

Abstract
The recent advent of genome‐wide mass‐marker technology has resulted in renewed optimism to unravel the genetic architecture of psychotic disorders. Genome‐wide association studies have identified a number of common polymorphisms robustly associated with schizophrenia, in ZNF804A, transcription factor 4, major histocompatibility complex, and neurogranin. In addition, copy number variants (CNVs) in 1q21.1, 2p16.3, 15q11.2, 15q13.3, 16p11.2, and 22q11.2 were convincingly implicated in schizophrenia risk. Furthermore, these studies have suggested considerable genetic overlap with bipolar disorder (particularly for common polymorphisms) and neurodevelopmental disorders such as autism (particularly for CNVs). The influence of these risk variants on relevant intermediate phenotypes needs further study. In addition, there is a need for etiological models of psychosis integrating genetic risk with environmental factors associated with the disorder, focusing specifically on environmental impact on gene expression (epigenetics) and convergence of genes and environment on common biological pathways bringing about larger effects than those of genes or environment in isolation (gene–environment interaction). Collaborative efforts that bring together expertise in statistics, genetics, epidemiology, experimental psychiatry, brain imaging, and clinical psychiatry will be required to succeed in this challenging task.
Keywords: Cannabis, Gene–environment interaction, GWAS, Psychosis, Trauma
Introduction
Schizophrenia is a heterogeneous psychotic syndrome, which, narrowly defined, affects about 1% of the population worldwide [1]. The DSM‐IV diagnostic criteria include psychotic and negative symptomatology severe enough to cause social and occupational dysfunction over a period of at least 6 months [2]. Although psychotic symptoms (hallucinations, delusions, disorganized speech, and behavior) and negative symptoms (e.g., flattened affect, avolition, social withdrawal) are widely recognized as core symptoms of schizophrenia, there also is consensus on the marked heterogeneity in clinical presentation [3]. Recent nosological approaches put forward the suggestion of incorporating dimensional representations of psychopathology in addition to more classic categorical models of classification [4].
Despite substantial clinical heterogeneity, twin studies have shown that schizophrenia is characterized by a high heritability, estimated around 80%[5]. A large body of work has addressed the association of several a priori hypothesized candidate genes with schizophrenia and it is likely that some of these candidate genes are true susceptibility factors, as suggested in a number of excellent articles specifically reviewing the evidence for these individual candidate genes (e.g., [6, 7, 8]). For none of these candidate genes, however, does the association with schizophrenia remain undisputed, and serious methodological concerns with regard to the candidate gene approach have been formulated [9, 10, 11, 12]. The recent advent of technology to simultaneously assess 1 million single nucleotide polymorphisms (SNPs) has resulted in renewed optimism to unravel the genetic architecture of psychotic disorders. This article will focus on the genetic findings emerging from genome‐wide studies and how they can be integrated with existing knowledge on the importance of environmental factors in the developmental pathway to psychosis and with evidence for gene–environment interaction (GxE).
Recent Genome‐Wide Studies
Genome‐wide association (GWA) studies contrast large numbers of genetic variants in patients and healthy control subjects. Since a large number of polymorphisms are investigated (100 thousand–1 million), and the anticipated effect sizes are small (odds ratios [OR] in the order of 1.10), large sample sizes are required in order to find even the smallest number of convincingly associated polymorphisms. It was estimated that a sample of about 12,000 cases and 12,000 controls will be needed to detect a common risk allele with a frequency of 0.2 and an additive OR of 1.15 at the genome‐wide threshold of significance of P < 7.2 × 10−8[13, 14].
Several GWA studies of increasingly larger samples of patients with schizophrenia have now been published and researchers across the globe are working together to further increase sample size and thus, statistical power [15]. In the first GWA studies in schizophrenia [16, 17], sample sizes were smaller than 1000 subjects and unsurprisingly, no genome‐wide significant findings were reported, although the smallest GWA study of only 178 cases and 144 controls did find suggestive evidence for a marker between “colony stimulating factor 2 receptor alpha” (CSF2RA) and “short stature homeobox isoform b” (SHOX) (P= 3.7 × 10−7), each gene being about 350 kb from the associated marker [16]. The relevance of this initial finding is unclear as it was not confirmed in larger GWA studies. Nevertheless, these initial studies demonstrated the low probability of finding common variants with a moderate‐to‐large effect (i.e., an OR above 2).
