

Abstract
Alzheimer's disease (AD) is a neurodegenerative disorder that affects more than 37 million people worldwide. Current drugs for AD are only symptomatic, but do not interfere with the underlying pathogenic mechanisms of the disease. AD is characterized by the presence of ß‐amyloid (Aβ) plaques, neurofibrillary tangles, and neuronal loss. The identification of the molecular determinants underlying AD pathogenesis is a fundamental step to design new disease‐modifying drugs. Recently, a specific impairment of transforming‐growth‐factor‐β1 (TGF‐β1) signaling pathway has been demonstrated in AD brain. The deficiency of TGF‐β1 signaling has been shown to increase both Aβ accumulation and Aβ‐induced neurodegeneration in AD models. The loss of function of TGF‐ß1 pathway seems also to contribute to tau pathology and neurofibrillary tangle formation. Growing evidence suggests a neuroprotective role for TGF‐β1 against Aβ toxicity both in vitro and in vivo models of AD. Different drugs, such as lithium or group II mGlu receptor agonists are able to increase TGF‐β1 levels in the central nervous system (CNS), and might be considered as new neuroprotective tools against Aβ‐induced neurodegeneration. In the present review, we examine the evidence for a neuroprotective role of TGF‐β1 in AD, and discuss the TGF‐β1 signaling pathway as a new pharmacological target for the treatment of AD.
Keywords: Alzheimer's disease, Apoptosis, β‐amyloid, Cell cycle, Neurofibrillary tangles, Neuroprotection, Lithium, Transforming‐growth‐factor‐β1
Introduction
Alzheimer's disease (AD) is the most common cause of dementia in the elderly, affecting approximately 10% of individuals by 65 years of age and 47% by 85 years of age. It is mainly characterized by memory loss, with disoriented behavior and impairments in language, comprehension, and spatial skills also characterizing this disorder. Neuropsychiatric symptoms, such as agitation and psychosis are also frequent in people with AD, and are a common precipitant of institutional care [1]. The economic burden of AD is massive; in the United States alone, the annual cost care for patients with AD is approximately 150 billion of USD [2]. However, the number of therapeutic options for AD remains severely limited. Currently marketed drugs for AD (i.e., the acetylcholinesterase inhibitors, donepezil, rivastigmine, and galantamine, and the NMDA receptor antagonist, memantine) provides mainly symptomatic short‐term benefit, without affecting the underlying pathogenic mechanisms [3]. Therefore, much effort is now directed to find treatments that effectively counteract the progression of AD. The comprehension of the molecular mechanisms underlying AD is therefore an essential step for the identification of new targets and the design of disease‐modifying drugs able to slow down or even stop the degenerative processes and the resulting memory loss.
AD is characterized by the presence of extracellular aggregates of β‐amyloid (Aß) in the senile plaques, intracellular aggregate of tau protein in the neurofibrillary tangles (NFT), and progressive neuronal loss. Different hypotheses have been proposed to understand the role of Aβ or tau protein in the pathophysiology of AD. The expression pattern of NFT in AD brain strongly correlates with the clinical onset and severity of dementia, but molecular genetics supports a primary role for Aß in the cascade of events leading to neuronal death in AD [4]. Oligomeric species composed of aggregated Aβ are believed to exert toxic effects on synaptic and cellular functions, finally leading to neurodegeneration.
In vitro studies have shown that Aß causes neuronal death via multiple mechanisms, which include membrane ion channel opening [5], radical oxygen species formation [6], amplification of NMDA toxicity [7, 8], and cell cycle activation in differentiated neurons [9, 10, 11, 12, 13]. Furthermore, Aβ is known to promote the phosphorylation of tau protein and the subsequent formation of NFT through the activation of the two kinases, that is, the cyclin‐dependent kinase 5 (CDK5) and the glycogen synthase kinase 3β (GSK‐3β) [14, 15, 16]. In this scenario, tau hyperphosphorylation and NFT formation might be placed within the same molecular cascade initiated by Aβ, which leads to progressive synaptic loss, and, finally, to neuronal death.
In vivo studies have also been carried out for the analysis of neurotoxicity by Aß, but evidence is less consistent as compared to in vitro studies. Only a few transgenic mice overexpressing Aß show neuronal loss, perhaps because the lifespan of mice is too short for a full development of the death cascade [17]. Injection of Aß into the rodent brain causes a damage restricted to the injection site at best [18, 19, 20]. We can hypothesize that neurotoxicity of Aßin vivo is limited by the presence of endogenous protective factors that may be lacking in the AD brain [21, 22, 23]. One possible candidate is transforming‐growth‐factor β1 (TGF‐β1). Transgenic mice lacking TGF‐β1 show enhanced neuronal susceptibility to different neurotoxic insults [24].
In the present review, we examine the role of TGF‐β1 in AD pathogenesis prior to discussing the rationale for considering TGF‐β1signaling as a new target for neuroprotection in AD.
TGF‐β1 Signaling Pathway: Smad and Non‐Smad Dependent Pathways
TGF‐β1 is a member of TGF‐beta superfamily, which consists of several groups of highly conserved multifunctional cell–cell signaling proteins of key importance in the control of tissue homeostasis [25].
The TGF‐β subfamily includes three isoforms in mammals, TGF‐β1, 2, and 3, which are important modulators of cell survival, inflammation, and apoptosis [26], and also exert a central role in immune suppression, and repair after injury [27]. The three TGFβs are all synthesized as homodimeric proproteins (proTGFβ) that are around 400 amino acids in size and products of separate genes. The proTGFβs are cleaved intracellularly by furin into a larger C‐terminal proregion also known as latency‐associated peptide (LAP), and a shorter N‐terminal active peptide, which forms the mature homodimers (25 kDa). LAP remains noncovalently associated with the mature TGFβ 25‐kDa dimer before the complex is secreted [28]. The association between the TGF‐β1, 2, and 3 prodomains (LAPs) and the corresponding mature growth factors prevents signaling through the TGF‐β high affinity receptors [29]. Thus, TGF‐bioactivity requires dissociation from LAP, a process termed latent TGF‐β activation.
Extracellular activation of TGF‐β is a critical but incompletely understood process in vivo. In particular, an important and unresolved issue in TGF‐β biology regards the connection between matrix incorporation and activation of the latent TGF‐β. A variety of molecules, from protons to different proteases, such as plasmin and trombospondin, have been described as latent TGFβ activators [30]. It seems that inactive TGF‐β stored in tissues can be activated in response to injury and subsequent extracellular matrix perturbations. After TGF‐β is released from its latency‐associated peptide, it becomes able to initiate its diverse cellular responses by binding to, and activating specific cell surface receptors that have intrinsic serine/threonine kinase activity.
All three TGF‐β isoforms interact with a high‐affinity transmembrane receptor complex consisting of the activin‐like kinase 5 (ALK5)/TGF‐β type I receptor and the TGF‐β type II receptor (TβRII) subunits [25] (see Figure 2). Several studies have demonstrated that ligand binding to TβRII induces the assembly of type I and type II receptors into complexes with the subsequent phosphorylation and activation of ALK5, which then propagates the signal inside the cell through the phosphorylation of receptor‐regulated Smads (R‐Smads: Smad2, Smad3, Smad5, and Smad8). The interaction between R‐Smads and (ALK5)/TGF‐β type I receptor is facilitated by the Smad anchor for receptor activation (SARA) [31]. Phosphorylated R‐Smads form heteromeric complexes with Smad4. These complexes accumulate in the nucleus, where they regulate gene expression in a cell‐type‐specific and ligand dose‐dependent manner through interactions with transcription factors and specific promoter elements of target genes.
Figure 2.
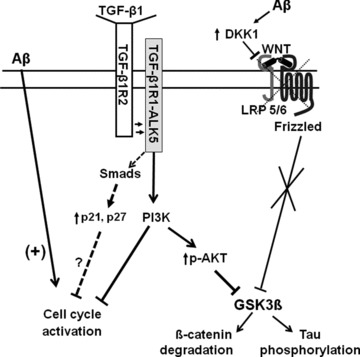
Putative mechanisms underlying the neuroprotective effects of TGF‐β1 against Aβ‐induced neurodegeneration. Aβ induces neuronal death via an early activation of cell cycle, and a late induction of DKK1 leading to an inhibition of the canonical Wnt pathway with ensuing activation of GSK‐3β. TGF‐β1 inhibits cell cycle activation and rescues the Wnt pathway via a direct activation of the PI3K pathway. Activation of the classical Smad‐dependent pathway leading to an enhanced expression of cyclin‐dependent kinase inhibitors (p21, p27) and cell cycle arrest is also shown (dotted). Whether this pathway may also contribute to the protective effect of TGF‐β1 against Aβ‐induced neurodegeneration is unknown.
