

SUMMARY
The cannabinoid (CB) system is widespread in the central nervous system and is crucial for controlling a range of neurophysiological processes such as pain, appetite, and cognition. The endogenous CB molecules, anandamide, and 2‐arachidonoyl glycerol, interact with the G‐protein coupled CB receptors, CB1 and CB2. These receptors are also targets for the phytocannabinoids isolated from the cannabis plant and synthetic CB receptor ligands. The CB system is emerging as a key regulator of neuronal cell fate and is capable of conferring neuroprotection by the direct engagement of prosurvival pathways and the control of neurogenesis. Many neurological conditions feature a neurodegenerative component that is associated with excitotoxicity, oxidative stress, and neuroinflammation, and certain CB molecules have been demonstrated to inhibit these events to halt the progression of neurodegeneration. Such properties are attractive in the development of new strategies to treat neurodegenerative conditions of diverse etiology, such as Alzheimer's disease, multiple sclerosis, and cerebral ischemia. This article will discuss the experimental and clinical evidence supporting a potential role for CB‐based therapies in the treatment of certain neurological diseases that feature a neurodegenerative component.
Keywords: Alzheimer's disease, Cannabinoid, Cerebral ischemia, Multiple sclerosis, Neurodegeneration, Parkinson's disease
Introduction
The use of the Cannabis sativa plant as a medicinal preparation is referred to in ancient Asian pharmacopoeia but it was the Irishman Sir William B. O'Shaugnessey who conducted the first series of formal experiments into the potential medicinal use of Cannabis sativa and the publication of his findings in 1842 brought the use of cannabis for the treatment of pain, spasticity, and rheumatism to clinicians throughout Europe [1]. However, difficulties in obtaining consistent preparations and the availability of more effective drugs marginalized the medicinal use of cannabis. A renewed revival in the cannabinoid (CB) field occurred in the 1960s upon the identification and synthesis of the active psychotropic component of cannabis, Δ9‐Tetrahydrocannabinol (Δ9‐THC) [2]. In addition to the phytocannabinoid molecules, such as Δ9‐THC and cannabidiol (CBD), that have been extracted from Cannabis sativa, recent years have witnessed a marked advance in endocannabinoid biology that has propelled it to the forefront of biomedical research. The CB system is now recognized as an important physiological modulator of various central nervous system processes including pain [3, 4], appetite [5, 6], motor function [7, 8], synaptic plasticity [9], neuroinflammation [10, 11], and neural cell fate [12, 13, 14]. The current therapeutic applications of CB‐based medications, such as Dronabinol and Nabilone, include the treatment of anorexia and emesis [15]. The oromucosal spray, Sativex, which contains the phytocannabinoids, Δ9‐THC, and cannabinidiol (Figure 1), is effective in treating the spasticity and pain experienced by patients with multiple sclerosis (MS) [16], and Δ9‐THC has been demonstrated to exert antitumoral activity in clinical studies [17]. There is also a growing interest in developing a CB‐based approach to treat other disorders, which are associated with neuroinflammation and neurodegeneration. The development of such therapeutic strategies will rely on a more detailed understanding of the role of the CB system in the disease pathology in order to exploit that knowledge and circumvent the disease process.
Figure 1.
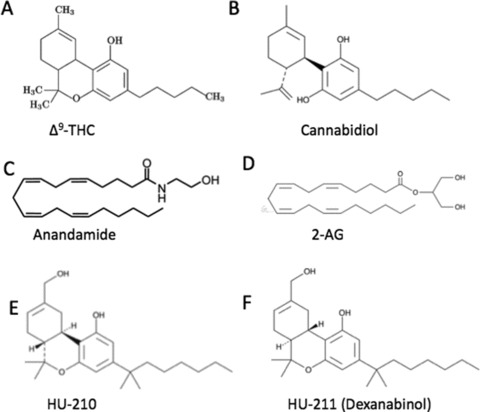
Chemical structure of cannabinoids. The chemical structure of the phytocannabinoids (A)Δ9‐tetrahydrocannabinol (Δ9‐THC) and (B) cannabidiol; the endocannabinoids (C) anandamide and (D) 2‐arachidonoyl glycerol (2‐AG); the synthetic cannabinoids (E) HU‐210 and (F) HU‐211.
The CB System
The endogenous CB signaling system in the brain is composed of the endocannabinoid molecules, 2‐arachidonoyl glycerol (2‐AG), and anandamide (AEA), which interact with the G‐protein‐coupled CB receptors, CB1 and CB2. The CB1 receptor is abundantly expressed in the central nervous system (CNS) [18] while expression of the CB2 receptor is mainly associated with the peripheral immune system. CB2 receptor expression has been observed in the CNS, albeit confined to neurones within the brainstem [19] and on microglia during neuroinflammation [20]. Endocannabinoids are synthesized on demand and travel in a retrograde manner across the postsynaptic membrane to activate presynaptic CB1 receptors resulting in the depression of neurotransmitter release. Endocannabinoid levels are controlled by the action of two enzymes: fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) which degrade AEA and 2‐AG, respectively. Endocannabinoid pharmacology is constantly evolving with new cannabimetic ligands [21] and putative CB receptors [22] being proposed. For example, novel orphan receptors such as GRP55 [23, 24] and GPR18 [25] are sensitive to CBs and have been demonstrated to regulate the recruitment and activation of microglia and neuropathic pain. The TRPV1 receptor is also regulated by CBs and is involved in pain and inflammation [26]. These proteins may represent interesting targets for drug development to treat neurological conditions, which are associated with neuroinflammation and neuropathic pain.
Activation of the canonical CB receptor inhibits adenylate cyclase resulting in a decreased level of the second messenger cyclic adenosine monophosphate (cAMP). This second messenger regulates a number of intracellular signaling cascades essential to a number of cellular functions such as the regulation of cell fate [27]. CB receptor activation also couples to ion channel regulation such as inhibition of voltage‐dependant Ca2+ channels and activation of inwardly rectifying K+ channels [28, 29, 30]. The modulation of voltage‐dependent ion channels is thought to underlie CB‐induced depression of excitatory neurotransmission [31]. Currently, the potential neuroprotective effects of CBs in acute brain injury such as stroke and in chronic neurodegenerative diseases such as Alzheimer's disease (AD) have been attributed, in part, to their proclivity to reduce neuronal damaging events such as excessive glutamatergic transmission, prolonged Ca2+ influx, and oxidative stress [32]. There is now abundant experimental and clinical evidence demonstrating that the endocannabinoid system is altered in common neurodegenerative conditions and that CB molecules can confer neuroprotection through an array of mechanisms. The CB system is therefore an opportunistic target for therapeutic exploitation, especially for those diseases with a neurodegenerative component for which effective treatment is lacking.
CBs and AD
AD is an age‐related neurodegenerative disease and with the demographic shift toward an increasingly aged population, an increase in the prevalence of AD is to be expected. AD is defined by the progressive deterioration of cognition and memory and is the most common form of dementia among the elderly [33]. The pathological hallmarks of AD include neurofibrillary tangles, amyloid‐beta (Aβ) plaques, and synaptic degeneration resulting in the preferential neuronal loss in the hippocampus and surrounding areas of the cerebral cortex. This disease is also characterized by the prolonged activation of microglia in plaque‐bearing areas which give rise to an environment profuse in inflammation [34, 35] and oxidative stress [36].