Given these power considerations, a third smaller GWA study used a two‐step approach [18]. In this study, the authors conducted a genome‐wide analysis of 479 cases and 2937 controls. None of the markers achieved genome‐wide significance, but 12 surpassed the P < 10−5 threshold. Subsequently, these 12 markers were followed up in a replication sample of more than 6600 cases and about 9900 controls. In the replication stage, three markers –“zinc‐finger binding protein” (ZNF804A) and two SNPs in intergenic regions at 11p14.1 and 16p13.12 – were strongly associated with schizophrenia in the follow‐up sample (P < 5 × 10−4). These P‐values are relatively convincing since only 12 instead of several hundreds of thousands of markers were interrogated, thus requiring less stringent control for multiple testing. Another two markers, in “opioid binding protein/cell adhesion molecule‐like” (OPCML) and the ciliary gene RPGRIP1L, showed modest evidence for association. The strongest associated marker, an SNP in an intron of ZNF804A, did not reach genome‐wide statistical significance (P= 1.61 × 10−7) when considering schizophrenia alone, but interestingly, it met criteria for genome‐wide significance when the affected phenotype included bipolar disorder (9.96 × 10−9). Since the original report, supportive evidence for association with ZNF804A was found in another genome‐wide study (P= 0.029) [19] and two case‐control studies [20, 21].
A number of smaller GWA studies have additionally been published [22, 23, 24, 25], none of them finding SNPs with genome‐wide significance, although one study found a female‐specific association with reelin, a neurodevelopmental gene previously associated with schizophrenia [23]. Follow‐up in four additional samples were in support of the initial finding and meta‐analysis of the combined samples suggested an estimated relative risk in women carrying the common genotype of 1.58 with a P‐value approaching genome‐wide statistical significance (P= 8.8 × 10−7).
The most recent contribution to the literature consisted of three back‐to‐back papers reporting genome‐wide data [19, 26, 27]. Combined analysis of their samples (12,945 cases; 34,591 controls) revealed genome‐wide significant associations with multiple SNPs across the major histocompatibility complex (MHC) at chromosome 6, with a marker located upstream of the neurogranin gene on 11q24.2 and a marker in an intron of transcription factor 4 (TCF4) [26]. One of these three studies suggested that at least 30% of schizophrenia liability may be explained by common polymorphisms [19].
Copy Number Variants (CNVs)
Genome‐wide data, however, cannot only help to provide insights into the genetic contribution of polymorphic variation but are also able to demonstrate the presence of deletions and duplications with a size of 1 or more kb, so‐called CNVs. The involvement of CNVs in schizophrenia has been known for many years, as a deletion in 22q11 is associated with a 25 times increased risk for schizophrenia [28]. Now there is strong evidence, however, that the 22q11 deletion is not the only CNV that is implicated in schizophrenia. Several studies have clearly shown an increased overall burden of rare CNVs (frequency <1%) in patients with schizophrenia [29, 30, 31, 32], although there were marked differences in the reported burden estimates. One study found an excess burden especially in early‐onset cases [29], whereas another study found that de novo CNVs were particularly common in families without a history of psychosis [32].
For now, the most convincing evidence exists for deletions in 1q21.1 and 15q13.3 that have been described in two large studies [31, 33]. These deletions are individually rare (frequency <1%) but confer large effects on disease risk, with estimated OR around 10 [30, 33]. An additional CNV suggested to be associated with schizophrenia, which probably has a smaller effect size (OR 3), is located in 15q11.2 [30, 33]. Recently, McCarthy and colleagues reported that, in a cohort of 1906 cases of schizophrenia and 3971 controls, microduplications in 16p11.2 conferred large effects on schizophrenia risk (OR 25.8), a finding that was replicated in an independent sample of 2645 cases and 2420 healthy controls (OR 8.3, combined OR 14.5) [34]. Furthermore, a meta‐analysis was conducted on a sample of 8590 individuals with schizophrenia, 2172 with autism, 4822 with bipolar disorder, and 30,492 controls. In the meta‐analysis, the 16p11.2 duplication was strongly associated with schizophrenia (P= 4.8 × 10−7), autism (P= 1.9 × 10−7), and to a lesser extent, bipolar disorder (P= 0.017), whereas the 16p11.2 microdeletion was only associated with developmental disorders or autism (P= 2.3 × 10−13) [34]. Also at chromosome 16, Ingasson and colleagues found an excess of duplications of 16p13.1 in cases with schizophrenia [35]. CNVs in this region were previously also reported in cases with autism and mental retardation [36, 37].