Smad6 and Smad7 are inhibitory Smads, which are known to counteract the signaling of R‐Smads through different mechanisms [25]. Inhibitory Smads bind to activated type I receptors, thus inhibiting the phosphorylation and the following nuclear translocation of R‐Smads. Furthermore, they can recruit E3‐ubiquitin ligases targeting the receptor complex to the ubiquitin degradation pathway with the following inhibition of TGF‐β/Smad signaling cascade.
Recent evidence suggests that TGF‐β1 can also exert its biological effects through the activation of smad‐independent pathways such as the extracellular‐regulated kinase (ERK) pathways [32, 33], the nuclear factor κB (NF‐κB) pathway [34], and the phosphatidylinositol‐3‐kinase (PI3K)/Akt pathway [35].
Role of TGF‐β1 in the Brain and in AD
In the central nervous system (CNS) TGF‐β2 and 3 isoforms account for almost all the TGF‐β immunoreactivity, while TGF‐β1 expression has been found to be constitutive only in the meninges and choroid plexus and, most importantly, in some specific brain regions such as the hippocampus and the cortex [36]. Interestingly, TGF‐β1 expression and release increase significantly in response to CNS lesions. Astrocytes and microglia seem to be the major sources of TGF‐β1 in the injured brain [37], and several studies have shown that TGF‐β1 induction during injury exerts a central role in preventing neurodegeneration [24, 36].
An increased expression of TGF‐β1 has been observed with age [37], and a protective role has been suggested for this neurotrophic factor in longevity [38]. Aging is characterized by an increased level of proinflammatory markers such as IL‐6, TNF‐α or IL‐1β[38, 39]. This state of subclinical chronic inflammation has been called “inflamm‐ageing,” and seems to be involved in the pathogenesis of several age‐related disorders such as cancer, diabetes, cardiovascular pathologies, and AD [39]. The protective role of TGF‐β in aging and longevity has been suggested by in vitro and in vivo studies [40, 41]. Increased plasma levels of bio‐active TGF‐β1 have been found in both male and female centenarians as compared to younger control subjects [41]. Similar results have been obtained by Forsey et al. [42] in octogenarian and nonagenarian subjects. Salvioli et al. [38] have also proposed that this age‐related increase of TGF‐β1 might counteract the proinflammatory status observed during aging, thus preventing the development of age‐related disorders such as cancer and AD.
Changes in TGF‐β1 serum and cerebrospinal fluid (CSF) levels have also been analyzed in AD. In particular, increased TGF‐β1 levels have been found in CSF of AD patients [43, 44], whereas a reduction of both its active (25 kDa) and inactive (50 kDa) forms has been reported in AD plasma [45].
Recently, a single nucleotide polymorphisms (SNPs) at codon +10 (T(C) and +25 (G/C) that affects the levels of expression of TGF‐β1 has been associated with an increased conversion of mild cognitive impairment (MCI) in AD [46]. We have recently investigated the same polymorphism in healthy controls (HC) and AD patients. Preliminary data suggest that both the +10 C allele and the CC genotype are overrepresented in AD when compared to HC, and, that CC genotype might act as a risk factor for the development of late‐onset AD (LOAD), independently of apolipoprotein status (unpublished results).
Many reports also describe a significant impairment of TGF‐β1 signaling in AD brain [23, 47, 48, 49, 50]. The study by Tesseur et al. [48] strongly points to a causal role for of TGF‐β signaling dysfunction in age‐dependent neurodegeneration and AD pathogenesis (Figure 1). The authors found that the expression of TGF‐β type II receptor (TβRII) by neurons is reduced very early in the course of AD, and this alteration seemed to be specific for AD and was not observed in other neurodegenerative conditions such as Parkinson's disease, frontotemporal dementia, or Lewy body dementia. The authors also found that a deficiency of TGF‐β signaling, in a mouse model of AD, promoted both Aβ deposition and neuronal loss [48]. Moreover, Tesseur et al. [48] have shown that the impairment of TGF‐β signaling in neuroblastoma cells resulted in neuritic dystrophy and increased levels of secreted Aβ and β‐secretase‐cleaved soluble amyloid precursor protein. These data suggest that a deficiency of TGF‐β/TβRII signaling axis might exert a pathogenetic role in AD, depriving cortical neurons of trophic support, and finally promoting Aβ‐induced neurodegeneration (see Figure 1).
Figure 1.
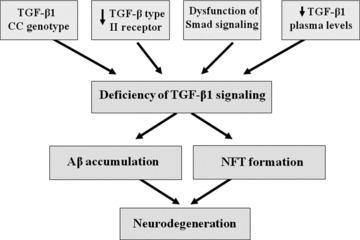
Hypothetical role of TGF‐ß1 in AD pathogenesis. Alterations of TGF‐β1 signaling in AD: (1) a reduced expression of the neuronal TGF‐β type II receptor, (2) a dysfunction of Smad signaling, (3) a reduction in TGF‐β1 plasma levels, (4) an increased occurrence of TGF‐ß1 CC genotype which can promote the conversion of MCI into AD. All alterations might lead to Aβ accumulation and neurofibrillary tangles (NFT) formation with ensuing neurodegeneration.
However, the role of TGF‐ß1 in AD pathophysiology is not unequivocal, and conflicting results have been reported recently. TGF‐ß1 is known to induce the expression of the APP gene in several different cell culture systems [51, 52] and might thus increase Aß production. The co‐expression of TGF‐ß1 in transgenic AD mice accelerates the deposition of Aß in cerebral blood vessels [53], and transgenic mice overexpressing TGF‐ß1 develop AD‐like vascular alterations [54]. In addition, vessel‐derived TGF‐ß1 has been suggested to contribute to inflammatory processes in the AD brain [55, 56]. Town et al. [57] have recently found that blocking TGF‐β‐Smad 2/3 signaling reduces cerebrovascular β‐amyloid deposits and Aβ abundance in Tg2576 mice, and these events result in promotion of Smad1/5/8 signaling with increased infiltration of Aβ‐containing peripheral macrophages around cerebral vessels and β‐amyloid plaques.
Overall data from the literature seem to suggest that TGF‐ß1 can promote Aβ deposition in cerebral blood vessels, but reduces Aβ accumulation in the brain parenchyma [23]. In particular, it has been demonstrated that a modest increase in astroglial TGF‐β1 production in aged transgenic mice expressing the human beta‐amyloid precursor protein (hAPP) results in a 50% reduction of Aβ load in the hippocampus, and a decrease in the number of dystrophic neurites [58].
Deficiency of TGF‐ß1 signaling is also involved in tau pathology and NFT formation. Luterman et al. [59] found that low levels of TGF‐ß1 mRNA negatively correlated with NFT in the AD brain, thus suggesting that a deficiency of TGF‐ß1 might also contribute to the cascade of events that result in the development of NFT‐bearing neurons. The relationship between tau hyperphosphorylation and TGF‐ß1 signaling has been recently studied in the temporal lobe in AD [50]. Interestingly, NFT can sequester phosphorylated Smad3 in AD brain, thus preventing its translocation into the nucleus and the induction of gene transcription [50].
Other groups report an impairment of Smad‐dependent TGF‐ß1 signaling in AD brain [47, 49], with an aberrant localization of phosphorylated Smad2 to the cytoplasm rather than the nucleus of hippocampal neurons and a specific colocalization with amyloid plaques and NFT. These data suggest a dysfunction of Smad signaling in AD brain, and, interestingly, a recent in vitro study has demonstrated that Aβ can inhibit TGF‐ß1 signaling by inducing the expression of Smad 7 [60].
Taken together, these data might explain the paradox observed in the AD brain, where TGF‐ß1 levels in CSF are found to be high; however, this neurotrophic factor might not exert its neuroprotective action for an impairment of Smad signaling (Figure 1).
Smad proteins are also implicated in initiation and maintenance of neuronal differentiation and synaptic plasticity, and TGF‐ß1 is a well‐known inhibitor of cell proliferation that may contribute to keep postmitotic neurons in a differentiated state [32]. The reduced function of TGF‐ß1 signaling in AD might therefore contribute to a re‐expression of cell cycle proteins in neurons, and the resulting activation of the cell cycle, which is considered as an early event in AD pathogenesis [61, 62, 63, 64].