The high density of CB1 receptors in the hippocampus and cerebral cortex [37] was an early indication that the endocannabinoid system might play a role in AD pathogenesis. In the AD brain, microglial CB1 and CB2 receptor expression is significantly increased, principally in plaque‐bearing areas [38], while neuronal CB1 receptor expression is reduced, particularly in the hippocampus and basal ganglia [38, 39]. In the AD brain CB receptors are nitrosylated [38], thus affecting G protein coupling and rendering the receptors functionally impaired. An upregulation of the endocannabinoid‐metabolizing enzyme, FAAH, on plaque‐associated astrocytes has also been reported in the AD brain [40]. Given that AEA and, at least partially 2‐AG, are metabolized by FAAH to produce arachidonic acid, the increased presence of FAAH on astrocytes links the endocannabinoid system to the destructive inflammatory process that accompanies AD. These AD‐related alterations provide evidence that components of the endocannabinoid system may be critically intertwined with the progression of AD pathogenesis.
The preclinical data have highlighted some beneficial effects of CBs in inhibiting certain neuropathological features of relevance to AD [38]. Van Der Stelt and colleagues demonstrated that in an animal model of AD‐like pathology, the Aβ‐induced hippocampal degeneration, gliosis, and cognitive decline result in a concomitant increase in 2‐AG [41]. The conclusions drawn from that study proposed that endocannabinoid tone may be enhanced in the diseased brain as a protective mechanism against Aβ‐induced damage. Several studies have supported neuroprotective attributes of the endocannabinoid system against a wide range of insults, including those that play prominent roles in the AD brain, for example, excitotoxicity, inflammation, and oxidative stress. These underlying pathologies are the target of many current AD therapies, though none of the drugs used in AD patients have had any curative or lasting effect. With regards to the endocannabinoid system, its therapeutic attraction is based on the fact that it has widespread effects, in some cases mimicking the results of current drugs approved by the Food and Drug Administration (FDA), and in most cases alleviating symptoms with superior potency. For example, the drug memantine, a noncompetitive low affinity antagonist of the N‐methyl D‐aspartate (NMDA) receptor, acts to reduce Ca2+ influx and limit excitotoxicity. In a similar fashion, the synthetic CB, HU‐211 (Dexanabinol) acts as a stereoselective inhibitor of NMDA receptors, and thus protects cells from NMDA‐induced neurotoxicity [42, 43]. Endocannabinoid‐mediated neuroprotection against excitotoxicity is a widely established phenomenon, and is therapeutically beneficial given that it can be achieved by a number of different mechanisms, including inhibition of presynaptic glutamate release [44], blockage of voltage‐dependent N–, P/Q–, and L‐type calcium channels [45, 46, 47], and inhibition of Ca2+ release from ryanodine sensitive stores [48]. Also, akin to the effect of memantine, CB1 receptor stimulation increases levels of brain‐derived neurotrophic factor (BDNF) [49], a neurotrophin required for adult neurogenesis [50], that is decreased in the AD brain [51, 52].
Aβ toxicity triggers neuroinflammatory processes, which are closely associated with the neuropathological and cognitive symptoms of AD [53]. For this reason antiinflammatory drugs, such as nonsteroidal antiinflammatory drugs, are thought to reduce the incidence of AD [54]. Ehrhart and colleagues have reported that stimulation of CB2 receptors suppresses microglial activation and subsequent production of TNF‐α and nitric oxide [55]. Similarly, in an animal model of AD‐like pathology, administration of the synthetic CB, WIN55,212–2, prevents Aβ‐induced cognitive impairment and neuronal loss [38] while in vitro analysis demonstrated that the CB1 and CB2 receptor agonist, HU‐210, prevents Aβ‐induced increases in microglia activation and TNF‐α release [38]. Thus, the upregulation of microglial CB2 receptors observed in the AD brain [38, 40] may in fact be a neuroprotective response by the endocannabinoid system to limit neuroinflammation. CB1 receptors, which are increased on glial cells in the AD brain [38], play a role in the modulation of several inflammatory cytokines [56].
The majority of drugs currently in use for the treatment of AD, such as tacrine (Cognex), act as acetylcholine esterase (AChE) inhibitors which prevent or slow the breakdown of ACh, a neurotransmitter that is reduced in the AD brain. Eubanks and colleagues have demonstrated that the phytocannabinoid, Δ9‐THC, has the proclivity to competitively inhibit AChE, thus increasing ACh levels [57]. Moreover, Δ9‐THC is capable of preventing Aβ peptide aggregation, which may hinder plaque formation [57]. These represent additional aspects of CB action that may be beneficial in AD.
The nonpsychoactive phytocannabinoid, CBD, has several features that could be exploited for the treatment of AD, including the prevention of glutamate‐induced excitotoxicity [58], reduction of proinflammatory mediators [59], and the ability to scavenge reactive oxygen species (ROS) and reduce lipid peroxidation [60]. CBD, by reducing the phosphorylation of glycogen synthase kinase‐3β, inhibits tau protein hyperphosphorylation, an additional neuropathological feature of AD [61].
Thus, manipulation of the CB system has diverse and effective consequences against a wide range of pathologies prominent in the AD brain. While the studies discussed here focus on recent preclinical data, the clinical data, albeit limited to a small number of studies, have also provided positive feedback on the usefulness of CB‐based drugs in AD. Dronabinol, an oil‐based solution of Δ9‐THC, is an antiemetic and appetite‐stimulator in AD patients [62], while Δ9‐THC also reduces the agitation that is common in patients with severe AD [63]. Despite these promising outcomes, the usefulness of CB‐based drugs for the treatment of AD awaits the results of rigorous clinical trials and the identification of novel molecular neurobiological affects of CBs [64].
CBs in Multiple Sclerosis
MS is an autoimmune disease that promotes demyelination of neurones and subsequent aberrant neuronal firing that contributes to spasticity and neuropathic pain. MS patients have significant neuroinflammation [65] and the disease is associated with excitotoxicity [66] and chronic neurodegeneration [67]. These pathological features of MS share similarities with other neurodegenerative conditions including AD and cerebral ischemia; disorders which may obtain benefit from compounds that target the endocannabinoid system. Indeed, there is evidence that MS patients gain symptomatic relief from cannabis extracts [68]. Furthermore, Sativex, an oral‐mucosal spray containing a 1:1 ratio of Δ9‐THC and CBD, has antispasmodic and analgesic properties with efficacy in MS patients [69]. The neuropathic pain associated with MS is reduced by dronabinol, an oral preparation of the Δ9‐THC analog [16, 70] and a meta‐analysis has revealed that CB‐based medications are superior to placebo in the treatment of MS‐related neuropathic pain [71]. The ability to modify pain may be attributed to a CB‐mediated regulation of supraspinal GABAergic and glutamatergic neurones [72]. Such analgesic properties of CBs represent an additional feature of CB‐based drugs that offer benefit to patients with MS.
The mechanisms underlying the efficacy of CB‐based treatments in MS may be addressed in the experimental autoimmune encephalomyelitis (EAE) animal model, which displays demyelination and neurological dysfunction consistent with the human disease [73]. The neurodegeneration associated with EAE is a complex series of events initiated by neuroinflammation and the associated infiltration of immune cells into the CNS, followed by a dysfunction of neural activity and neuronal loss [74]. Changes in the endocannabinoid system are a feature of EAE, as well as human MS. In EAE, brain endocannabinoid levels are downregulated [75], which may alter the immune‐regulatory function of the endocannabinoid system, while plasma endocannabinoid levels are increased in MS patients [76]. CB receptors are distributed in human immune cells including macrophages, lymphocytes, and natural killer (NK) cells [77] and mediate the effects of CBs on the activation, differentiation, and migration of these cells. Thus, the synthetic CB, HU‐211, reduces clinical disease severity in an EAE model in parallel with a reduction in inflammatory cell infiltration [78]. The CB1 and CB2 receptor agonist, WIN55,212–2, reduces the differentiation of T cells into Th1 effector cells, thereby reducing the production of inflammatory mediators and disease severity [79]. The CB2 receptor is notable for its role in the regulation of inflammation associated with EAE with CB2 deficiency associated with a more pronounced disease, increased mononuclear cell infiltration, and enhanced production of proinflammatory cytokines [80]. In MS patients increased CB2 immunoreactivity has been observed in the spinal cord [81], which may represent an attempt to reduce the neuroinflammation occurring during the disease.