While these CNV findings may help to provide insights in the molecular etiological mechanisms underlying schizophrenia, this may not be an easy mission, especially since most CNVs span multiple genes. The difficulty of pinpointing etiological mechanisms associated with CNVs spanning multiple genes is illustrated by the 22q11.2 deletion, for which a consistent mechanism explaining the increased schizophrenia risk has yet to emerge, despite the presence of promising candidate genes such as catechol‐O‐methyltransferase (COMT) and proline dehydrogenase(PRODH) in this region.
One exception to the rule that CNVs span multiple genes is the deletion in the neurexin gene at 2p16.3. Several studies have shown that rare deletions of the neurexin gene, particularly those that disrupt exons, confer an increased risk of schizophrenia [22, 29, 30, 38, 39, 40, 41]. Together, these results provide good evidence that the neurexin CNV confers a considerable increase in risk for schizophrenia (see [42] for review). These results would implicate a neurodevelopmental pathway related to deficits in synaptic transmission in the etiology of schizophrenia, or at least in a subgroup of patients diagnosed with the disorder [43].
Genetic Heterogeneity
As previously mentioned, GWA studies contrast a large number of genetic variants in patients with that of healthy control subjects. Comparing individuals with schizophrenia versus healthy controls is feasible, at least as a first step, since studies have demonstrated the heritability of a schizophrenia diagnosis [5]. Nevertheless, few will disagree that in individuals diagnosed with schizophrenia, clinical heterogeneity is the rule rather than the exception. Schizophrenia is not a disease but a cluster of symptoms that usually, but not always and to a varying degree, is present in patients having the diagnosis.
Recent GWA studies have now convincingly shown that this clinical heterogeneity is underpinned by considerable genetic heterogeneity, given the evidence that possibly thousands of common polymorphisms contribute to schizophrenia risk [19] and evidence implicating multiple individually rare CNVs in several different genomic areas to risk for the disorder [29, 31, 32, 33, 34, 35].
Perhaps even more important, however, is the considerable genetic overlap with other neuropsychiatric conditions reported in recent GWA studies. One study showed that a combined set of schizophrenia “score alleles,” that is, alleles that were numerically more frequent in patients with schizophrenia in the International Schizophrenia Consortium discovery sample at five thresholds of increasingly relaxed significance (P < 0.10, P < 0.20, P < 0.30, P < 0.40, P < 0.50), together were also significantly associated with the risk for bipolar disorder in two independent samples [19]. Furthermore, several CNVs not only seem to increase risk for schizophrenia but also that of other neuropsychiatric conditions such as autism, epilepsy, and mental retardation, suggesting overlapping pathways related to neurodevelopment in these disorders (see [44] for review).
From Mere Association to Etiological Mechanisms: Genes to Phenes
Understanding the mechanisms tying identified risk variants to an increased risk for schizophrenia is the next, and perhaps most challenging step. Indeed, the absolute risk associated with most of the variants is very small, apart from a few rare CNVs with large effect size.
A sensible way to investigate the effects of common variants on disease risk in humans is to study their relative contribution to intermediate phenotypes relevant for psychosis. The importance of this approach is exemplified by a recent study by Esslinger and coworkers, who found that the novel schizophrenia risk polymorphism located in ZNF804A was associated with neural connectivity [45]. More specifically, the A risk allele was associated with decreased interhemispheric connectivity between the left and right prefrontal cortex. Furthermore, it was associated with increased prefrontal–hippocampal and prefrontal–amygdala connectivity, which – the authors suggest – could be hypothesized to result in increased sensitivity to stressful environments [45].
This finding, although in urgent need of replication, together with the GWA between schizophrenia and polymorphisms in MHC – an important region with respect to the bodily reactions to stress and infection – once again point to the importance of considering more than just the genetic data in order to understand the biological mechanisms underlying the development of psychotic disorders. Indeed, a considerable body of evidence now suggests that “genes” and “environments” operate in interplay to produce schizophrenia, rather than in isolation [46].
Genes and Environments
Which environmental exposures contribute to psychosis risk, and how they may do so, has been reviewed in numerous previous publications (e.g., [46, 47, 48, 49]). A summary of different environmental factors for which gene–environment interplay has been suggested, with the respective level of evidence, can be found in Table 1. An excellent description of how to select and measure relevant environmental factors can also be found elsewhere [50].
Table 1.