We believe that a deficiency in TGF‐ß1 signaling might exert a central role in AD pathogenesis via different mechanisms that finally lead to Aβ accumulation and/or NFT formation with an ensuing neurodegeneration (Figure 1).
Neuroprotective Effects of TGF‐β1 against Aβ‐Induced Neurodegeneration
TGF‐ß1 is known to protect neurons against a diverse number of insults, including excitotoxicity, hypoxia, ischemia, and deprivation of trophic factors [23, 36, 65, 66]. Several studies have suggested that TGF‐ß1 also exerts a neuroprotective role against Aβ toxicity by selectively interfering with different steps of the Aß‐induced death cascade. In cultured neurons, estrogen‐stimulated release of TGF‐ß1 from glial cells [67] or application of recombinant TGF‐ß1 [68, 69, 70, 71] reduces Aß‐induced neurodegeneration.
We have recently studied the neuroprotective effects of endogenous TGF‐β1 signaling in the rat brain after intracerebral injection of synthetic Aß[35]. Aß injection into the dorsal hippocampus produced only a small extent of neuronal loss in the pyramical layer of the CA1 region. However, Aß neurotoxicity was amplified by i.c.v. injection of SB431542, which behaves as a selective inhibitor of the activin‐like kinase 5 (ALK5) TGF‐β type I receptor [72].
Different molecular mechanisms have been implicated in the neuroprotective effects of TGF‐β1 against Aß toxicity. TGF‐β1 receptors are expressed both in glial cells and neurons [65], and, therefore TGF‐β1 might exert its protective effects by acting on both cell types.
TGF‐β1 has a constitutive role in the suppression of inflammation, and appears to control the degree of microglial activation in the CNS [24]. Inhibition of TGF‐β1 in different models of neurodegenerative disorders is associated with local inflammation mediated by macrophage/microglia and T cells [24, 73]. Inflammatory responses elicited by elevated Aβ peptides play an important role in the progression of AD, and microglia activation is an early event in AD pathogenesis and can be already detected in patients with MCI [74]. Aß can activate microglia to release proinflammatory cytokines such as IL‐1β, IL‐6, and TNF‐α[75], which can contribute to neuronal death in the AD brain. Interestingly, several studies have demonstrated that TGF‐β1 reduces microglia activation and promotes the degradation of Aβ by the microglia [58, 76, 77].
TGF‐β1 might also affect neuronal survival through other mechanisms because it acts synergistically with other neurotrophins and is required for a full neuroprotective activity of nerve growth factor (NGF), brain‐derived neurotrophic factor (BDNF), and glial‐derived neurotrophic factor (GDNF) [78, 79, 80]. The levels of BDNF and its receptor, tropomyosin receptor kinase B (TRKB), are reduced in the AD brain, and deficiency of BDNF signaling has been related to neurodegeneration and cognitive dysfunction in AD [81, 82]. Interestingly, TGF‐β1 enhances the expression of BDNF and TrkB in rat neuronal cultures [83].
It might be possible that the contemporary failure of both BDNF and TGF‐β1 signaling in the AD brain enhances neuronal vulnerability to Aß, thus accelerating the progression of AD.
Finally, a component of the neuroprotective action of TGF‐β1 is mediated by the activation of neuronal TGF‐β receptors. TGF‐β1 is known to prevent apoptotic cell death in neurons through the inhibition of caspase‐3 activation [84]. In addition, TGF‐β1 maintains mitochondrial membrane potential and increases the expression of antiapoptotic proteins, such as Bcl‐2 and Bcl‐xl [68]. TGF‐β1 can also activate the extracellular‐regulated kinase (ERK) pathway in hippocampal neurons, thus promoting the phosphorylation and subsequent inhibition of the proapoptotic protein, Bad [85]. Furthermore, TGF‐β1 can increase the transcriptional activity of the antiapoptotic transcriptional factor, NF‐kappaB, through the PI3K/Akt and ERK signaling pathways [86].
TGF‐β1 Interferes with the Death Triggered by Aβ in Neurons: From Cell Cycle Inhibition to the Rescue of the Wnt Pathway
The process of neuronal death triggered by Aß proceeds along an aberrant re‐activation of the cell cycle [10, 11, 13]. Cell cycle in proliferating cells depends on the sequential activation of cyclin (Cyc)/cyclin dependent protein kinases (CDKs), which control the transition through the different phases of the cycle [87]. Aß activates the cell cycle in neurons by inducing the sequential expression of different cell cycle proteins usually functioning in proliferating cells, such as cyclin D1, phosphorylated retinoblastoma protein (ppRB), cyclin E, and cyclin A, which are necessary for G1/S transition, and S phase progression [10]. Reactivation of the cell cycle is an obligatory step in the apoptotic pathway evoked by Aß, suggesting that an ectopic S phase triggers the signal for neuronal death. DNA replication has also been demonstrated in neurons from AD brains [88], providing the in vivo counterpart of in vitro findings.
Recent studies suggest that cell cycle activation in neurons leads to a pathological DNA replication performed by a noncanonical enzymatic machinery, which finally contributes to generation of a death signal in neurons [11, 13]. As opposed to proliferating cells, neurons that enter the S phase in response to Aβ fail to express DNA polymerase‐α (DNA pol‐α), which has an essential role in the canonical DNA synthesis [11], but overexpress DNA polymerase‐β (DNA pol‐β), a repair enzyme that only occasionally performs de novo DNA synthesis. DNA pol‐β is an error‐prone enzyme and, therefore, the aberrant DNA synthesis induced by Aβ might contribute to the signal to trigger neuronal apoptosis. The extension of DNA replication performed by DNA pol‐β is critical for the activation of a death signal that is mediated by an increased expression of p53, a major sensor of DNA damage in eukaryotic cells [11, 12, 13, 89].
The increased expression of p53 in Aβ‐treated neurons in response to DNA damage can promote neuronal death through different pathways. The activation of a p53/DNA damage‐dependent pathway triggers the execution phase of apoptotic death via the induction of the proapoptotic protein Bax and the downregulation of the antiapoptotic protein Bcl‐2 [90]. On the other hand, p53 induction in Aβ‐treated neurons might also activate a slow degenerative process, which finally leads to NFT formation. Several genes are under the control of p53 in eukaryotic cells [89], and p53 has been shown to induce, in cultured neurons challenged with Aβ, the expression of Dickkopf (Dkk‐1), a specific antagonist of the Wnt signaling pathway [91]. Wnt signaling has an established role in maintaining neuronal homeostasis, and GSK‐3ß, the main enzyme involved in tau hyperphosphorylation [15], is a key component of the Wnt pathway. Activation of the Wnt pathway leads to the inhibition of GSK‐3ß through a cascade of intracellular reactions, which involve adaptor proteins such as disheveled (Dvl), and a multiprotein complex containing GSK‐3ß, β‐catenin, axin, and adenomatous polyposis coli (APC) [92]. Inhibition of GSK3ß prevents tau phosphorylation and also phosphorylation of ß‐catenin, which thus escapes degradation and translocates to the nucleus where it drives the expression of different genes involved in the regulation of neuronal survival, such as Bcl‐2 and survivin [93]. Accumulated β‐catenin can also be targeted to synapses, where it modulates synaptic strength in response to depolarization [94, 95].
In this scenario, the p53‐dependent induction of Dkk‐1 observed both in AD models and in the AD brain, may determine a loss of Wnt function, with the following activation of GSK‐3ß and NFT formation [91, 93]. Accordingly, Dkk‐1 knockdown prevents tau hyperphosphorylation and β‐catenin degradation in Aß‐treated neurons [91].
Interestingly, we recently found that TGF‐ß1 applied to cultured cortical neurons challenged with Aβ prevents the abnormal DNA replication, enhances the levels of Ser9‐phosphorylated (inhibited) GSK‐3β, and prevents β‐catenin degradation and tau hyperphosphorylation, thus attenuating neuronal death. All these effects were abrogated by the PI3K inhibitor, LY294402, suggesting that TGF‐ß1 is protective against Aβ neurotoxicity via the activation of the PI3K pathway (Figure 2). Interestingly, a defect in the PI3K pathway has been associated with AD [96, 97]. Figure 2 also shows the classical Smad‐dependent pathway leading to cell cycle inhibition in response to TGF‐β. Whether this pathway contributes to cell cycle arrest and neuroprotection under conditions of a defective PI3K activation (as may occur in AD) is unknown.