In CB1 receptor‐deficient animals induced with EAE the development of neurodegeneration is more rapid [82] indicating an important role for CB1 in the provision of neuroprotection, which may be via an inhibition of excitotoxicity and/or oxidative stress [83, 84]. The neurodegenerative component of EAE has been demonstrated to involve glutamate‐induced excitotoxicity, which has been verified using glutamate receptor antagonists [85]. However, adverse effects of glutamate receptor antagonists have been reported in humans [86] and this has prompted investigation into alternative approaches to reduced excitotoxicity. In this regard, upregulation of endocannabinoid tone using UCM707, an inhibitor of endocannabinoid uptake, protects neurones from excitotoxicity in parallel with a therapeutic effect in a mouse model of MS [87]. This approach may therefore augment CB activity at sites of injury without the necessity for direct CB1 receptor activation, thereby reducing possible psychoactive side effects.
The CB System in Cerebral Ischemia
Cerebral ischemia is an acute neurodegenerative insult that may arise as a consequence of stroke, trauma, or cardiac arrest. There are mounting in vitro and in vivo data to suggest that CBs have neuroprotective effects following brain injury [88]. Indeed, the formation of endocannabinoids is enhanced after brain injury and there is evidence that these compounds reduce the secondary damage incurred after the initial injury [89, 90, 91]. Studies have shown increased AEA levels following controlled interruptions of blood flow in vivo[92] due to a decrease in AEA catabolism since FAAH expression and activity were found to be decreased [93]. Furthermore, AEA levels are significantly and preferentially (above other N‐aceylethanolamines) increased in middle cerebral artery occlusion (MCAO) ischemic models that include a reperfusion period compared to occlusion only [93, 94]. These data have been observed in one human study where increased AEA levels were found in a patient with a hemispheric stroke [95]. Although 2‐AG levels are increased following physical trauma such as concussive head injury and seizures [90, 96], a reduction in 2‐AG levels were observed in mice following MCAO [94, 97]. Studies have demonstrated that reperfusion injury is associated with a rapid increase in CB1 receptor expression on resident cells [98, 99] while increased CB2 receptor expression is seen on astrocytes, microglia, and infiltrating macrophages in the ischemic penumbra following transient MCAO [100]. Treatment with CB receptor agonists WIN55,212–2 and CP 55940 prior and up to 30 min postischemic insult prevents neuronal death and reduces infarct volumes in a CB1‐dependant manner [101]. These results have been supported in experiments using CB1 receptor knockout mice which show increased mortality following MCOA, thus reinforcing a role for the CB1 receptor in preventing ischemia‐induced neuronal loss [102]. Furthermore, the nonpsychoactive phytocannabinoid, CBD, results in the dramatic reversal of brain damage in hypoxic‐ischemic newborn piglets, in addition to other extracerebral benefits [103]. The control of the cerebral vasculature is an important consideration in ischemia/reperfusion injury and in this regard the CB1 receptor antagonist, SR141716A, in combination with the CB2 receptor agonist, 0–1966, has been found to increase blood flow to the brain in an animal model of stroke [104]. The proclivity of O‐1966 to attenuate neuroinflammation represents an additional beneficial effect of this molecule in cerebral ischemia [104]. In experiments where endocannabinoid catabolism is inhibited a neuroprotective effect against ischemia was observed [105]. However, the upregulation of the endocannabinoid system can manifest a dual response since some studies show that inhibition of the CB1 receptor is neuroprotective against ischemia [106, 107] while other studies report an exacerbation of brain injury following activation of the CB system [108].
Dexanabinol (HU‐211), the synthetic noncompetitive NMDA antagonist, exerts long‐term cerebroprotective effects in models of focal cerebral ischemia [109, 110] and combines its NMDA receptor antagonistic activity with free radical‐scavenging abilities to confer neuroprotection [111]. This multiplicity of action supports the potential use of dexanabinol in conditions such as stroke where oxidative stress and excitotoxicity feature. Both CBD and Δ9‐THC are neuroprotective following MCAO [112]. The effects of CBD have been attributed to its potent antioxidant activity and, notably, tolerance does not develop to the neuroprotective actions of CBD. Also, since the actions of CBD are CB1 receptor‐independent, and thus devoid of psychoactivity, CBD is an attractive therapeutic possibility for the treatment of cerebrovascular disorders. CBD is also a ligand at GPR55 [113] and an inhibitor of the nucleoside transporter resulting in an enhancement of adenosine‐mediated antiinflammatory effects [114]. Such non‐CB receptor targets and subsequent antiinflammatory actions may also be of potential benefit in the future. Although no consensus has been reached as to the exact nature of neuroprotective mechanism of endocannabinoids in ischemia, the application of CBs to the treatment of brain injury and stroke is extremely relevant and deserving of further investigations.
Parkinson's Disease
Parkinson's disease (PD) is a degenerative condition affecting dopaminergic neurotransmission in the basal ganglia resulting in hypokinesia. The disease may be precipitated by environmental factors such as pesticides and neuroleptic drugs or mutations in genes encoding several proteins (e.g., α‐synuclein, parkin, PINK1). The disease is associated with the intracellular accumulation of misfolded proteins and Lewy bodies which lead to neurodegeneration. Oxidative stress, excitotoxicity, and neuroinflammation are additional features of the disease, which share commonality with other neurodegenerative conditions. Current therapeutic strategies aim to augment dopaminergic transmission in the basal ganglia through administration of dopamine precursors such as L‐DOPA, however in a proportion of patients the efficacy of the treatment declines over time. The endocannabinoid system may serve as a useful target for the treatment of motor dysfunction since the endocannabinoid system is expressed in the basal ganglia where it regulates neurotransmitter release [115] and motor activity [116]. In PD patients endocannabinoid levels in the cerebrospinal fluid are increased [117] and basal ganglia CB1 receptor mRNA is upregulated in a rodent model of PD. Despite the upregulation of the CB system that is observed at intermediate‐late stages of the disease process, in the presymptomatic phase of the disease CB1 receptors are desensitized [118] which may render the basal ganglia more vulnerable to the cytotoxic milieu of the PD brain and promote excitotoxicity due to the loss of CB1‐mediated presynaptic inhibition of glutamate release [119].
Since CB receptor agonists promote hypokinesia [116], CB1 receptor antagonists are a preferable option for the treatment of PD in order to counter the consequences of an upregulation of the CB system that occurs at the advanced stage of the disease. Preclinical studies have demonstrated that the CB1 receptor antagonist, rimonabant, reduces the hypokinesia in an animal model of PD in a manner that is particularly effective at low dose [120]. Notwithstanding the hypokinetic profile of CB agonists, which is undesirable in the treatment of PD, certain CB agonists have neuroprotective properties which would halt the neurodegenerative aspect to this disease. Δ9‐THC and CBD have been found to protect nigrostriatal dopaminergic neurones from the neurotoxin 6‐hydroxydopamine via a nonreceptor, antioxidant action [121]. The CB‐based medicine, Sativex, which contains both Δ9‐THC and CBD, may therefore be an interesting candidate for future clinical investigation in PD.