Published environmental exposures for psychosis for which GxE has been suggested
| Environmental variables with likely impact in fetal life: |
| 1. M+: Maternal pregnancy complications, in particular fetal hypoxia and proxies for fetal folate deficiency |
| 2. M+/−: Prenatal maternal infection, prenatal maternal stress, prenatal maternal folate deficiency |
| 3. M+: Paternal age |
| 4. M−: Prenatal exposure to chemical agents (e.g., lead) |
| Environmental variables with likely impact in early life: |
| 5. M−: Quality of early rearing environment (institutional care, school, parents) |
| 6. M+/−: Childhood trauma (abuse or neglect) |
| Environmental variables with likely impact in middle childhood/adolescence: |
| 7. M+: Urban environment during development: A variable indicating the level of population density, or size of a city within a country, of the place where the individual was growing up (between the ages of 5–15 years). |
| 8. M+: Cannabis use |
| 9. M+: Migration |
| 10. M+/−: Stressful life events |
| 11. M−: Traumatic brain injury |
| Measures of the wider social environment: |
| 12. M−: Neighborhood measures of social fragmentation, social capital, and social deprivation |
| Measures of the microenvironment in the flow of daily life: |
| 13. M−: Small daily life stressors, assessed using momentary assessment technology, subtly impacting on affect, salience, and reward |
M+, at least one positive meta‐analytic estimate;
M+/−, inconclusive meta‐analytic estimate;
M−, no meta‐analytic estimate available.
Although mechanisms underlying gene–environment interactions probably differ as a function of the specific environmental factor implied and the developmental period in which exposure took place, in general, environmental measures may interact with genetic variation in at least two different ways [46].
First, genetic variation may result in differences in biological functionality; these differences may be advantageous or disadvantageous in certain environments. In case of GxE, functional aspects related to variation in the DNA sequence and to a particular environmental factor converge on common biological pathways and their combined effect is larger than the added effects of both genes and environment in isolation. As such, the combination of particular genetic variation and environment may result in a substantially increased risk for the disorder (Figure 1A). An often cited example of GxE in psychosis genetics is the possible interaction between COMT Val158Met and cannabis [51, 52, 53]. Here, the COMT Val allele results in lower dopamine availability in the prefrontal cortex, which could be disadvantageous in the context of cannabis use, although it has to be noted that studies examining the effects of cannabis on striatal dopamine release have produced contradictory results [54, 55] and evidence is inconclusive that COMT Val158Met increases risk for schizophrenia [56, 57, 58], despite high rates of cannabis use in patient samples [59, 60].
Figure 1.
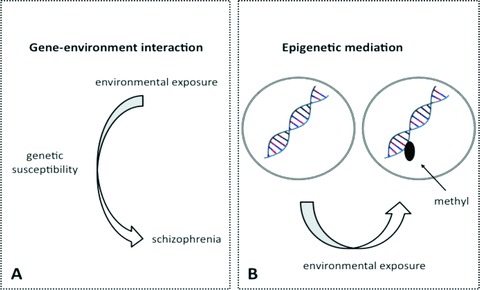
Mechanisms of gene–environment interplay: gene–environment interaction (Figure 1A) and epigenetic mediation (Figure 1B).
A second way how environments can influence disease risk in conjunction with genetic variation is by impacting on gene expression through so‐called epigenetic mechanisms[61]. Epigenetics refers to the reversible regulation of various genomic functions, occurring independently of DNA sequence, mediated through changes in DNA methylation and chromatin structure [62]. Such epigenetic mechanisms have essential functions during development, and allow the long‐term regulation of gene function and may mediate environmental effects on gene function, for example, by altering DNA methylation (Figure 1B). Although DNA methylation and histone modifications are the most frequently studied epigenetic mechanisms, other epigenetic processes are also known to regulate gene function (e.g., X‐inactivation). Findings of typical monozygotic twin discordance, risk‐increasing effects of prenatal factors, and possible risk‐increasing effects of developmental trauma in schizophrenia are consistent with a role for epigenetic mechanisms in the developmental pathway of psychotic disorder [63, 64]. For example, early maternal behavior in animals can affect offspring stress‐sensitivity through altered DNA methylation of key neuronal receptor genes involved in the stress response. The first epigenetic studies in schizophrenia patients suggest altered DNA methylation in genetic loci with essential roles in brain development and the stress response system [65]. Importantly, evidence for transgenerational transmission of epigenetic mechanisms has blurred the demarcation between epigenetic‐ and DNA sequence‐based inheritance [66, 67, 68], and challenges the assumption that the “heritable” component to schizophrenia, and other complex disorders, is entirely based on variations in the sequence of the genome. The phenomenon that certain genotypes may be associated with differential DNA methylation may be of interest as well. Differential DNA methylation based on genotype has, for example, been shown for BDNF Val66Met, Val homozygotes displaying significantly more exonic DNA methylation [65]. If these differences in methylation indeed are caused by a differential genotype sensitivity to epigenetic changes, this may potentially blur the distinction between “GxE” and “epigenetics” and complicate statistical models of GxE that assume a nonchanging, predictable effect of the respective alleles of a functional polymorphism on protein expression and function.