Pharmacological Perspectives
The knowledge of the molecular processes underlying AD pathogenesis is a fundamental step for the development of disease‐modifying drugs able to counteract the degenerative processes in AD.
According to the evidence discussed in the present review, the deficiency of TGF‐ß1 signaling seems to be an early event in AD pathogenesis, which contributes to Aß‐induced neurodegeneration and NFT formation in the AD brain (see Figure 1). We therefore suggest that the rescue of TGF‐ß1 signaling might be a new strategy to promote neuroprotection in AD.
Neurotrophic factor therapy represents a difficult challenge for CNS drug discovery, because protein growth factors do not cross the blood–brain barrier and require intracerebral administration to be effective. The implant of autologous fibroblasts genetically modified to express human growth factors into selected areas of CNS has been proposed as a new strategy for the treatment of AD. This approach has been adopted for NGF in AD, and a phase I clinical trial has shown promising results [98]. Recently, BDNF gene delivery has also been found to exert substantial protective effects in AD models and has been proposed as a new potential therapy for AD [99]. However, central delivery of neurotrophic factors can be proposed only for a subset of AD patients and cannot be considered as a suitable approach for general medical practice.
The strong neuroprotective activity of neurotrophic factors has stimulated the search for small‐molecules drugs that activate neurotrophic factor receptors or potentiate the action of growth factors by affecting their signaling pathway [100, 101]. Along this line, small‐molecule drugs that selectively activate specific elements of the TGF‐β1 signaling pathway have been studied for the identification of drugs that can be beneficial in AD [102]. A more feasible approach would be the use of centrally available drugs that are able to increase the local production of TGF‐ß1. Several drugs are known to induce the synthesis and the release of TGF‐ß1 in vitro and in vivo (Table 1).
Table 1.
Potential pharmacological approaches to rescue TGF‐β1 signaling in AD
| Drug | Mechanism | References |
|---|---|---|
| TGF‐β1 mimetics | Activation of TGF‐β1 receptors | [102] |
| Estrogens | Increased secretion of TGF‐ß1 from astrocytes | [67] |
| Aspirin | Increased TGF‐ß1 plasma levels in vivo | [111, 113] |
| Statins | Stimulated TGF‐ß1 synthesis and secretion from monocytes | [112, 113] |
| Glatiramer | Induction of TGF‐ß1 synthesis from Th2 type cells and glial cells | [116] |
| Sertraline, paroxetine | Increased TGF‐ß1 plasma levels in vivo | [118, 124] |
| Venlafaxine | Stimulated TGF‐ß1 production and release from glial cells | [119] |
| Lithium | Increased release of TGF‐ß1 from astrocytes | [See Figure 3] |
| mGlu2/3 receptor agonists | Increased synthesis and secretion of TGF‐ß1 from astrocytes | [130, 131] |
Estrogen treatment has been shown to reduce the risk of AD when administered at the time of the menopause and continued over several years [103]. Estrogens exert strong neuroprotective effects in hippocampal neurons when administered before Aβ treatment [104]. Interestingly, estrogens act as neuroprotectants via an increased secretion of TGF‐ß1 from astrocytes [67]. TGF‐ß1 released from astrocytes exposed to 17β‐estradiol prevents Aß toxicity in pure neuronal cultures by preventing the unscheduled activation of the cell cycle [67]. These data suggest a possible role for TGF‐ß1 in the neuroprotective activity of estrogen therapy in AD, but further evidence is needed to confirm this hypothesis.
Some cardiovascular drugs, such as aspirin and statins, can promote TGF‐ß1 synthesis and release [105] and, interestingly, both of drugs are known to reduce the risk of developing AD [106, 107, 108, 109]. Aspirin inhibits vascular smooth muscle cell proliferation via the TGF‐ß1 pathway [110], and higher levels of active TGF‐ß1 have been found in patients with coronary artery disease treated with aspirin [111]. Pravastatin has been found to increase TGF‐ß1 synthesis and secretion in plaque monocytes from atherosclerotic patients [112]. In addition, the combination of atorvastatin with aspirin in patients undergoing coronary artery bypass grafting (CABG) has been shown to decrease the risk of major adverse cardiac events via the suppression of inflammatory responses and an increased production of TGF‐ß1 [113]. Unfortunately, no studies have been carried out on the effects of these drugs on TGF‐ß1 synthesis in the CNS.
Interestingly, some drugs used for the treatment of CNS disorders are known to promote TGF‐ß1 synthesis in the brain. Glatiramer (GA) is a synthetic amino acid copolymer currently approved for the treatment of multiple sclerosis (MS) that reduces both relapse rate and progression of disability [114]. Different mechanisms of action have been postulated for this drug in humans. Arnon and Aharoni [115] have demonstrated that glatiramer in mice induces specific suppressor cells of the T helper (Th2) type that migrate to the brain where they express antiinflammatory cytokines such as IL‐10 and TGF‐ß1 in addition to BDNF [116]. Furthermore, GA‐specific cells increase the expression of TGF‐ß1 from glial cells in the cerebral cortex and hippocampus, two brain regions that are strongly implicated in the pathophysiology of AD. It could be interesting to examine the effects of glatiramer treatment on amyloid and tau pathology in AD models.
Different antidepressants, including tianeptine [117], sertraline [118], and venlafaxine [119], can increase TGF‐ß1 production. Interestingly, therapeutic concentrations of venlafaxine prevent microglial activation, reduce proinflammatory cytokine secretion, and finally increase the release of TGF‐ß1 in an astroglia–microglia coculture model [119]. These data suggest that glial cells can mediate the antiinflammatory effects of antidepressant drugs, but the potential neuroprotective activity of these compounds has been only partially explored in AD models. Presymptomatic treatment with the antidepressant paroxetine reduces both amyloid and tau pathology and also reverses memory impairment in the 3xTgAD mouse model of AD [120]. This study suggests that antidepressants are neuroprotective activity and may retard the development of AD. Accordingly, a history of major depression early in life has been considered as a risk factor for later development of AD [121]. Furthermore, the presence of depressive symptoms significantly increases the conversion of MCI into AD [122]. Plasma TGF‐ß1 levels are reduced in major depressed patients and show a significant negative correlation with the Hamilton Depression Rating Scale (HDRS) [123]. Interestingly, different antidepressant drugs, including venlafaxine, paroxetine, and sertraline, significantly increase circulating TGF‐ß1 levels in major depressed patients [118, 124]. A recent study by Kessing et al. (2009) shows that continued long‐term antidepressants treatment is associated with a reduction in the rate of AD [125], suggesting that it might be worth to assess whether TGF‐β1 signaling is a common target for both depression and AD.
Other psychotropic drugs such as lithium ions can also influence TGF‐ß1 production. Recent evidence suggests that lithium is neuroprotective against a variety of neurodegenerative conditions, including AD [126]. Prevalence of AD is lower in patients treated with lithium [127], and lithium reduces the risk of developing AD in elderly patients with bipolar disorder [128]. Different molecular mechanisms have been suggested to explain the neuroprotective effects of lithium in AD, such as the reduction of Aβ production or, more important, the inhibition of GSK‐3β, which might counteract the loss of Wnt signaling observed in the AD brain [35, 126]. Recently, we have found that lithium strongly induces the release of TGF‐ß1 from rat cortical astrocytes, as assessed by immunoblotting and ELISA assay (unpublished results; see Figure 3). Hence, our own data suggest that the very broad neuroprotective activity of lithium might be related to the induced release of TGF‐ß1 from glial cells.
Figure 3.
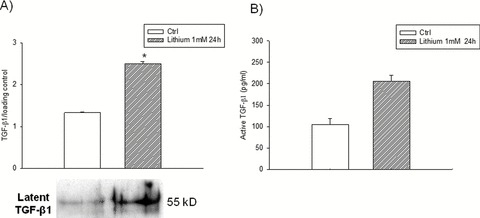
Lithium stimulates TGF‐β1 release from rat cortical astrocytes. Rat cortical astrocytes were exposed to 1 mM lithium chloride for 24 h, and the incubation medium was collected for western blot analysis (A) and ELISA assay (B). (A) Representative immunoblot of latent TGF‐β1 (about 55 kDa). Protein loading was checked by staining membrane‐transferred proteins with Ponceau's solution. Values are the means ± SEM of 3 determinations; P < 0.05 (by one‐way ANOVA + Fisher's LSD test) versus control (*). (B) Each bar represents the mean ± SEM of active TGF‐β1 protein levels in the incubation medium. Data are from three different experiments; P < 0.05 (by one‐way ANOVA + Fisher's LSD test) versus control (*).
