Lee JP, Chang KA, Kim HS, Kim SS, Jeong SJ, Suh YH. APP carboxyl‐terminal fragment without or with Aβ domain equally induces cytotoxicity in differentiated PC12 cells and cortical neurons. J Neurosci Res 2000;60:565–570. [DOI] [PubMed] [Google Scholar]
- 77. Saura CA, Chen G, Malkani S, et al Conditional inactivation of presenilin 1 prevents amyloid accumulation and temporarily rescues contextual and spatial working memory impairments in amyloid precursor protein transgenic mice. J Neurosci 2005;25:6755–6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in presenilin‐1‐deficient mice. Cell 1997;89:629–639. [DOI] [PubMed] [Google Scholar]
- 79. Lewis HD, Pérez Revuelta BI, Nadin A, Neduvelil JG, Harrison T, Pollack SJ, Shearman MS. Catalytic site‐directed γ‐secretase complex inhibitors do not discriminate pharmacologically between Notch S3 and beta‐APP cleavages. Biochemistry 2003;42:7580–7586. [DOI] [PubMed] [Google Scholar]
- 80. Maillard I, Adler SH, Pear WS. Notch and the immune system. Immunity 2003;19:781–791. [DOI] [PubMed] [Google Scholar]
- 81. Stanger BZ, Datar R, Murtaugh LC, Melton DA. Direct regulation of intestinal fate by Notch. Proc Natl Acad Sci USA 2005;102:12,443–12,448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Schouwey K, Beermann F. The Notch pathway: Hair graying and pigment cell homeostasis. Histol Histopathol 2008;23:609–619. [DOI] [PubMed] [Google Scholar]
- 83. Searfoss GH, Jordan WH, Calligaro DO, et al Adipsin, a biomarker of gastrointestinal toxicity mediated by a functional γ‐secretase inhibitor. J Biol Chem 2003;278:46,107–46,116. [DOI] [PubMed] [Google Scholar]
- 84. Samimy A, Wales M, Patel R. Alzheimer's research: Progressing toward investability? UBS 2007 November 15.
- 85. Elan and Wyeth plan to amend bapineuzumab phase 3 protocols. Elan Corp plc PRESS RELEASE 2009 April 02.
- 86. Leuchter B, Ho MK, Malloy M. Alzheimer's disease: Drug market poised for expansion. Goldman Sachs 2008 October 12.