Another mechanism potentially complicating statistical models that assume stable and predictable effects of individual SNPs is epistasis, or interaction between genes. Epistatic interactions are not picked up by traditional “main effect” approaches and could partly explain the relatively low variance explained by common polymorphisms (around 30%, as described above) despite high heritability, although other, not necessarily mutually exclusive explanations such as individually rare markers with large effect size, are also possible.
Altogether, the available data suggest that more dynamic, integrative genetic models of psychosis, incorporating copy‐number variation (i.e., CNVs), DNA sequence variation and genetically determined sensitivity to environmental factors – mediated through different but not necessarily independent mechanisms – may be required.
Integrative Genetic Models of Psychosis
The large amount of genetic data acquired by genome‐wide platforms poses a considerable challenge to researchers examining genetic main effects, especially in terms of dealing with the problem of multiple testing and distinguishing true from false associations; by definition, more complex models involving environmental factors and interactions will pose even greater challenges. Until recently, most GxE studies in psychiatry have investigated functional SNPs (resulting in a codon that codes for a different amino acid, thus changing the protein and its function) situated within a given gene of interest, guided by an excellent opinion paper describing how to sensibly investigate GxE [50]. However, although analyses of functional polymorphisms have yielded valuable insights, they are associated with low prior probability especially in the absence of detectable genetic main effects, thus increasing the risk of type I errors. Although the most logical rationale for picking a GxE candidate gene is indeed its role in the reactivity to the environmental factor rather than its association with disorder as argued by Moffitt and colleagues [50], it seems implausible that genetic variants of unknown functionality but with reliable association with the disorder would not affect reactivity to the environment, especially since increased reactivity to the (stressful) environment is a key feature of most psychiatric disorders, psychosis included [69, 70, 71]. Therefore, recent efforts have focused on finding approaches to study GxE using genome‐wide data, so‐called Gene–Environment Wide Interaction Studies (GEWIS) [72]. It is obvious that GEWIS pose formidable conceptual and statistical challenges. Traditional epidemiological tools and methodologies are not equipped for the mass‐marker agnostic approach of GWAS, and the scale, cost, and precision of environmental measurements differ radically from those used in molecular genetics. In addition, new statistical approaches need to be developed beyond interaction as departure from additive or multiplicative joint effects while guarding against noninterpretable flooding of false positive signals from GEWIS [73]. Traditional GxE approaches focusing on candidate genes therefore will likely remain valuable, especially when investigating SNPs identified by GWA studies, as these will be associated with higher prior probability given relatively robust evidence of gene‐to‐disorder association.
In addition, an elegant approach to examine clustering of associations within functional pathways based on genome‐wide data was recently developed [74]. This approach may have considerable relevance for GxE as well, as it would allow identifying functional pathways interacting with specific environmental factors, thus facilitating focus and the development of novel, more specific GxE hypotheses with higher prior probability.
Conclusion
In conclusion, GWA studies are identifying novel common and rare genetic variants associated with psychotic disorder. The integration of these genetic variants in integrative genetic models of psychosis by examining the influence on relevant intermediate phenotypes and interactions with environmental factors is the next and perhaps most difficult step. Collaborative efforts that bring together expertise in statistics, genetics, epidemiology, experimental psychiatry, brain imaging, and clinical psychiatry, which were recently initiated [75], will be required to succeed in this challenging task.
Conflict of Interest
All authors declare that they have no conflicts of interest.