Additional levels of interaction between TGF‐ß1 signaling and lithium‐regulated pathways have been proposed, including lithium's inhibition of Smad3/4‐dependent TGF‐ß1 signaling in neurons [129]. This inhibition of Smad‐regulated gene transcription seems to be useful under specific pathological conditions (e.g., mood disorders) that may benefit from the suppression of plasminogen activator inhibitor type‐1 (PAI‐1) transcription [129]. The evidence that lithium's inhibitory effects on Smad3/4 transcriptional activity are due, at least in part, to the activation of PI3‐K/AKT signaling [129], suggests that in neurons the activation of the PI3‐K/AKT pathway might be mutually exclusive with the activation of the Smad‐dependent pathway. In other words, the activation of Smad/non‐Smad signaling pathways by TGF‐ß1 might be fine‐tuned in a context‐dependent manner to result unchangingly into neuroprotection.
Finally, the production of TGF‐ß is enhanced by agonists of group II metabotropic glutamate (mGlu) receptors both in cultured astrocytes [130] and in the mouse brain [131]. In addition, these drugs protect neurons grown in the presence of astrocytes against Aß toxicity [132]. It is noteworthy that highly potent and centrally available group II mGlu receptor agonists, such as LY404039, are under clinical development for the treatment of schizophrenia [133]. We suggest that these drugs are potential candidates as neuroprotectant agents in AD, and might be also useful for the treatment of psychosis in AD (PAD) as an alternative to antipsychotic drugs, which may increase the risk of cerebrovascular events in demented patients [134].
Conflict of Interest
All authors declare that no potential conflict of interest exists, including all relevant financial interests in any company or institution that might benefit from the publication.
Acknowledgments
This work was supported by PRIN 2007 from the Italian Ministry of University and Research, and by Progetto MUR 105/04.
References
- 1. Ballard C, Day S, Sharp S, Wing G, Sorensen S. Neuropsychiatric symptoms in dementia: Importance and treatment considerations. Int Rev Psychiatry 2008;20:396–404. [DOI] [PubMed] [Google Scholar]
- 2. Mount C, Downton C. Alzheimer's disease: Progress or profit? Nature Med 2006;12:780–784. [DOI] [PubMed] [Google Scholar]
- 3. Klafki HW, Staufenbiel M, Kornhuber J, Wiltfang J. Therapeutic approaches to Alzheimer's disease. Brain 2006;129:2840–2855. [DOI] [PubMed] [Google Scholar]
- 4. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science 2002;297:353–356. [DOI] [PubMed] [Google Scholar]
- 5. Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 2005;280:17294–17300. [DOI] [PubMed] [Google Scholar]
- 6. Butterfield DA, Reed T, Newman SF, Sultana R. Roles of amyloid beta‐peptide‐associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment. Free Radic Bio Med 2007;43:658–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. beta‐Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci 1992;12:376–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hynd MR, Scott HL, Dodd PR. Glutamate‐mediated excitotoxicity and neurodegeneration in Alzheimer's disease. Neurochem Int 2004;45:583–595. [DOI] [PubMed] [Google Scholar]
- 9. Giovanni A, Wirtz‐Brugger F, Keramaris E, Slack R, Park DS. Involvement of cell cycle elements, cyclin‐dependent kinases, pRb, and E2F‐DP, in β‐amyloid‐induced neuronal death. J Biol Chem 1999;274:19011–19016. [DOI] [PubMed] [Google Scholar]
- 10. Copani A, Condorelli F, Caruso A, et al Mitotic signaling by beta‐amyloid causes neuronal death. FASEB J 1999;13:2225–2234. [PubMed] [Google Scholar]
- 11. Copani A, Sortino MA, Caricasole A, et al Erratic expression of DNA polymerases by beta‐amyloid causes neuronal death. FASEB J 2002;16:2006–2008. [DOI] [PubMed] [Google Scholar]
- 12. Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: Cell cycle events as triggers of nerve cell death. J Neurosci 2004;24:9232–9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Copani A, Hoozemans JJ, Caraci F, et al DNA polymerase‐ß is early expressed in neurons of Alzheimer's Disease brain and is loaded into DNA replication forks in neurons challenged with ß‐amyloid. J Neurosci 2006;26:10949–10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Otth C, Concha II, Arendt T, et al AbetaPP induces cdk5‐dependent tau hyperphosphorylation in transgenic mice Tg2576. J Alzheimers Dis 2002;4:417–430. [DOI] [PubMed] [Google Scholar]
- 15. Takashima A, Honda T, Yasutake K, et al Activation of tau protein kinase I/glycogen synthase kinase‐3beta by amyloid beta peptide (25–35) enhances phosphorylation of tau in hippocampal neurons. Neurosci Res 1998;31:317–323. [DOI] [PubMed] [Google Scholar]
- 16. Lucas JJ, Hernández F, Gómez‐Ramos P, Morán MA, Hen R, Avila J. Decreased nuclear beta‐catenin, tau hyperphosphorylation and neurodegeneration in GSK‐3beta conditional transgenic mice. EMBO J 2001;20:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hock BJ Jr, Lamb BT. Transgenic mouse models of Alzheimer's disease. Trends Genet 2001;17:S7–S12. [DOI] [PubMed] [Google Scholar]
- 18. Malin DH, Crothers MK, Lake JR, et al Hippocampal injections of amyloid beta‐peptide 1–40 impair subsequent one‐trial/day reward learning. Neurobiol Learn Mem 2001;76:125–137. [DOI] [PubMed] [Google Scholar]
- 19. Miguel‐Hidalgo JJ, Cacabelos R. Beta‐amyloid(1–40)‐induced neurodegeneration in the rat hippocampal neurons of the CA1 subfield. Acta Neuropathol (Berl) 1998;95:455–465. [DOI] [PubMed] [Google Scholar]
- 20. Morimoto K, Yoshimi K, Tonohiro T, Yamada N, Oda T, Kaneko I. Co‐injection of beta‐amyloid with ibotenic acid induces synergistic loss of rat hippocampal neurons. Neuroscience 1998;84:479–487. [DOI] [PubMed] [Google Scholar]
- 21. Connor B, Dragunow M. The role of neuronal growth factors in neurodegenerative disorders of the human brain. Brain Res Brain Res Rev 1998;27:1–39. [DOI] [PubMed] [Google Scholar]
- 22. Castren E, Tanila H. Neurotrophins and dementia‐keeping in touch. Neuron 2006;51:1–3. [DOI] [PubMed] [Google Scholar]
- 23. Wyss‐Coray T. Tgf‐Beta pathway as a potential target in neurodegeneration and Alzheimer's. Curr Alzh Res 2006;3:191–195. [DOI] [PubMed] [Google Scholar]
- 24. Brionne TC, Tesseur I, Masliah E, Wyss Coray T. Loss of TGF‐β1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron 2003;40:1133–1145. [DOI] [PubMed] [Google Scholar]
- 25. Ten Dijke P, Hill CS. New insights into TGF‐beta‐Smad signalling. Trends Biochem Sci 2004;29:265–273. [DOI] [PubMed] [Google Scholar]
- 26. Taipale J, Saharinen J, Keski‐Oja J. Extracellular matrixassociated transforming growth factor‐beta: Role in cancer cell growth and invasion. Adv Cancer Res 1998;75:87–134. [DOI] [PubMed] [Google Scholar]
- 27. Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor‐β regulation of immune responses. Annu Rev Immunol 2006;24:99–146. [DOI] [PubMed] [Google Scholar]
- 28. Dubois CM, Laprise MH, Blanchette F, Gentry LE, Leduc R. Processing of transforming growth factor beta 1 precursor by human furin convertase. J Biol Chem 1995;270:10618–10624. [DOI] [PubMed] [Google Scholar]
- 29. Lawrence DA, Pircher R, Kryceve‐Martinerie C, Jullien P. Normal embryo fibroblasts release transforming growth factors in a latent form. J Cell Physiol 1984;121:184–188. [DOI] [PubMed] [Google Scholar]
- 30. Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci 2003;116:217–224. [DOI] [PubMed] [Google Scholar]