CBs and Brain Repair: Implications for Huntington's Disease
Regardless of the diverse etiology, there are common features of neurodegenerative disorders, such as neuroinflammation and oxidative stress, which contribute to the neuronal cell loss. While CB‐based drugs can target these processes to confer neuroprotection, the ability to directly evoke pathways associated with cell survival is another interesting facet of CB action. Neuronal survival is dependent upon the local concentration gradients of growth factors and neuronal viability may be enhanced by augmenting the availability of neurotrophic factors. BDNF is crucial for sustaining neuronal survival and neuronal sensitivity to BDNF is enhanced by the endocannabinoid, 2‐AG, which may represent an important regulator of neural repair [122]. Also, BDNF gene transcription is upregulated by CB1 receptor activation in striatal excitotoxic lesions to rescue striatal neurones from degeneration [123]. That study may be of particular relevance to Huntington's disease where BDNF has been implicated in the pathophysiology of that disease. However, in Huntington's disease there is a reduction in CB1 receptor expression and since CB1 agonists can trigger undesirable psychoactive effects, the search for alternative CB targets for neurodegenerative conditions is gaining momentum. In this regard, the CB2 receptor may be an interesting candidate since the CB2 receptor activation is neuroprotective in models of Huntington's disease by virtue of its ability to reduce microglial activation [124]. An additional physiological process that is crucial for sustaining neural function is the formation of new neurones by neurogenesis and in neurodegenerative conditions such as AD and Huntington's disease neurogenesis is impaired [125, 126]. The age‐related reduction in neurogenesis is reversed by the CB1 and CB2 agonist, WIN55,212–2 [10] and the CB1 receptor has been implicated in neural precursor proliferation and neurogenesis in a model of excitotoxicity [127]. The CB2 receptor has also been identified as a physiological regulator of neural progenitor cell proliferation [128]. CB2 receptor agonists and the pharmacological inhibition of FAAH can stimulate neurogenesis in the adult mouse and since this proneurogenic effect was also seen in aged animals, it has been proposed that the CB system may help to counteract the decline in adult neurogenesis that occurs in aging [129]. Targeting this pathway in the future may help to replenish neurones in neurodegenerative diseases.
Both acute and chronic neurodegeneration occurs upon a platform of neuroinflammation and, as such, the antiinflammatory aspect of CB action may be useful for the treatment of a number of brain disorders. In terms of the antiinflammatory mechanisms of action, Δ9‐THC and CBD inhibit activation of the inflammatory transcription factors, NFκB and STAT1 in a CB receptor‐independent manner [130]. The upregulation of antiinflammatory cytokines, such as IL‐10, is induced by AEA via engagement of CB2 receptor signaling [131]. Since IL‐10 is a negative regulator of microglial activation, the pharmacological modulation of AEA uptake and degradation may prove useful for the treatment of diseases with a neuroinflammatory component.
Outlook
The CB1 receptor is likely to have limited usefulness as a drug target due to its psychoactive activity, together with the fact that the CB1 receptor is downregulated in certain neurodegenerative states and the important physiological role that CB1 plays in processes such as appetite, pain, and cognition. Alternative targets that may be more fruitful include the development of CB2 receptor ligands since this receptor is crucial in regulating neural inflammation and neurogenesis, as well as being devoid of psychoactive side effects. Also, CBD, the nonpsychotropic component of cannabis, is considered to be a promising drug for neurodegenerative disorders due to its antiinflammatory and antioxidant profile [132]. Alternatively, neuroprotection may be achieved by enhancing endogenous CB tone or via allosteric modulation of CB receptors [133, 134].
In summary, the CB system exerts multiplicity of actions, which may be harnessed in the development of new therapeutic approaches to treat neurodegenerative conditions (Figure 2). The antiinflammatory, antioxidant, and proneurogenic properties of the CBs are characteristics that may be relevant to the treatment of a number of neurodegenerative conditions. The existing challenges are to develop drugs that lack psychoactive side effects and, in that regard, targeting the nonpsychotropic CB2 receptor offers particular promise.
Figure 2.
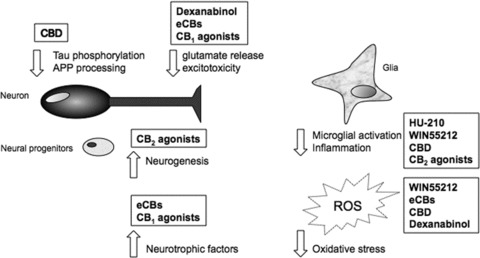
Summary of cannabinoid targets that are relevant to the treatment of neurodegenerative disease. Cannabinoids can limit neurodegenerative processes via a direct influence on neurones to reduce glutamate release and subsequent excitotoxicity, as well as inducing neurotrophic factors and stimulating neurogenesis. Disease‐specific pathology such as amyloid precursor protein (APP) processing and tau hyperphosphorylation in Alzheimer's disease is reduced by cannabinoids. Microglial activation and the production of proinflammatory cytokines is attenuated by cannabinoids and their antioxidant properties also contribute to improving neuronal viability. CBD, cannabidiol; eCBs, endocannabinoids; ROS, reactive oxygen species.
Conflict of Interest
The authors have no conflict of interest.
References
- 1. O'Shaugnessey WB. On the preparation of the Indian hemp, or Gunjah (Cannabis Indica) and their effects on the animal system in health and their utility in the treatment of tetanus and other convulsive disorders. Trans Med Phys Soc Calcutta 1842;8:421–461. [Google Scholar]
- 2. Mechoulam R, Gaoni Y. A total synthesis of Dl‐Delta‐1‐tetrahydrocannabinol, the active constituent of hashish. J Am Chem Soc 1965;87:3273–3275. [DOI] [PubMed] [Google Scholar]
- 3. Sagar DR, Gaw AG, Okine BR, Woodhams SG, Wong A, Kendall DA, Chapman V. Dynamic regulation of the endocannabinoid system: Implications for analgesia. Mol Pain 2009;5:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Butler RK, Rea K, Lang Y, Gavin AM, Finn DP. Endocannabinoid‐mediated enhancement of fear‐conditioned analgesia in rats: Opioid receptor dependency and molecular correlates. Pain 2008;140:491–500. [DOI] [PubMed] [Google Scholar]
- 5. Jesudason D, Wittert G. Endocannabinoid system in food intake and metabolic regulation. Curr Opin Lipidol 2008;19:344–348. [DOI] [PubMed] [Google Scholar]
- 6. Stoving RK, Andries A, Brixen K, Flyvbjerg A, Horder K, Frystyk J. Leptin, ghrelin, and endocannabinoids: Potential therapeutic targets in anorexia nervosa. J Psychiatr Res 2009;43:671–679. [DOI] [PubMed] [Google Scholar]