References
- 1. Jablensky A, Sartorius N, Ernberg G, et al Schizophrenia: Manifestations, incidence and course in different cultures. A World Health Organization ten‐country study. Psychol Med Monogr Suppl 1992;20:1–97. [DOI] [PubMed] [Google Scholar]
- 2. American Psychiatric Association . Diagnostic and statistical manual of mental disorders. Washington , DC : American Psychiatric Association, 1994. [Google Scholar]
- 3. McCormick L, Flaum M. Diagnosing schizophrenia circa 2005: How and why? Curr Psychiatry Rep 2005;7:311–315. [DOI] [PubMed] [Google Scholar]
- 4. Van Os J, Kapur S. Schizophrenia. Lancet 2009;374:635–645. [DOI] [PubMed] [Google Scholar]
- 5. Cardno AG, Gottesman II. Twin studies of schizophrenia: From bow‐and‐arrow concordances to star wars Mx and functional genomics. Am J Med Genet 2000;97:12–17. [PubMed] [Google Scholar]
- 6. Owen MJ, Craddock N, O’Donovan MC. Schizophrenia: Genes at last? Trends Genet 2005;21:518–525. [DOI] [PubMed] [Google Scholar]
- 7. Ross CA, Margolis RL, Reading SA, Pletnikov M, Coyle JT. Neurobiology of schizophrenia. Neuron 2006;52:139–153. [DOI] [PubMed] [Google Scholar]
- 8. Straub RE, Weinberger DR. Schizophrenia genes – famine to feast. Biol Psychiatry 2006;60:81–83. [DOI] [PubMed] [Google Scholar]
- 9. Collier DA. Schizophrenia: The polygene princess and the pea. Psychol Med 2008;38:1687–1691; discussion 1818–1620. [DOI] [PubMed] [Google Scholar]
- 10. Sullivan PF. The dice are rolling for schizophrenia genetics. Psychol Med 2008;38:1693–1696; discussion 1818–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. O’Donovan MC, Craddock N, Owen MJ. Schizophrenia: Complex genetics, not fairy tales. Psychol Med 2008;38:1697–1699; discussion 1818–1620. [DOI] [PubMed] [Google Scholar]
- 12. Crow TJ. The emperors of the schizophrenia polygene have no clothes. Psychol Med 2008;38:1681–1685. [DOI] [PubMed] [Google Scholar]
- 13. O’Donovan MC, Craddock NJ, Owen MJ. Genetics of psychosis; insights from views across the genome. Hum Genet 2009;126:3–12. [DOI] [PubMed] [Google Scholar]
- 14. Dudbridge F, Gusnanto A. Estimation of significance thresholds for genomewide association scans. Genet Epidemiol 2008;32:227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Psychiatric GWAS Consortium Coordinating Committee . Genomewide association studies: History, rationale, and prospects for psychiatric disorders. Am J Psychiatry 2009;166:540–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lencz T, Morgan TV, Athanasiou M, et al Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry 2007;12:572–580. [DOI] [PubMed] [Google Scholar]
- 17. Sullivan PF, Lin D, Tzeng JY, et al Genomewide association for schizophrenia in the CATIE study: Results of stage 1. Mol Psychiatry 2008;13:570–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. O’Donovan MC, Craddock N, Norton N, et al Identification of loci associated with schizophrenia by genome‐wide association and follow‐up. Nat Genet 2008;40:1053–1055. [DOI] [PubMed] [Google Scholar]
- 19. Purcell SM, Wray NR, Stone JL, et al Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009;460:748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Steinberg S, Mors O, Borglum AD, et al Expanding the range of ZNF804A variants conferring risk of psychosis. Mol Psychiatry 2010. doi: 0.1038/mp.2009.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Riley B, Thiselton D, Maher BS, et al Replication of association between schizophrenia and ZNF804A in the Irish Case‐Control Study of Schizophrenia sample. Mol Psychiatry 2010;15:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Need AC, Ge D, Weale ME, et al A genome‐wide investigation of SNPs and CNVs in schizophrenia. PLoS Genet 2009;5:e1000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shifman S, Johannesson M, Bronstein M, et al Genome‐wide association identifies a common variant in the reelin gene that increases the risk of schizophrenia only in women. PLoS Genet 2008;4:e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kirov G, Zaharieva I, Georgieva L, et al A genome‐wide association study in 574 schizophrenia trios using DNA pooling. Mol Psychiatry 2009;14:796–803. [DOI] [PubMed] [Google Scholar]
- 25. Mah S, Nelson MR, Delisi LE, et al Identification of the semaphorin receptor PLXNA2 as a candidate for susceptibility to schizophrenia. Mol Psychiatry 2006;11:471–478. [DOI] [PubMed] [Google Scholar]