- 31. Shi Y, Massagué J. Mechanisms of TGF‐beta signaling from cell membrane to the nucleus. Cell 2003;113:685–700. [DOI] [PubMed] [Google Scholar]
- 32. Derynck R, Zhang YE. Smad‐dependent and Smad‐independent pathways in TGF‐beta family signalling. Nature 2003;425:577–584. [DOI] [PubMed] [Google Scholar]
- 33. Caraci F, Gili E, Calafiore M, et al TGF‐beta1 targets the GSK‐3beta/beta‐catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol Res 2008;57:274–282. [DOI] [PubMed] [Google Scholar]
- 34. König HG, Kögel D, Rami A, Prehn JH. TGF‐{beta}1 activates two distinct type I receptors in neurons: Implications for neuronal NF‐{kappa}B signaling. J Cell Biol 2005;168:1077–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Caraci F, Battaglia G, Busceti C, et al TGF‐beta 1 protects against Abeta‐neurotoxicity via the phosphatidylinositol‐3‐kinase pathway. Neurobiol Dis 2008;30:234–242. [DOI] [PubMed] [Google Scholar]
- 36. Vivien D, Ali C. Transforming growth factor‐β signalling in brain disorder. Cytokine Growth Factor Rev 2006;17:121–128. [DOI] [PubMed] [Google Scholar]
- 37. Finch CE, Laping NJ, Morgan TE, Nichols NR, Pasinetti GM. TGF‐β1 is an organizer of response to neurodegeneration. J Cell Biochem 1993;53:314–322. [DOI] [PubMed] [Google Scholar]
- 38. Salvioli S, Capri M, Bucci L, et al Why do centenarians escape or postpone cancer? The role of IGF‐1, inflammation and p53. Cancer Immunol Immunother 2009;58:1909–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Franceschi C, Bonafe M, Valensin S, et al Inflamm‐aging. An evolutionary perspective on immunosenescence. Ann NY Acad Sci 2000;908:244–254. [DOI] [PubMed] [Google Scholar]
- 40. Frippiat C, Chen QM, Zdanov S, Magalhaes JP, Remacle J, Toussaint O. Subcytotoxic H2O2 stress triggers a release of transforming growth factor‐beta 1, which induces biomarkers of cellular senescence of human diploid fibroblasts. J Biol Chem 2001;276:2531–2537. [DOI] [PubMed] [Google Scholar]
- 41. Carrieri G, Marzi E, Olivieri F, et al The G/C915 polymorphism of transforming growth factor beta1 is associated with human longevity: A study in Italian centenarians. Aging Cell 2004;3:443–448. [DOI] [PubMed] [Google Scholar]
- 42. Forsey RJ, Thompson JM, Ernerudh J, et al Plasma cytokine profiles in elderly humans. Mech Ageing Dev 2003;124:487–493. [DOI] [PubMed] [Google Scholar]
- 43. Tarkowski E, Issa R, Sjögren M, et al Increased intrathecal levels of the angiogenic factors VEGF and TGF‐beta in Alzheimer's disease and vascular dementia. Neurobiol Aging 2002;23:237–243. [DOI] [PubMed] [Google Scholar]
- 44. Chao CC, Ala TA, Hu S, et al Serum cytokine levels in patients with Alzheimer's disease. Clin Diagn Lab Immunol 1994;1:433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mocali A, Cedrola S, Della Malva N, et al Increased plasma levels of soluble CD40, together with the decrease of TGF beta 1, as possible differential markers of Alzheimer disease. Exp Gerontol 2004;39:1555–1561. [DOI] [PubMed] [Google Scholar]
- 46. Arosio B, Bergamaschini L, Galimberti L, et al +10 T/C polymorphisms in the gene of transforming growth factor‐beta1 are associated with neurodegeneration and its clinical evolution. Mech Ageing Dev 2007;128:553–557. [DOI] [PubMed] [Google Scholar]
- 47. Lee HG, Ueda M, Zhu X, Perry G, Smith MA. Ectopic expression of phospho‐Smad2 in Alzheimer's disease: Uncoupling of the transforming growth factor‐beta pathway? J Neurosci Res 2006;84:1856–1861. [DOI] [PubMed] [Google Scholar]
- 48. Tesseur I, Zou K, Esposito L, et al Deficiency in neuronal TGF‐beta signaling promotes neurodegeneration and Alzheimer's pathology. J Clin Invest 2006;116:3060–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ueberham U, Ueberham E, Gruschka H, Arendt T. Altered subcellular location of phosphorylated Smads in Alzheimer's disease. Eur J Neurosci 2006;24:2327–2334. [DOI] [PubMed] [Google Scholar]
- 50. Chalmers KA, Love S. Neurofibrillary tangles may interfere with Smad 2/3 signaling in neurons. J Neuropathol Exp Neurol 2007;66:158–167. [DOI] [PubMed] [Google Scholar]
- 51. Burton T, Liang B, Dibrov A, Amara F. Transforming growth factor‐beta‐induced transcription of the Alzheimer beta‐amyloid precursor protein gene involves interaction between the CTCFcomplex and Smads. Biochem Biophys Res Commun 2002;295:713–723. [DOI] [PubMed] [Google Scholar]
- 52. Lesne S, Docagne F, Gabriel C, et al Transforming growth factor‐beta 1 potentiates amyloid‐beta generation in astrocytes and in transgenic mice. J Biol Chem 2003;278:18408–18418. [DOI] [PubMed] [Google Scholar]
- 53. Wyss‐Coray T, Masliah E, Mallory M, et al Amyloidogenic role of cytokine TGF‐β1 in transgenic mice and Alzheimer's disease. Nature 1997;389:603–606. [DOI] [PubMed] [Google Scholar]
- 54. Gaertner RF, Wyss‐Coray T, Von Euw D, Lesne S, Vivien D, Lacombe P. Reduced brain tissue perfusion in TGF‐beta 1 transgenic mice showing Alzheimer's disease‐like cerebrovascular abnormalities. Neurobiol Dis 2005;19:38–46. [DOI] [PubMed] [Google Scholar]
- 55. Harris‐White ME, Chu T, Balverde Z, Sigel JJ, Flanders KC, Frautschy SA. Effects of transforming growth factor‐beta (isoforms 1–3) on amyloid‐beta deposition, inflammation, and cell targeting in organotypic hippocampal slice cultures. J Neurosci 1998;18:10366–10374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Grammas P, Ovase R. Cerebrovascular transforming growth factor‐beta contributes to inflammation in the Alzheimer's disease brain. Am J Pathol 2002;160:1583–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Town T, Laouar Y, Pittenger C, et al Blocking TGF‐beta‐Smad2/3 innate immune signaling mitigates Alzheimer‐like pathology. Nat Med 2008;14:681–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wyss‐Coray T, Lin C, Yan F, et al TGF‐beta1 promotes microglial amyloid‐beta clearance and reduces plaque burden in transgenic mice. Nat Med 2001;7:612–618. [DOI] [PubMed] [Google Scholar]
- 59. Luterman JD, Haroutunian V, Yemul S, et al Cytokine gene expression as a function of the clinical progression of Alzheimer disease dementia. Arch Neurol 2000;57:1153–1160. [DOI] [PubMed] [Google Scholar]
- 60. Lee EO, Kang JL, Chong YH. The amyloid‐beta peptide suppresses transforming growth factor‐beta1‐induced matrix metalloproteinase‐2 production via Smad7 expression in human monocytic THP‐1 cells. J Biol Chem 2005;280:7845–7853. [DOI] [PubMed] [Google Scholar]
- 61. Arendt T, Brückner MK. Linking cell‐cycle dysfunction in Alzheimer's disease to a failure of synaptic plasticity. Biochim Biophys Acta 2007;1772:413–421. [DOI] [PubMed] [Google Scholar]
- 62. Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer's disease? J Cell Biol 1996;132:413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: Cell cycle events as triggers of nerve cell death. J Neurosci 2004;24:9232–9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Copani A, Caraci F, Hoozemans JJ, Calafiore M, Sortino MA, Nicoletti F. The nature of the cell cycle in neurons: Focus on a “non‐canonical” pathway of DNA replication causally related to death. Biochim Biophys Acta 2007;1772:409–412. [DOI] [PubMed] [Google Scholar]
- 65. Flanders KC, Ren RF, Lippa CF. Transforming growth factor–βs in neurodegenerative disease. Prog Neurobiol 1998;54:71–85. [DOI] [PubMed] [Google Scholar]
- 66. Dhandapani KM, Hadman M, De Sevilla L, Wade MF, Mahesh VB, Brann DW. Astrocyte protection of neurons: Role of transforming growth factor‐beta signaling via a c‐Jun‐AP‐1 protective pathway. J Biol Chem 2003;278:43329–43339. [DOI] [PubMed] [Google Scholar]