- 7. Dowie MJ, Bradshaw HB, Howard ML, Nicholson LF, Faull RL, Hannan AJ, Glass M. Altered CB1 receptor and endocannabinoid levels precede motor symptom onset in a transgenic mouse model of Huntington's disease. Neuroscience 2009;163:456–465. [DOI] [PubMed] [Google Scholar]
- 8. Fernandez‐Ruiz J, Lastres‐Becker I, Cabranes A, Gonzalez S, Ramos JA. Endocannabinoids and basal ganglia functionality. Prostaglandins Leukot Essent Fatty Acids 2002;66:257–267. [DOI] [PubMed] [Google Scholar]
- 9. Heifets BD, Castillo PE. Endocannabinoid signaling and long‐term synaptic plasticity. Ann Rev Physiol 2009;71:283–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marchalant Y, Brothers HM, Norman GJ, Karelina K, DeVries AC, Wenk GL. Cannabinoids attenuate the effects of aging upon neuroinflammation and neurogenesis. Neurobiol Dis 2009;34:300–307. [DOI] [PubMed] [Google Scholar]
- 11. Burstein SH, Zurier RB. Cannabinoids, endocannabinoids, and related analogs in inflammation. AAPS J 2009;11:109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Campbell VA, Gowran A. Alzheimer's disease: Taking the edge off with cannabinoids? Br J Pharmacol 2007;152:655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guzman M, Sanchez C, Galve‐Roperh I. Cannabinoids and cell fate. Pharmacol Ther 2002;95:175–184. [DOI] [PubMed] [Google Scholar]
- 14. Guzman M. Neurons on cannabinoids: Dead or alive? Br J Pharmacol 2003;140:439–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Turcott D, Le Dorze J‐A, Esfahani F, Frost E, Gomori A, Namaka M. Examining the roles of cannabinoids in pain and other therapeutic indications: A review. Exp Opinion 2010;11:17–31. [DOI] [PubMed] [Google Scholar]
- 16. Wade DT, Makela P, Robson P, House H, Bateman C. Do cannabis‐based medicinal extracts have general or specific effects on symptoms in multiple sclerosis? A double‐blind, randomized, placebo‐controlled study on 160 patients. Mult Scler 2004;10:434–441. [DOI] [PubMed] [Google Scholar]
- 17. Guzman M, Duarte MJ, Blazquez C, et al A pilot clinical study of Δ9‐tetrahydrocannabinol in patients with recurrent glioblasoma multiforme. Br J Cancer 2006;95:197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J Neurosci 1991;11:563–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Van Sickle MD, Duncan M, Kingsley PJ, et al Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005;310:329–332. [DOI] [PubMed] [Google Scholar]
- 20. Nunez E, Benito C, Tolon RM, Hillard CJ, Griffin WS, Romero J. Glial expression of cannabinoid CB2 receptors and fatty acid amide hydrolase are beta amyloid‐linked events in Down's syndrome. Neuroscience 2008;151:104–110. [DOI] [PubMed] [Google Scholar]
- 21. Thakur GA, Tichkule R, Bajaj S, Makriyannis A. Latest advances in cannabinoid receptor agonists. Expert Opin Ther Pat 2009;19:1647–1673. [DOI] [PubMed] [Google Scholar]
- 22. Brown AJ. Novel cannabinoid receptors. Br J Pharmacol 2007;152:567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sharir H, Abood ME. Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacol Ther 2010;126:301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nevalainen T, Irving AJ. GPR55, a lysophosphatidylinositol receptor with cannabinoid sensitivity? Curr Top Med Chem 2010;10:799–813. [DOI] [PubMed] [Google Scholar]
- 25. McHugh D, Hu SS, Rimmerman N, Juknat A, Vogel Z, Walker JM, Bradshaw HB. N‐arachidononyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci 2010;11:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Costa B, Bettoni I, Petrosino S, Comelli F, Giagnoni G, Di Marzo V. The dual fatty acid amide hydrolase/TRPV1 blocker, arachidonoyl‐serotonin, relieves carrageenan‐induced inflammation and hyperalgesia in mice. Pharmacol Res 2010;61:537–546. [DOI] [PubMed] [Google Scholar]
- 27. Miyamoto N, Tanaka R, Zhang N, et al Crucial role for Ser133‐phosphorylated form of cyclic AMP‐responsive element binding protein signaling in the differentiation and survival of neural progenitors under chronic cerebral hypoperfusion. Neuroscience 2009;162:525–536. [DOI] [PubMed] [Google Scholar]
- 28. Hampson RE, Mu J, Deadwyler SA. Cannabinoid and kappa opioid receptors reduce potassium K+ current via activation of G(s) proteins in cultured hippocampal neurons. J Neurophysiol 2000;84:2356–2364. [DOI] [PubMed] [Google Scholar]
- 29. Mackie K, Devane WA, Hille B. Anandamide, an endogenous cannabinoid, inhibits calcium currents as a partial agonist in N18 neuroblastoma cells. Mol Pharmacol 1993;44:498–503. [PubMed] [Google Scholar]
- 30. Deadwyler SA, Hampson RE, Mu J, Whyte A, Childers S. Cannabinoids modulate voltage sensitive potassium A‐current in hippocampal neurons via a cAMP‐dependent process. J Pharmacol Exp Ther 1995;273:734–743. [PubMed] [Google Scholar]
- 31. Nicholson RA, Liao C, Zheng J, et al Sodium channel inhibition by anandamide and synthetic cannabimimetics in brain. Brain Res 2003;978:194–204. [DOI] [PubMed] [Google Scholar]
- 32. Hillard CJ. Role of cannabinoids and endocannabinoids in cerebral ischemia. Curr Pharm Des 2008;14:2347–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Minati L, Edginton T, Bruzzone MG, Giaccone G. Current concepts in Alzheimer's disease: A multidisciplinary review. Am J Alzheimers Dis Other Demen 2009;24:95–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bayer TA, Buslei R, Havas L, Falkai P. Evidence for activation of microglia in patients with psychiatric illnesses. Neurosci Lett 1999;271:126–128. [DOI] [PubMed] [Google Scholar]
- 35. Heneka MT, O’Banion MK. Inflammatory processes in Alzheimer's disease. J Neuroimmunol 2007;184:69–91. [DOI] [PubMed] [Google Scholar]
- 36. Pratico D. Evidence of oxidative stress in Alzheimer's disease brain and antioxidant therapy: Lights and shadows. Ann N Y Acad Sci 2008;1147:70–78. [DOI] [PubMed] [Google Scholar]
- 37. Riedel G, Davies SN. Cannabinoid function in learning, memory and plasticity. Handb Exp Pharmacol 2005;168:445–477. [DOI] [PubMed] [Google Scholar]
- 38. Ramirez BG, Blazquez C, Gomez del Pulgar T, Guzman M, de Ceballos ML. Prevention of Alzheimer's disease pathology by cannabinoids: Neuroprotection mediated by blockade of microglial activation. J Neurosci 2005;25:1904–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Westlake TM, Howlett AC, Bonner TI, Matsuda LA, Herkenham M. Cannabinoid receptor binding and messenger RNA expression in human brain: An in vitro receptor autoradiography and in situ hybridization histochemistry study of normal aged and Alzheimer's brains. Neuroscience 1994;63:637–652. [DOI] [PubMed] [Google Scholar]
- 40. Benito C, Nunez E, Tolon RM, Carrier EJ, Rabano A, Hillard CJ, Romero J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque‐associated glia in Alzheimer's disease brains. J Neurosci 2003;23:11136–11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Van Der Stelt M, Mazzola C, Esposito G, et al Endocannabinoids and beta‐amyloid‐induced neurotoxicity in vivo: Effect of pharmacological elevation of endocannabinoid levels. Cell Mol Life Sci 2006;63:1410–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nadler V, Mechoulam R, Sokolovsky M. Blockade of 45Ca2+ influx through the N‐methyl‐D‐aspartate receptor ion channel by the non‐psychoactive cannabinoid HU‐211. Brain Res 1993;622:79–85. [DOI] [PubMed] [Google Scholar]