- 26. Stefansson H, Ophoff RA, Steinberg S, et al Common variants conferring risk of schizophrenia. Nature 2009;460:744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shi J, Levinson DF, Duan J, et al Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 2009;460:753–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velo‐cardio‐facial syndrome. Arch Gen Psychiatry 1999;56:940–945. [DOI] [PubMed] [Google Scholar]
- 29. Walsh T, McClellan JM, McCarthy SE, et al Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 2008;320:539–543. [DOI] [PubMed] [Google Scholar]
- 30. Kirov G, Grozeva D, Norton N, et al Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum Mol Genet 2009;18:1497–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. International Schizophrenia Consortium . Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008;455:237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu B, Roos JL, Levy S, Van Rensburg EJ, Gogos JA, Karayiorgou M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet 2008;40:880–885. [DOI] [PubMed] [Google Scholar]
- 33. Stefansson H, Rujescu D, Cichon S, et al Large recurrent microdeletions associated with schizophrenia. Nature 2008;455:232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McCarthy SE, Makarov V, Kirov G, et al Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet 2009;41:1223–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ingason A, Rujescu D, Cichon S, et al Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Mol Psychiatry 2009. doi: 10.1038/mp.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ullmann R, Turner G, Kirchhoff M, et al Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum Mutat 2007;28:674–682. [DOI] [PubMed] [Google Scholar]
- 37. Behjati F, Shafaghati Y, Firouzabadi SG, et al M‐banding characterization of a 16p11.2p13.1 tandem duplication in a child with autism, neurodevelopmental delay and dysmorphism. Eur J Med Genet 2008;51:608–614. [DOI] [PubMed] [Google Scholar]
- 38. Vrijenhoek T, Buizer‐Voskamp JE, Van Der Stelt I, et al Recurrent CNVs disrupt three candidate genes in schizophrenia patients. Am J Hum Genet 2008;83:504–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kirov G, Gumus D, Chen W, et al Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum Mol Genet 2008;17:458–465. [DOI] [PubMed] [Google Scholar]
- 40. Ikeda M, Aleksic B, Kirov G, et al Copy number variation in schizophrenia in the Japanese population. Biol Psychiatry 2010;67:283–286. [DOI] [PubMed] [Google Scholar]
- 41. Rujescu D, Ingason A, Cichon S, et al Disruption of the neurexin 1 gene is associated with schizophrenia. Hum Mol Genet 2009;18:988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kirov G, Rujescu D, Ingason A, et al Neurexin 1 (NRXN1) deletions in schizophrenia. Schizophr Bull 2009;35:851–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008;455:903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sebat J, Levy DL, McCarthy SE. Rare structural variants in schizophrenia: One disorder, multiple mutations; one mutation, multiple disorders. Trends Genet 2009;25:528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Esslinger C, Walter H, Kirsch P, et al Neural mechanisms of a genome‐wide supported psychosis variant. Science 2009;324:605. [DOI] [PubMed] [Google Scholar]
- 46. Van Os J, Rutten BP, Poulton R. Gene‐environment interactions in schizophrenia: Review of epidemiological findings and future directions. Schizophr Bull 2008;34:1066–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Van Winkel R, Stefanis NC, Myin‐Germeys I. Psychosocial stress and psychosis. A review of the neurobiological mechanisms and the evidence for gene‐stress interaction. Schizophr Bull 2008;34:1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Henquet C, Di Forti M, Morrison P, Kuepper R, Murray RM. Gene‐environment interplay between cannabis and psychosis. Schizophr Bull 2008;34:1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Van Os J, Krabbendam L, Myin‐Germeys I, Delespaul P. The schizophrenia envirome. Curr Opin Psychiatry 2005;18:141–145. [DOI] [PubMed] [Google Scholar]
- 50. Moffitt TE, Caspi A, Rutter M. Strategy for investigating interactions between measured genes and measured environments. Arch Gen Psychiatry 2005;62:473–481. [DOI] [PubMed] [Google Scholar]
- 51. Caspi A, Moffitt TE, Cannon M, et al Moderation of the effect of adolescent‐onset cannabis use on adult psychosis by a functional polymorphism in the catechol‐O‐methyltransferase gene: Longitudinal evidence of a gene X environment interaction. Biol Psychiatry 2005;57:1117–1127. [DOI] [PubMed] [Google Scholar]