- 67. Sortino MA, Chisari M, Merlo S, et al Glia mediates the neuroprotective action of estradiol on beta‐amyloid‐induced neuronal death. Endocrinology 2004;145:5080–5086. [DOI] [PubMed] [Google Scholar]
- 68. Prehn JH, Bindokas VP, Jordan J, et al Protective effect of transforming growth factor‐beta 1 on beta‐amyloid neurotoxicity in rat hippocampal neurons. Mol Pharmacol 1996;49:319–328. [PubMed] [Google Scholar]
- 69. Ren RF, Flanders KC. Transforming growth factors‐beta protect primary rat hippocampal neuronal cultures from degeneration induced by beta‐amyloid peptide. Brain Res 1996;732:16–24. [DOI] [PubMed] [Google Scholar]
- 70. Ren RF, Hawver DB, Kim RS, Flanders KC. Transforming growth factor‐beta protects human hNT cells from degeneration induced by beta‐amyloid peptide: Involvement of the TGF‐beta type II receptor. Brain Res Mol Brain Res 1997;48:315–322. [DOI] [PubMed] [Google Scholar]
- 71. Kim ES, Kim RS, Ren RF, Hawver DB, Flanders KC. Transforming growth factor‐beta inhibits apoptosis induced by beta‐amyloid peptide fragment 25–35 in cultured neuronal cells. Brain Res Mol Brain Res 1998;62:122–130. [DOI] [PubMed] [Google Scholar]
- 72. Inman GJ, Nicolas FJ, Callahan JF, et al SB‐431542 is a potent and specific inhibitor of transforming growth factor‐beta superfamily type I activin receptor‐like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol 2002;62:65–74. [DOI] [PubMed] [Google Scholar]
- 73. Boche D, Cunningham C, Docagne F, Scott H, Perry VH. TGFbeta1 regulates the inflammatory response during chronic neurodegeneration. Neurobiol Dis 2006;22:638–650. [DOI] [PubMed] [Google Scholar]
- 74. Okello A, Edison P, Archer HA, et al Microglial activation and amyloid deposition in mild cognitive impairment: A PET study. Neurology 2009;72:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Maccioni RB, Rojo LE, Fernández JA, Kuljis RO. The role of neuroimmunomodulation in Alzheimer's disease. Ann N Y Acad Sci 2009;1153:240–246. [DOI] [PubMed] [Google Scholar]
- 76. Frautschy SA, Cole GM, Baird A. Phagocytosis and deposition of vascular β‐amyloid in rat brains injected with Alzheimer β‐amyloid. Am J Pathol 1992;140:1389–1399. [PMC free article] [PubMed] [Google Scholar]
- 77. Magnus T, Chan A, Linker RA, Toyka KV, Gold R. Astrocytes are less efficient in the removal of apoptotic lymphocytes than microglia cells: Implications for the role of glial cells in the inflamed central nervous system. J Neuropathol Exp Neurol 2002;61:760–766. [DOI] [PubMed] [Google Scholar]
- 78. Unsicker K, Krieglstein K. Co‐activation of TGF‐β and cytokine signaling pathways are required for neurotropic functions. Cytokine Growth Factor Rev 2000;11:97–102. [DOI] [PubMed] [Google Scholar]
- 79. Unsicker K, Krieglstein K. TGF‐betas and their roles in the regulation of neuron survival. Adv Exp Med Biol 2002;513:353–374. [DOI] [PubMed] [Google Scholar]
- 80. Schober A, Peterziel H, von Bartheld CS, Simon H, Krieglstein K, Unsicker K. GDNF applied to the MPTP‐lesioned nigrostriatal system requires TGF‐beta for its neuroprotective action. Neurobiol Dis 2007;25:378–391. [DOI] [PubMed] [Google Scholar]
- 81. Murer MG, Yan Q, Raisman‐Vozari R. Brain‐derived neurotrophic factor in the control human brain, and in Alzheimer's disease and Parkinson's disease. Prog Neurobiol 2001;63:71–124. [DOI] [PubMed] [Google Scholar]
- 82. Cotman CW. The role of neurotrophins in brain aging: A perspective in honor of Regino Perez‐Polo. Neurochem Res 2005;30:877–881. [DOI] [PubMed] [Google Scholar]
- 83. Sometani A, Kataoka H, Nitta A, Fukumitsu H, Nomoto H, Furukawa S. Transforming growth factor‐beta1 enhances expression of brain‐derived neurotrophic factor and its receptor, TrkB, in neurons cultured from rat cerebral cortex. J Neurosci Res 2001;66:369–376. [DOI] [PubMed] [Google Scholar]
- 84. Zhu Y, Ahlemeyer B, Bauerbach E, Krieglstein J. TGF‐beta1 inhibits caspase‐3 activation and neuronal apoptosis in rat hippocampal cultures. Neurochem Int 2001;38:227–235. [DOI] [PubMed] [Google Scholar]
- 85. Zhu Y, Yang G‐Y, Ahlemeyer B, et al Transforming growth factor‐b1 increases bad phosphorylation and protects neurons against damage. J Neurosci 2002;22:3898–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhu Y, Culmsee C, Klumpp S, Krieglstein J. Neuroprotection by transforming growth factor‐beta1 involves activation of nuclear factor‐kappaB through phosphatidylinositol‐3‐OH kinase/Akt and mitogen‐activated protein kinase‐extracellular‐signal regulated kinase1,2 signaling pathways. Neuroscience 2004;123:897–906. [DOI] [PubMed] [Google Scholar]
- 87. Hübscher U, Nasheuer HP, Syväoja JE. Eukaryotic DNA polymerases, a growing family. Trends Biochem Sci 2004;25:143–147. [DOI] [PubMed] [Google Scholar]
- 88. Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci 2001;21:2661–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Slee EA, O’Connor DJ, Lu X. To die or not to die: How does p53 decide? Oncogene 2004;23:2809–2818. [DOI] [PubMed] [Google Scholar]
- 90. Paradis E, Douillard H, Koutroumanis M, Goodyer C, LeBlanc A. Amyloid beta peptide of Alzheimer's disease downregulates Bcl‐2 and upregulates bax expression in human neurons. J Neurosci 1996;16:7533–7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Caricasole A, Copani A, Caraci F, et al Induction of Dickkopf‐1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer's brain. J Neurosci 2004;24:6021–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Willert K, Nusse R. 1998. Beta‐catenin: A key mediator of Wnt signaling. Curr Opin Genet Dev 1998;8:95–102. [DOI] [PubMed] [Google Scholar]
- 93. Fuentealba RA, Farias G, Scheu J, Bronfman M, Marzolo MP, Inestrosa NC. Signal transduction during amyloid‐beta‐peptide neurotoxicity: Role in Alzheimer disease. Brain Res Brain Res Rev 2004;47:275–289. [DOI] [PubMed] [Google Scholar]
- 94. Murase S, Mosser E, Schuman EM. Depolarization drives beta‐catenin into neuronal spines promoting changes in synaptic structure and function. Neuron 2002;35:91–105. [DOI] [PubMed] [Google Scholar]
- 95. Yu X, Malenka RC. Beta‐catenin is critical for dendritic morphogenesis. Nat Neurosci 2003;6:1169–1177. [DOI] [PubMed] [Google Scholar]
- 96. Liolitsa D, Powell J, Lovestone S. Genetic variability in the insulin signalling pathway may contribute to the risk of late onset Alzheimer's disease. J Neurol Neurosurg Psychiatry 2002;73:261–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Castri P, Iacovelli L, De Blasi A, et al Reduced insulin‐induced phosphatidylinositol‐3‐kinase activation in peripheral blood mononuclear leucocytes from patients with Alzheimer's disease. Eur J Neurosci 2007;26:2469–2472. [DOI] [PubMed] [Google Scholar]
- 98. Tuszynski MH, Thal L, Pay M, et al A phase I clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med 2005;11:551–555. [DOI] [PubMed] [Google Scholar]
- 99. Nagahara AH, Merrill DA, Coppola G, et al Neuroprotective effects of brain‐derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med 2009;15:331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Pollack SJ, Harper SJ. Small molecule Trk receptor agonists and other neurotrophic factor mimetics. Curr Drug Targets CNS Neurol Disord 2002;1:59–80. [DOI] [PubMed] [Google Scholar]