- 43. Eshhar N, Striem S, Biegon A. HU‐211, a non‐psychotropic cannabinoid, rescues cortical neurones from excitatory amino acid toxicity in culture. Neuroreport 1993;5:237–240. [DOI] [PubMed] [Google Scholar]
- 44. Marsicano G, Goodenough S, Monory K, et al CB1 cannabinoid receptors and on‐demand defense against excitotoxicity. Science 2003;302:84–88. [DOI] [PubMed] [Google Scholar]
- 45. Mackie K, Hille B. Cannabinoids inhibit N‐type calcium channels in neuroblastoma‐glioma cells. Proc Natl Acad Sci USA 1992;89:3825–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Twitchell W, Brown S, Mackie K. Cannabinoids inhibit N‐ and P/Q‐type calcium channels in cultured rat hippocampal neurons. J Neurophysiol 1997;78:43–50. [DOI] [PubMed] [Google Scholar]
- 47. Gebremedhin D, Lange AR, Campbell WB, Hillard CJ, Harder DR. Cannabinoid CB1 receptor of cat cerebral arterial muscle functions to inhibit L‐type Ca2+ channel current. Am J Physiol 1999;276(6 Pt 2):H2085–H2093. [DOI] [PubMed] [Google Scholar]
- 48. Zhuang SY, Bridges D, Grigorenko E, McCloud S, Boon A, Hampson RE, Deadwyler SA. Cannabinoids produce neuroprotection by reducing intracellular calcium release from ryanodine‐sensitive stores. Neuropharmacology 2005;48:1086–1096. [DOI] [PubMed] [Google Scholar]
- 49. Khaspekov LG, Brenz Verca MS, Frumkina LE, Hermann H, Marsicano G, Lutz B. Involvement of brain‐derived neurotrophic factor in cannabinoid receptor‐dependent protection against excitotoxicity. Eur J Neurosci 2004;19:1691–1698. [DOI] [PubMed] [Google Scholar]
- 50. Scharfman H, Goodman J, Macleod A, Phani S, Antonelli C, Croll S. Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp Neurol 2005;192:348–356. [DOI] [PubMed] [Google Scholar]
- 51. Peng S, Garzon DJ, Marchese M, et al Decreased brain‐derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer's disease. J Neurosci 2009;29:9321–9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee J, Fukumoto H, Orne J, et al Decreased levels of BDNF protein in Alzheimer temporal cortex are independent of BDNF polymorphisms. Exp Neurol 2005;194:91–96. [DOI] [PubMed] [Google Scholar]
- 53. Akiyama H, Barger S, Barnum S, et al Inflammation and Alzheimer's disease. Neurobiol Aging 2000;21:383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Andersen K, Launer LJ, Ott A, Hoes AW, Breteler MM, Hofman A. Do nonsteroidal anti‐inflammatory drugs decrease the risk for Alzheimer's disease? The Rotterdam Study. Neurology 1995;45:1441–1445. [DOI] [PubMed] [Google Scholar]
- 55. Ehrhart J, Obergon D, Mri T, et al Stimulation of CB2 suppresses microglial activation. J Neuroinflamm 2005;12:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Molina‐Holgado F, Pinteaux E, Moore JD, Molina‐Holgado E, Guaza C, Ribson RM, Rothwell NJ. Endogenous interleukin‐1 receptor antagonist mediates anti‐inflammatory and neuroprotective actions of cannabinoids in neurons and glia. J Neurosci 2003;23:6470–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eubanks LM, Rogers CJ, Beuscher AE, Koob GF, Olson AJ, Dickerson TJ, Janda KD. A molecular link between the active component of marijuana and Alzheimer's disease pathology. Mol Pharm 2006;3:773–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hampson AJ, Grimaldi M, Axelrod J, Wink D. Cannabidiol and (‐)Δ9‐tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci USA 1998;95:8268–8273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Esposito G, Scuderi C, Savani C, et al Cannabidiol in vivo blunts beta‐amyloid induced neuroinflammation by suppressing IL‐1ß and iNOS expression. Br J Pharmacol 2007;151:1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Iuvone T, Esposito G, Esposito R, Santamaria R, Di Rosa M, Izzo AA. Neuroprotective effect of cannabidiol, a non‐psychoactive component from Cannabis sativa, on ß‐amyloid‐induced toxicity in PC12 cells. J Neurochem 2004;89:134–141. [DOI] [PubMed] [Google Scholar]
- 61. Esposito G, De Filippis D, Carnuccio R, Izzo AG, Iuvone T. The marijuana component, cannabidiol, inhibits ß‐amyloid‐induced tau protein hyperphosphorylation through Wnt/ß‐catenin pathway rescue in PC12 cells. J Mol Med 2006;84:253–258. [DOI] [PubMed] [Google Scholar]
- 62. Volicer L, Stelly M, Morris J, McLaughlin J, Volicer BJ. Effects of dronabinol on anorexia and disturbed behavior in patients with Alzheimer's disease. Int J Geriatr Psychiatry 1997;12:913–919. [PubMed] [Google Scholar]
- 63. Walther S, Mahlberg R, Eichmann U, Kunz D. Δ9‐tetrahydrocannabinol for nighttime agitation in severe dementia. Psychopharmacology (Berl) 2006;185:524–528. [DOI] [PubMed] [Google Scholar]
- 64. Krishnan S, Cairns R, Howard R. Cannabinoids for the treatment of dementia. Cochrane Database Syst Rev 2009;CD007204. doi: 10.1002/14651858. CD007204.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Buntinx M, Stinissen P, Steels P, Ameloot M, Raus J. Immune‐mediated oligodendrocyte injury in multiple sclerosis: Molecular mechanisms and therapeutic interventions. Crit Rev Immunol 2002;22:391–424. [PubMed] [Google Scholar]
- 66. Vercellino M, Merola A, Piacentino C, et al Altered glutamate reuptake in relapsing‐remitting secondary progressive multiple sclerosis cortex: Correleation with microglia infiltration, demyelination and neuronal and synaptic damage. J Neuropathol Exp Neurol 2007;66:732–739. [DOI] [PubMed] [Google Scholar]
- 67. Docagne F, Muneton V, Clemente D, et al Excitotoxicity in a chronic model of multiple sclerosis: Neuroprotective effects of cannabinoids through CB1 and CB2 receptor activation. Mol Cell Neurosci 2007;34:551–561. [DOI] [PubMed] [Google Scholar]
- 68. Lakhan SE, Rowland M. Whole plant cannabis extracts in the treatment of spasticity in multiple sclerosis: A systematic review. BMC Neurol 2009;9:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rog DJ, Nurmikko TJ, Young CA. Oromucosal Δ9‐tetrahydrocannabinol/cannabidiol for neuropathic pain associated with multiple sclerosis: An uncontrolled, open‐label, 2‐year extension trial. Clin Ther 2007;29:2068–2079. [DOI] [PubMed] [Google Scholar]
- 70. Zajicek J, Fox P, Sanders H, Wright DE, Vickery PJ, Nunn AJ, Thompson AJ. Cannabinoids for treatment of spasticity and other symptoms related to multiple sclerosis (CAMS study): Multicentre randomised placebo‐controlled trial. Lancet 2003;362:1517–1526. [DOI] [PubMed] [Google Scholar]
- 71. Iskedjian M, Bereza B, Gordon A, Piwko C, Einarson TR. Meta‐analysis of cannabis based treatments for neuropathic and multiple sclerosis‐related pain. Curr Med Res Opin 2007;23:17–24. [DOI] [PubMed] [Google Scholar]