- 52. Henquet C, Rosa A, Krabbendam L, et al An experimental study of catechol‐o‐methyltransferase Val158Met moderation of delta‐9‐tetrahydrocannabinol induced effects on psychosis and cognition. Neuropsychopharmacology 2006;31:2748–2757. [DOI] [PubMed] [Google Scholar]
- 53. Henquet C, Rosa A, Delespaul P, Papiol S, Fananas L, Van Os J, Myin‐Germeys I. COMT ValMet moderation of cannabis‐induced psychosis: A momentary assessment study of ‘switching on’ hallucinations in the flow of daily life. Acta Psychiatr Scand 2009;119:156–160. [DOI] [PubMed] [Google Scholar]
- 54. Stokes PR, Mehta MA, Curran HV, Breen G, Grasby PM. Can recreational doses of THC produce significant dopamine release in the human striatum? Neuroimage 2009;48:186–190. [DOI] [PubMed] [Google Scholar]
- 55. Bossong MG, Van Berckel BN, Boellaard R, et al Delta 9‐tetrahydrocannabinol induces dopamine release in the human striatum. Neuropsychopharmacology 2009;34:759–766. [DOI] [PubMed] [Google Scholar]
- 56. Allen NC, Bagade S, McQueen MB, et al Systematic meta‐analyses and field synopsis of genetic association studies in schizophrenia: The SzGene database. Nat Genet 2008;40:827–834. [DOI] [PubMed] [Google Scholar]
- 57. Shi J, Gershon ES, Liu C. Genetic associations with schizophrenia: Meta‐analyses of 12 candidate genes. Schizophr Res 2008;104:96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Okochi T, Ikeda M, Kishi T, et al Meta‐analysis of association between genetic variants in COMT and schizophrenia: An update. Schizophr Res 2009;110:140–148. [DOI] [PubMed] [Google Scholar]
- 59. Fergusson DM, Poulton R, Smith PF, Boden JM. Cannabis and psychosis. BMJ 2006;332:172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Koskinen J, Lohonen J, Koponen H, Isohanni M, Miettunen J. Rate of cannabis use disorders in clinical samples of patients with schizophrenia: A meta‐analysis. Schizophr Bull 2009. doi: 10.1093/schbul/sbp031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet 2007;8:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jaenisch R, Bird A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet 2003;33(Suppl):245–254. [DOI] [PubMed] [Google Scholar]
- 63. Oh G, Petronis A. Environmental studies of schizophrenia through the prism of epigenetics. Schizophr Bull 2008;34:1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rutten BP, Mill J. Epigenetic mediation of environmental influences in major psychotic disorders. Schizophr Bull 2009;35:1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mill J, Tang T, Kaminsky Z, et al Epigenomic profiling reveals DNA‐methylation changes associated with major psychosis. Am J Hum Genet 2008;82:696–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sandovici I, Kassovska‐Bratinova S, Loredo‐Osti JC, et al Interindividual variability and parent of origin DNA methylation differences at specific human Alu elements. Hum Mol Genet 2005;14:2135–2143. [DOI] [PubMed] [Google Scholar]
- 67. Rakyan V, Whitelaw E. Transgenerational epigenetic inheritance. Curr Biol 2003;13:R6. [DOI] [PubMed] [Google Scholar]
- 68. Richards EJ. Inherited epigenetic variation–revisiting soft inheritance. Nat Rev Genet 2006;7:395–401. [DOI] [PubMed] [Google Scholar]
- 69. Myin‐Germeys I, Delespaul P, Van Os J. Behavioural sensitization to daily life stress in psychosis. Psychol Med 2005;35:733–741. [DOI] [PubMed] [Google Scholar]
- 70. Myin‐Germeys I, Van Os J, Schwartz JE, Stone AA, Delespaul PA. Emotional reactivity to daily life stress in psychosis. Arch Gen Psychiatry 2001;58:1137–1144. [DOI] [PubMed] [Google Scholar]
- 71. Van Winkel R, Henquet C, Rosa A, et al Evidence that the COMT(Val158Met) polymorphism moderates sensitivity to stress in psychosis: An experience‐sampling study. Am J Med Genet B Neuropsychiatr Genet 2008;147:10–17. [DOI] [PubMed] [Google Scholar]
- 72. Murcray CE, Lewinger JP, Gauderman WJ. Gene‐environment interaction in genome‐wide association studies. Am J Epidemiol 2009;169:219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Van Os J, Rutten BP. Gene‐environment‐wide interaction studies in psychiatry. Am J Psychiatry 2009;166:964–966. [DOI] [PubMed] [Google Scholar]
- 74. Holmans P, Green EK, Pahwa JS, et al Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am J Hum Genet 2009;85:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. European Network of Schizophrenia Networks for the Study of Gene‐Environment Interactions (EU‐GEI). Schizophrenia aetiology: Do gene‐environment interactions hold the key ? Schizophr Res 2008;102:21–26. [DOI] [PubMed] [Google Scholar]