- 101. Skaper SD. The biology of neurotrophins, signalling pathways, and functional peptide mimetics of neurotrophins and their receptors. CNS Neurol Disord Drug Targets 2008;7:46–62. [DOI] [PubMed] [Google Scholar]
- 102. Zhang H, Zou K, Tesseur I, Wyss‐Coray T. Small molecule tgf‐beta mimetics as potential neuroprotective factors. Curr Alzheimer Res 2005;2:183–186. [DOI] [PubMed] [Google Scholar]
- 103. Brinton RD. Impact of estrogen therapy on Alzheimer's disease: A fork in the road? CNS Drugs 2004;18:405–422. [DOI] [PubMed] [Google Scholar]
- 104. Chen S, Nilsen J, Brinton RD. Dose and temporal pattern of estrogen exposure determines neuroprotective outcome in hippocampal neurons: Therapeutic implications. Endocrinology 2006;147:5303–5313. [DOI] [PubMed] [Google Scholar]
- 105. Redondo S, Santos‐Gallego CG, Tejerina T. TGF‐beta1: A novel target for cardiovascular pharmacology. Cytokine Growth Factor Rev 2007;18:279–286. [DOI] [PubMed] [Google Scholar]
- 106. Broe GA, Grayson DA, Creasey HM, et al Anti‐inflammatory drugs protect against Alzheimer disease at low doses. Arch Neurol 2000;57:1586–1591. [DOI] [PubMed] [Google Scholar]
- 107. Nilsson SE, Johansson B, Takkinen S, et al Does aspirin protect against Alzheimer's dementia? A study in a Swedish population‐based sample aged > or = 80 years. Eur J Clin Pharmacol 2003;59:313–319. [DOI] [PubMed] [Google Scholar]
- 108. Li G, Larson EB, Sonnen JA, et al Statin therapy is associated with reduced neuropathologic changes of Alzheimer disease. Neurology 2007;69:878–885. [DOI] [PubMed] [Google Scholar]
- 109. Sparks DL, Kryscio RJ, Sabbagh MN, Connor DJ, Sparks LM, Liebsack C. Reduced risk of incident AD with elective statin use in a clinical trial cohort. Curr Alzheimer Res 2008;5:416–421. [DOI] [PubMed] [Google Scholar]
- 110. Redondo S, Santos‐Gallego CG, Ganado P, García M, Rico L, Del Rio M, Tejerina T. Acetylsalicylic acid inhibits cell proliferation by involving transforming growth factor‐beta. Circulation 2003;107:626–629. [DOI] [PubMed] [Google Scholar]
- 111. Grainger DJ, Kemp PR, Metcalfe JC, et al The serum concentration of active transforming growth factor‐beta is severely depressed in advanced atherosclerosis. Nat Med 1995;1:174–179. [DOI] [PubMed] [Google Scholar]
- 112. Porreca E, Di Febbo C, Baccante G, Di Nisio M, Cuccurullo F. Increased transforming growth factor‐beta(1) circulating levels and production in human monocytes after 3‐hydroxy‐3‐methyl‐glutarylcoenzyme a reductase inhibition with pravastatin. J Am Coll Cardiol 2002;39:1752–1757. [DOI] [PubMed] [Google Scholar]
- 113. Nakamura K, Masuda H, Kariyazono H, et al Effects of atorvastatin and aspirin combined therapy on inflammatory responses in patients undergoing coronary artery bypass grafting. Cytokine 2006;36:201–210. [DOI] [PubMed] [Google Scholar]
- 114. Simpson D, Noble S, Perry C. Glatiramer acetate: A review of its use in relapsing‐remitting multiple sclerosis. CNS Drugs 2002;16:825–850. [DOI] [PubMed] [Google Scholar]
- 115. Arnon R, Aharoni R. Mechanism of action of glatiramer acetate in multiple sclerosis and its potential for the development of new applications. Proc Natl Acad Sci U S A 2004;101(Suppl 2):14593–14598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Aharoni R, Eilam R, Domev H, Labunskay G, Sela M, Arnon R. The immunomodulator glatiramer acetate augments the expression of neurotrophic factors in brains of experimental autoimmune encephalomyelitis mice. Proc Natl Acad Sci U S A 2005;102:19045–19050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Breivik T, Gundersen Y, Myhrer T, et al Enhanced susceptibility to periodontitis in an animal model of depression: Reversed by chronic treatment with the anti‐depressant tianeptine. J Clin Periodontol 2006;33:469–477. [DOI] [PubMed] [Google Scholar]
- 118. Sutcigil L, Oktenli C, Musabak U, et al Pro‐ and anti‐inflammatory cytokine balance in major depression: Effect of sertraline therapy. Clin Dev Immunol 2007; doi: 10.1155/2007/76396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Vollmar P, Haghikia A, Dermietzel R, Faustmann PM. Venlafaxine exhibits an anti‐inflammatory effect in an inflammatory co‐culture model. Int J Neuropsychopharmacol 2008;11:111–117. [DOI] [PubMed] [Google Scholar]
- 120. Nelson RL, Guo Z, Halagappa VM, et al Prophylactic treatment with paroxetine ameliorates behavioral deficits and retards the development of amyloid and tau pathologies in 3xTgAD mice. Exp Neurol 2007;205:166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ownby RL, Crocco E, Acevedo A, John V, Loewenstein D. Depression and risk for Alzheimer disease: Systematic review, meta‐analysis, and metaregression analysis. Arch Gen Psychiatry 2006;63:530–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Modrego PJ, Ferrández J. Depression in patients with mild cognitive impairment increases the risk of developing dementia of Alzheimer type: A prospective cohort study. Arch Neurol 2004;61:1290–1293. [DOI] [PubMed] [Google Scholar]
- 123. Myint AM, Leonard BE, Steinbusch HW, Kim YK. Th1, Th2, and Th3 cytokine alterations in major depression. J Affect Disord 2005;88:167–173. [DOI] [PubMed] [Google Scholar]
- 124. Lee KM, Kim YK. The role of IL‐12 and TGF‐beta1 in the pathophysiology of major depressive Disorder. Int Immunopharmacol 2006;6:1298–1304. [DOI] [PubMed] [Google Scholar]
- 125. Kessing LV, Søndergård L, Forman JL, Andersen PK. Antidepressants and dementia. J Affect Disord 2009;117:24–29. [DOI] [PubMed] [Google Scholar]
- 126. Wada A, Yokoo H, Yanagita T, Kobayashi H. Lithium: Potential therapeutics against acute brain injuries and chronic neurodegenerative diseases. J Pharmacol Sci 2005;99:307–321. [DOI] [PubMed] [Google Scholar]
- 127. Yeh HL, Tsai SJ. Lithium may be useful in the prevention of Alzheimer's disease in individuals at risk of presenile familial Alzheimer's disease. Med Hypotheses 2008;71:948–951. [DOI] [PubMed] [Google Scholar]
- 128. Nunes PV, Forlenza OV, Gattaz WF. Lithium and risk for Alzheimer's disease in elderly patients with bipolar disorder. Br J Psychiatry 2007;190:359–360. [DOI] [PubMed] [Google Scholar]
- 129. Liang MH, Wendland JR, Chuang DM. Lithium inhibits Smad 3/4 transactivation via increased CREB activity induced by enhanced PKA and AKT signaling. Mol Cell Neurosc 2008;37:440–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Bruno V, Battaglia G, Casabona G, Copani A, Caciagli F, Nicoletti F. Neuroprotection by glial metabotropic glutamate receptors is mediated by transforming growth factor‐beta. J Neurosci 1998;18:9594–9600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. D’Onofrio M, Cuomo L, Battaglia G, et al Neuroprotection mediated by glial group‐II metabotropic glutamate receptors requires the activation of the MAP kinase and the phosphatidylinositol‐3‐kinase pathways. J Neurochem 2001;78:435–445. [DOI] [PubMed] [Google Scholar]
- 132. Copani A, Bruno V, Battaglia G, et al Activation of metabotropic glutamate receptors protects cultured neurons against apoptosis induced by beta‐amyloid peptide. Mol Pharmacol 1995;47:890–897. [PubMed] [Google Scholar]
- 133. Patil ST, Zhang L, Martenyi F, et al Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: A randomized Phase 2 clinical trial. Nat Med 2007;13:1102–1107. [DOI] [PubMed] [Google Scholar]
- 134. Gill SS, Rochon PA, Herrmann N, et al Atypical antipsychotic drugs and risk of ischaemic stroke: Population based retrospective cohort study. BMJ 2005; doi: 10.1136/bmj.38330.470486.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]