- 72. Rea K, Roche M, Finn DP. Supraspinal modulation of pain by cannabinoids: The role of GABA and glutamate. Br J Pharmacol 2007;152:633–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cassan C, Liblau RS. Immune tolerance and control of CNS autoimmunity: From animal models to MS patients. J Neurochem 2007;100:883–892. [DOI] [PubMed] [Google Scholar]
- 74. Skunric DS. Experimental models of relapsin‐remitting multiple sclerosis: Current concepts and perspective. Curr Neurovasc Res 2005;2:349–362. [DOI] [PubMed] [Google Scholar]
- 75. Jean‐Gilles L, Feng S, Tench CR, Chapman V, Kendall DA, Barrett DA, Constantinescu CS. Plasma endocannabinoid levels in multiple sclerosis. J Neurol Sci 2009;287:212–215. [DOI] [PubMed] [Google Scholar]
- 76. Centonze D, Bari M, Rossi S, et al The endocannabinoid system is dysregulated in multiple sclerosis and in experimental autoimmune encephalomyelitis. Brain 2007;130:2543–2553. [DOI] [PubMed] [Google Scholar]
- 77. Galiegue S, Mary S, Merchand J, et al Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem 1995;232:54–61. [DOI] [PubMed] [Google Scholar]
- 78. Achiron A, Miron S, Lavie V, Margalit R, Biegon A. Dexanabinol (HU‐211) effect on experimental autoimmune encephalomyelitis: Implications for the treatment of acute relapses of multiple sclerosis. J Neuroimmunol 2000;102:26–31. [DOI] [PubMed] [Google Scholar]
- 79. Croxford JL, Miller SD. Immunoregulation of a viral model of multiple sclerosis using the synthetic cannabinoid R+WIN55,212. J Clin Invest 2003;111:1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Maresz K, Pryce G, Ponmarev ED, et al Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nat Med 2007;13:492–497. [DOI] [PubMed] [Google Scholar]
- 81. Yiangou Y, Facer P, Durrenberger P. COX‐2, CB2 and P2X7‐ immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol 2006;6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pryce G, Ahmed Z, Hankey DJ, et al Cannabinoids inhibit neurodegeneration in models of multiple sclerosis. Brain 2003;126:2191–2202. [DOI] [PubMed] [Google Scholar]
- 83. Marsican G, Moosmann B, Herman H, Lutz B, Behl C. Neuroprotective properties of cannabinoids against oxidative stress: Role of the cannabinoid receptor CB1. J Neurochem 2002;80:448–456. [DOI] [PubMed] [Google Scholar]
- 84. Croxford JL, Pryce G, Jackson SJ, et al Cannabinoid‐mediated neuroprotection, not immunosuppression, may be more relevant to multiple sclerosis. J Neuroimmunol 2008;193:120–129. [DOI] [PubMed] [Google Scholar]
- 85. Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med 2000;6:67–70. [DOI] [PubMed] [Google Scholar]
- 86. Hoyte L, Barber PA, Buchan AM, Hill MD. The rise and fall of NMDA antagonists for ischemic stroke. Curr Mol Med 2004;4:131–136. [DOI] [PubMed] [Google Scholar]
- 87. Loria F, Petrosino S, Hernangómez M, et al An endocannabinoid tone limits excitotoxicity in vitro and in a model of multiple sclerosis. Neurobiol Dis 2010;37:166–176. [DOI] [PubMed] [Google Scholar]
- 88. Mechoulam R, Panikashvili D, Shohami E. Cannabinoids and brain injury: Therapeutic implications. Trends Mol Med 2002;8:58–61. [DOI] [PubMed] [Google Scholar]
- 89. Hansen HH, Schmid PC, Bittigau P, et al Anandamide, but not 2‐arachidonoylglycerol, accumulates during in vivo neurodegeneration. J Neurochem 2001;78:1415–1427. [DOI] [PubMed] [Google Scholar]
- 90. Panikashvili D, Simeonidou C, Ben‐Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E. An endogenous cannabinoid (2‐AG) is neuroprotective after brain injury. Nature 2001;413:527–531. [DOI] [PubMed] [Google Scholar]
- 91. Schmid HH, Schmid PC, Natarajan V. The N‐acylation‐phosphodiesterase pathway and cell signalling. Chem Phys Lipids 1996;80:133–142. [DOI] [PubMed] [Google Scholar]
- 92. Berger C, Schmid PC, Schabitz WR, Wolf M, Schwab S, Schmid HH. Massive accumulation of N‐acylethanolamines after stroke. Cell signalling in acute cerebral ischemia? J Neurochem 2004;88:1159–1167. [DOI] [PubMed] [Google Scholar]
- 93. Amantea D, Spagnuolo P, Bari M, et al Modulation of the endocannabinoid system by focal brain ischemia in the rat is involved in neuroprotection afforded by 17beta‐estradiol. FEBS J 2007;274:4464–4775. [DOI] [PubMed] [Google Scholar]
- 94. Franklin A, Parmentier‐Batteur S, Walter L, Greenberg DA, Stella N. Palmitoylethanolamide increases after focal cerebral ischemia and potentiates microglial cell motility. J Neurosci 2003;23:7767–7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Schabitz WR, Giuffrida A, Berger C, Aschoff A, Schwaninger M, Schwab S, Piomelli D. Release of fatty acid amides in a patient with hemispheric stroke: A microdialysis study. Stroke 2002;33:2112–2114. [DOI] [PubMed] [Google Scholar]
- 96. Sugiura T, Yoshinaga N, Kondo S, Waku K, Ishima Y. Generation of 2‐arachidonoylglycerol, an endogenous cannabinoid receptor ligand, in picrotoxin‐administered rat brain. Biochem Biophys Res Commun 2000;271:654–658. [DOI] [PubMed] [Google Scholar]
- 97. Muthian S, Rademacher DJ, Roelke CT, Gross GJ, Hillard CJ. Anandamide content is increased and CB1 cannabinoid receptor blockade is protective during transient, focal cerebral ischemia. Neuroscience 2004;129:743–750. [DOI] [PubMed] [Google Scholar]
- 98. Jin KL, Mao XO, Goldsmith PC, Greenberg, DA . CB1 cannabinoid receptor induction in experimental stroke. Ann Neurol 2000;48:257–261. [PubMed] [Google Scholar]
- 99. Zhang M, Martin BR, Adler MW, Razdan RK, Ganea D, Tuma RF. Modulation of the balance between cannabinoid CB(1) and CB(2) receptor activation during cerebral ischemic/reperfusion injury. Neuroscience 2008;152:753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ashton JC, Rahman RM, Nair SM, Sutherland BA, Glass M, Appleton I. Cerebral hypoxia‐ischemia and middle cerebral artery occlusion induce expression of the cannabinoid CB2 receptor in the brain. Neurosci Lett 2007;412:114–117. [DOI] [PubMed] [Google Scholar]
- 101. Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, Greenberg DA. Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci 1999;19:2987–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Parmentier‐Batteur S, Jin K, Mao XO, Xie L, Greenberg DA. Increased severity of stroke in CB1 cannabinoid receptor knock‐out mice. J Neurosci 2002;22:9771–9775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Alvarez FJ, Lafuente H, Rey‐Santano MC, et al Neuroprotective effects of the nonpsychoactive cannabinoid cannabidiol in hypoxic‐ischemic newborn piglets. Pediatr Res 2008;64:653–658. [DOI] [PubMed] [Google Scholar]
- 104. Zhang M, Martin BR, Adler MW, Razdan RJ, Kong W, Ganea D, Tuma RF. Modulation of cannabinoid receptor activation as a neuroprotective strategy for EAE and stroke. J Neuroimmun Pharmacol 2009;4:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Degn M, Lambertsen KL, Petersen G, et al Changes in brain levels of N‐acylethanolamines and 2‐arachidonoylglycerol in focal cerebral ischemia in mice. J Neurochem 2007;103:1907–1916. [DOI] [PubMed] [Google Scholar]
- 106. Hansen HH, Azcoitia I, Pons S, et al Blockade of cannabinoid CB(1) receptor function protects against in vivo disseminating brain damage following NMDA‐induced excitotoxicity. J Neurochem 2002;82:154–158. [DOI] [PubMed] [Google Scholar]
- 107. Louw DF, Yang FW, Sutherland GR. The effect of Δ9‐tetrahydrocannabinol on forebrain ischemia in rat. Brain Res 2000;857:183–187. [DOI] [PubMed] [Google Scholar]
- 108. Mathew RJ, Wilson WH, Davis R. Postural syncope after marijuana: A transcranial Doppler study of the hemodynamics. Pharmacol Biochem Behav 2003;75:309–318. [DOI] [PubMed] [Google Scholar]
- 109. Lavie G, Teichner A, Shohami E, Ovadia H, Leker RR. Long term cerebroprotective effects of dexanabinol in a model of focal cerebral ischemia. Brain Res 2001;901:195–201. [DOI] [PubMed] [Google Scholar]
- 110. Durmaz R, Oxden H, Kanbak G, Aral E, Arslan OC, Kartkaya K, Uzuner K. The protective effect of dexanabinol (HU‐211) on nitric oxide and cysteine protease‐mediated neuronal death in focal cerebral ischemia. Neurochem Res 2008;33:1683–1691. [DOI] [PubMed] [Google Scholar]
- 111. Eshhar N, Striem S, Kohen R, Tirosh O, Biegon A. Neuroprotective and antioxidant activities of HU‐211, a novel NMDA receptor antagonist. Eur J Pharmacol 1995;283:19–29. [DOI] [PubMed] [Google Scholar]
- 112. Hayakawa K, Mishia K, Nozako M, et al Repeated treatment with cannabidiol but not Delta9‐tetrahydrocannabinol has a neuroprotective effect without the development of tolerance. Neuropharmacol 2007;52:1079–1087. [DOI] [PubMed] [Google Scholar]
- 113. Ryberg E, Larsson N, Sjogren S, et al The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 2007;152:1092–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Carrier EJ, Auchampach JA, Hillard CJ. Inhibition of an equilibrative nucleoside transporter by cannabidiol: A mechanism of cannabinoid immunosuppression. Proc Natl Acad Sci USA 2006;103:7895–7900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Köfalvi A, Rodrigues RJ, Ledent C, Mackie K, Vizi ES, Cunha RA, Sperlágh B. Involvement of cannabinoid receptors in the regulation of neurotransmitter release in the rodent striatum: A combined immunochemical and pharmacological analysis. J Neurochem 2005;25:2874–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Fernandez‐Ruiz J. The endocannabinoid system as a target for the treatment of motor dysfunction. Br J Pharmacol 2009;156:1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Pisani A, Fezza F, Galati S, et al High endogenous cannabinoid levels in cerebrospinal fluid of untreated Parkinson's disease patients. Ann Neurol 2005;57:777–779. [DOI] [PubMed] [Google Scholar]
- 118. Garcia‐Arencibia M, Garcia C, Kurz A, et al Cannabinoid CB1 receptors are early downregulated followed by a further upregulation in the basal ganglia of mice with deletion of PARK genes. J Neural Transm 2009;73:269–275. [DOI] [PubMed] [Google Scholar]
- 119. Van Der Stelt M, Veldhuis WB, Maccarrone M, et al Acute neuronal injury, excitotoxicity and the endocannabinoid system. Mol Neurobiol 2002;26:317–346. [DOI] [PubMed] [Google Scholar]
- 120. Gonzalez S, Scorticati C, Garcia‐Arencibia M, de Miguel R, Ramos JA, Fernandez‐Ruiz J. Effects of rimonabant, a selective cannabinoid CB1 receptor antagonist, in a rat model of Parkinson's disease. Brain Res 2006;1073:209–219. [DOI] [PubMed] [Google Scholar]
- 121. Lastres‐Becker I, Molina‐Holgado F, Ramos JA, Mechoulam R, Fernandez‐Ruiz J. Cannabinoids provide neuroprotection against 6‐hydroxydopamine toxicity in vivo and in vitro: Relevance to Parkinson's disease. Neurobiol Dis 2005;19:97–107. [DOI] [PubMed] [Google Scholar]
- 122. Maison P, Walker DJ, Walsh FS, Williams G, Doherty P. BDNF regulates neuronal sensitivity to endocannabinoids. Neurosci Lett 2009;467:90–94. [DOI] [PubMed] [Google Scholar]
- 123. De March Z, Zuccato C, Giampá C, et al Cortical expression of brain derived neurotrophic factor and type‐1 cannabinoid receptor after striatal excitotoxic lesions. Neuroscience 2008;152:734–740. [DOI] [PubMed] [Google Scholar]
- 124. Palazuelos J, Aguado T, Pazos MR, et al Microglial CB2 cannabinoid receptors are neuroprotective in Huntington's disease excitotoxicity. Brain 2009;132:3152–3164. [DOI] [PubMed] [Google Scholar]
- 125. Crews L, Rockenstein E, Marliah E. APP transgenic modeling of Alzheimer's disease: Mechanisms of neurodegeneration and aberrant neurogenesis. Brain Struct Funct 2010;214:111–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Molero AE, Gokhan S, Gonzalez S, Feig JL, Alexandre LC, Mehler MF. Impairment of developmental stem cell‐mediated striatal neurogenesis and pluripotency genes in a knock‐in model of Huntington's disease. Proc Natl Acad Sci USA 2009;106:21900–21905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Aguado T, Romero E, Monory K, et al The CB1 cannabinoid receptor mediates excitotoxicity‐induced neural progenitor proliferation and neurogenesis. J Biol Chem 2007;282:23892–23898. [DOI] [PubMed] [Google Scholar]
- 128. Palazuelos J, Aguado T, Egia A, Mechoulam R, Guzmán M, Galve‐Roperh I. Non‐Psychoactive CB2 cannabinoid agonists stimulate neural progenitor proliferation. FASEB J 2006;20:2405–2407. [DOI] [PubMed] [Google Scholar]
- 129. Goncalves MB, Suetterlin P, Yip P, et al A diacylglycerol lipase‐CB2 cannabinoid pathway regulates adult subventricular zone neurogenesis in an age‐dependent manner. Mol Cell Neurosci 2008;38:526–536. [DOI] [PubMed] [Google Scholar]
- 130. Kozela E, Pietr M, Juknat A, Rimmerman N, Levy R, Vogel Z. Cannabinoids Delta (9)‐tetrahydrocannabinol and cannabidiol differentially inhibit the lipopolysaccharide‐activated NF‐kappaB and interferon‐beta/STAT proinflammatory pathways in BV‐2 microglial cells. J Biol Chem 2010;285:1616–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Correa F, Hernangómez M, Mestre L, et al Anandamide enhances IL‐10 production in activated microglia by targeting CB(2) receptors: Roles of ERK1/2, JNK, and NF‐kappaB. Glia 2010;58:135–147. [DOI] [PubMed] [Google Scholar]
- 132. Iuvone T, Esposito G, De Filippis D, Scuderi C, Steardo L. Cannabidiol: A promising drug for neurodegenerative disorders? CNS Neurosci Ther 2009;15:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Hwang J, Adamson C, Butler D, Janero DR, Makriyannis A, Bahr BA. Enhancement of endocannabinoid signaling by fatty acid amide hydrolase inhibition: A neuroprotective therapeutic modality. Life Sci 2010;86:615–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Pertwee RG. The therapeutic potential of drugs that target cannabinoid receptors or modulate the tissue levels or actions of endocannabinoids. AAPS J 2005;7:625–654. [DOI] [PMC free article] [PubMed] [Google Scholar]